

Familial Hypercholesterolemia Screening in a Cardiac Rehabilitation Program After Myocardial Infarction

 ,
,  , , , , , , , and
, , , , , , , and Highlights
- Simplified Dutch Lipid Clinic Network Scores (sDLCNS) for familial hypercholesterolemia (FH) were calculated in a prospective cohort of 245 patients studied in a Cardiac Rehabilitation Program (CRP) after myocardial infarction (MI) occurance.
- A third of the cohort was categorized with likelihoods rated as “possible” (n = 72, 29.4%) and “probable” (n = 11, 4.5%) for FH by sDLCNS.
- The rate of genetic testing for FH is low (n = 4, 1.6%), even in this high-risk subgroup of patients.
- Strategies to improve screening for FH should be prospectively implemented during CRP after MI.
- The implications of these strategies in enhancing lipid control in secondary prevention and cascade screening in first-degree relatives should be further studied.
Abstract
1. Introduction
2. Materials and Methods
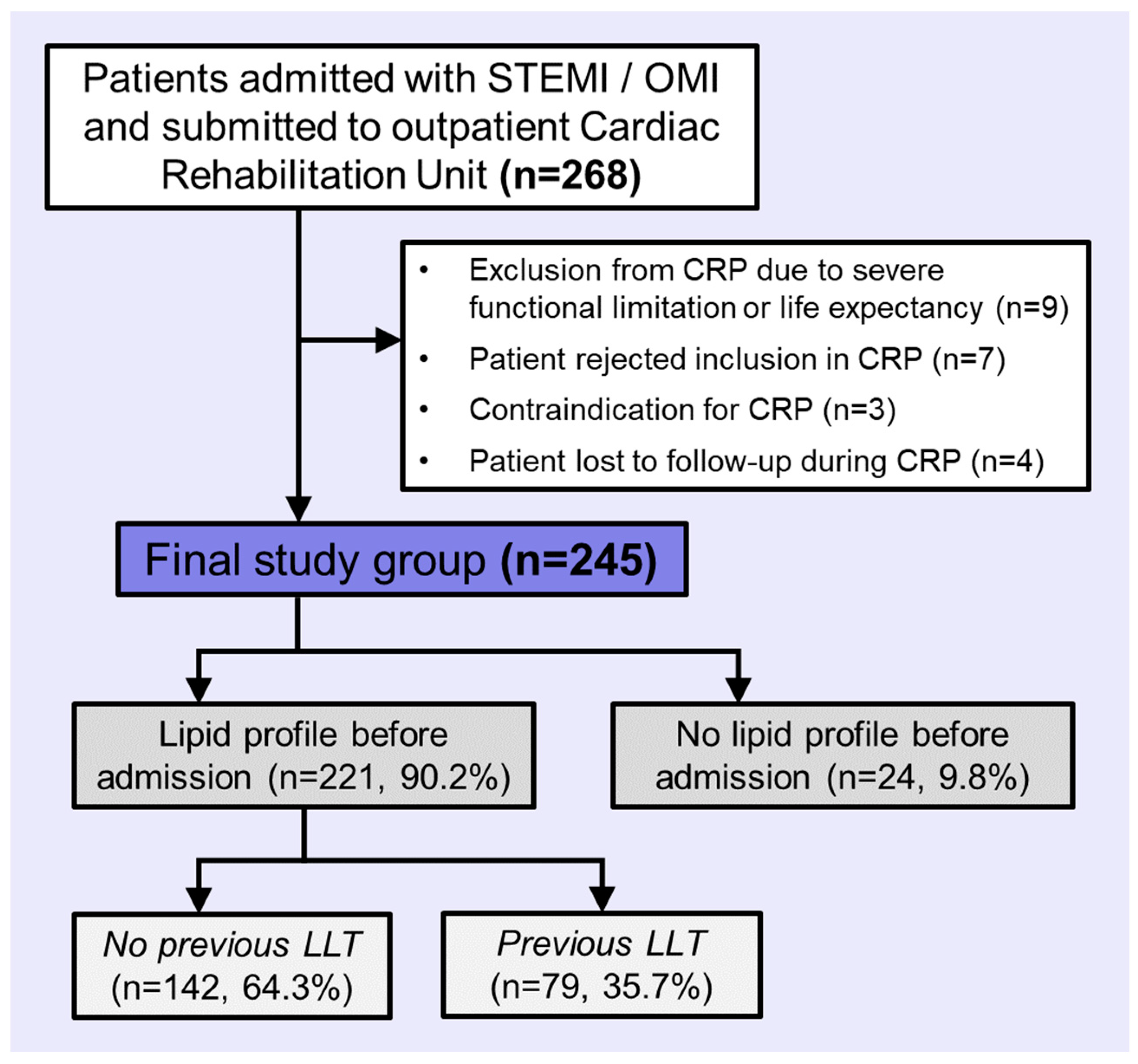
2.1. Population
2.2. Lipid Profile and Basal LDL-C
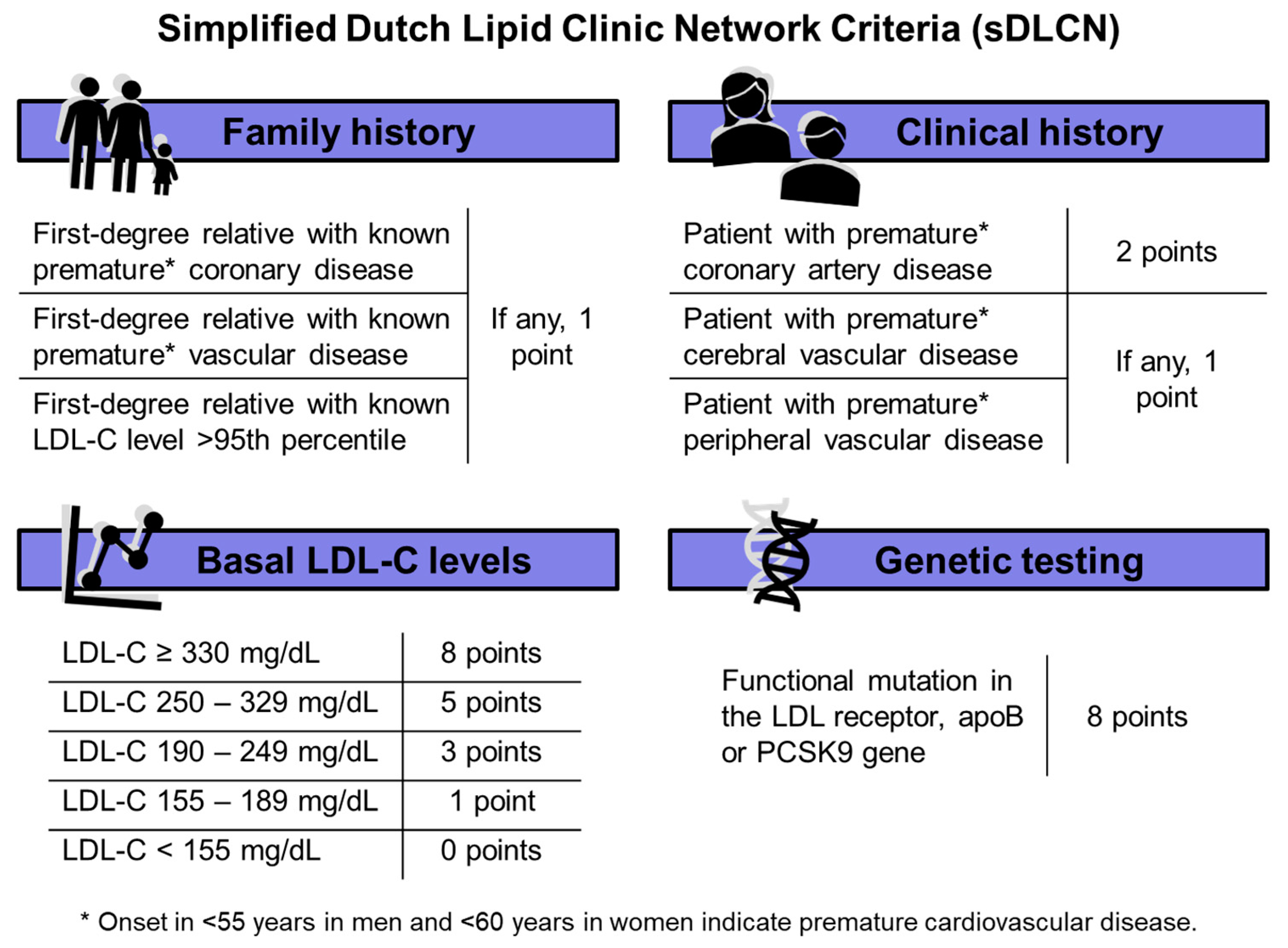
2.3. Screening for Familial Hypercholesterolemia
2.4. Genetic Testing for Familial Hypercholesterolemia
2.5. Ethics
2.6. Statistical Analysis
3. Results
3.1. Cohort Description
3.2. Screening for FH
3.3. Genetic Testing for FH
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sniderman, A.D.; Glavinovic, T.; Thanassoulis, G. Key Questions About Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2022, 79, 1023–1031. [Google Scholar] [CrossRef]
- Fularski, P.; Hajdys, J.; Majchrowicz, G.; Stabrawa, M.; Młynarska, E.; Rysz, J.; Franczyk, B. Unveiling Familial Hypercholesterolemia—Review, Cardiovascular Complications, Lipid-Lowering Treatment and Its Efficacy. Int. J. Mol. Sci. 2024, 25, 1637. [Google Scholar] [CrossRef] [PubMed]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Hauguel-Moreau, M.; Aïdan, V.; Hergault, H.; Beauchet, A.; Pépin, M.; Prati, G.; Pillière, R.; Ouadahi, M.; Josseran, L.; Rodon, C.; et al. Prevalence of Familial Hypercholesterolaemia in Patients Presenting with Premature Acute Coronary Syndrome. Arch. Cardiovasc. Dis. 2022, 115, 87–95. [Google Scholar] [CrossRef]
- Singh, A.; Gupta, A.; Collins, B.L.; Qamar, A.; Monda, K.L.; Biery, D.; Lopez, J.A.G.; De Ferranti, S.D.; Plutzky, J.; Cannon, C.P.; et al. Familial Hypercholesterolemia Among Young Adults with Myocardial Infarction. J. Am. Coll. Cardiol. 2019, 73, 2439–2450. [Google Scholar] [CrossRef]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Wang, C.; Yu, P.; Hu, L.; Liang, M.; Mao, Y.; Zeng, Q.; Wang, X.; Huang, K.; Yan, J.; Xie, L.; et al. Prevalence and Prognosis of Molecularly Defined Familial Hypercholesterolemia in Patients with Acute Coronary Syndrome. Front. Cardiovasc. Med. 2022, 9, 921803. [Google Scholar] [CrossRef] [PubMed]
- Umans-Eckenhausen, M.A.; Defesche, J.C.; Sijbrands, E.J.; Scheerder, R.L.; Kastelein, J.J. Review of First 5 Years of Screening for Familial Hypercholesterolaemia in the Netherlands. Lancet 2001, 357, 165–168. [Google Scholar] [CrossRef]
- Hadfield, S.G.; Horara, S.; Starr, B.J.; Yazdgerdi, S.; Marks, D.; Bhatnagar, D.; Cramb, R.; Egan, S.; Everdell, R.; Ferns, G.; et al. Family Tracing to Identify Patients with Familial Hypercholesterolaemia: The Second Audit of the Department of Health Familial Hypercholesterolaemia Cascade Testing Project. Ann. Clin. Biochem. Int. J. Lab. Med. 2009, 46, 24–32. [Google Scholar] [CrossRef]
- Séguro, F.; Bongard, V.; Bérard, E.; Taraszkiewicz, D.; Ruidavets, J.-B.; Ferrières, J. Dutch Lipid Clinic Network Low-Density Lipoprotein Cholesterol Criteria Are Associated with Long-Term Mortality in the General Population. Arch. Cardiovasc. Dis. 2015, 108, 511–518. [Google Scholar] [CrossRef]
- Ruel, I.; Brisson, D.; Aljenedil, S.; Awan, Z.; Baass, A.; Bélanger, A.; Bergeron, J.; Bewick, D.; Brophy, J.M.; Brunham, L.R.; et al. Simplified Canadian Definition for Familial Hypercholesterolemia. Can. J. Cardiol. 2018, 34, 1210–1214. [Google Scholar] [CrossRef]
- McGowan, M.P.; Hosseini Dehkordi, S.H.; Moriarty, P.M.; Duell, P.B. Diagnosis and Treatment of Heterozygous Familial Hypercholesterolemia. J. Am. Heart Assoc. 2019, 8, e013225. [Google Scholar] [CrossRef]
- Bellows, B.K.; Khera, A.V.; Zhang, Y.; Ruiz-Negrón, N.; Stoddard, H.M.; Wong, J.B.; Kazi, D.S.; De Ferranti, S.D.; Moran, A.E. Estimated Yield of Screening for Heterozygous Familial Hypercholesterolemia With and Without Genetic Testing in US Adults. J. Am. Heart Assoc. 2022, 11, e025192. [Google Scholar] [CrossRef]
- Taylor, R.S.; Dalal, H.M.; McDonagh, S.T.J. The Role of Cardiac Rehabilitation in Improving Cardiovascular Outcomes. Nat. Rev. Cardiol. 2021, 19, 1–15. [Google Scholar] [CrossRef]
- Lee, W.-J.; Chuang, H.-N.; Hsiao, T.-H.; Lee, W.-L.; Wu, J.-P.; Sheu, W.H.-H.; Liang, K.-W. Prevalence and Prognosis of Genetically Proven Familial Hypercholesterolemia in Subjects with Coronary Artery Disease and Reduced Ejection Fraction. Sci. Rep. 2023, 13, 16942. [Google Scholar] [CrossRef]
- Marcos-Garcés, V.; Merenciano-González, H.; Martínez Mas, M.L.; Palau, P.; Climent Alberola, J.I.; Perez, N.; López-Bueno, L.; Esteban Argente, M.C.; Valls Reig, M.; Muñoz Alcover, R.; et al. Short-Course High-Intensity Statin Treatment during Admission for Myocardial Infarction and LDL-Cholesterol Reduction—Impact on Tailored Lipid-Lowering Therapy at Discharge. J. Clin. Med. 2024, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.T.; Sharabiani, M.T.A.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef]
- Humphries, S.E.; Cooper, J.A.; Seed, M.; Capps, N.; Durrington, P.N.; Jones, B.; McDowell, I.F.W.; Soran, H.; Neil, H.A.W. Coronary Heart Disease Mortality in Treated Familial Hypercholesterolaemia: Update of the UK Simon Broome FH Register. Atherosclerosis 2018, 274, 41–46. [Google Scholar] [CrossRef]
- Williams, R.R.; Hunt, S.C.; Schumacher, M.C.; Hegele, R.A.; Leppert, M.F.; Ludwig, E.H.; Hopkins, P.N. Diagnosing Heterozygous Familial Hypercholesterolemia Using New Practical Criteria Validated by Molecular Genetics. Am. J. Cardiol. 1993, 72, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Wang, M.; Patel, A.P.; Ajufo, E.; Maamari, D.J.; Aragam, K.G.; Brockman, D.G.; Vosburg, T.; Ellinor, P.T.; Ng, K.; et al. Association of the Interaction Between Familial Hypercholesterolemia Variants and Adherence to a Healthy Lifestyle With Risk of Coronary Artery Disease. JAMA Netw. Open 2022, 5, e222687. [Google Scholar] [CrossRef]
- Mszar, R.; Mszar, E. Familial Hypercholesterolemia and Our Family’s Heart History: From Atherosclerosis and Angina to Awareness and Advocacy. Circ. Cardiovasc. Qual. Outcomes 2024, 17, e010864. [Google Scholar] [CrossRef] [PubMed]
- Mirzai, S.; Chevli, P.A.; Rikhi, R.; Shapiro, M.D. Familial Hypercholesterolemia: From Clinical Suspicion to Novel Treatments. Rev. Cardiovasc. Med. 2023, 24, 311. [Google Scholar] [CrossRef]
- Campbell-Salome, G.; Walters, N.L.; Ladd, I.G.; Sheldon, A.; Ahmed, C.D.; Brangan, A.; McMinn, M.N.; Rahm, A.K.; Schwartz, M.L.B.; Tricou, E.; et al. Motivating Cascade Testing for Familial Hypercholesterolemia: Applying the Extended Parallel Process Model for Clinician Communication. Transl. Behav. Med. 2022, 12, 800–809. [Google Scholar] [CrossRef]
- Marquina, C.; Morton, J.I.; Lloyd, M.; Abushanab, D.; Baek, Y.; Abebe, T.; Livori, A.; Dahal, P.; Watts, G.F.; Ademi, Z. Cost-Effectiveness of Screening Strategies for Familial Hypercholesterolaemia: An Updated Systematic Review. PharmacoEconomics 2024, 42, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Ademi, Z.; Watts, G.F.; Pang, J.; Sijbrands, E.J.G.; Van Bockxmeer, F.M.; O’Leary, P.; Geelhoed, E.; Liew, D. Cascade Screening Based on Genetic Testing is Cost-Effective: Evidence for the Implementation of Models of Care for Familial Hypercholesterolemia. J. Clin. Lipidol. 2014, 8, 390–400. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.-H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; Van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef]
- Amor-Salamanca, A.; Castillo, S.; Gonzalez-Vioque, E.; Dominguez, F.; Quintana, L.; Lluís-Ganella, C.; Escudier, J.M.; Ortega, J.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Genetically Confirmed Familial Hypercholesterolemia in Patients with Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2017, 70, 1732–1740. [Google Scholar] [CrossRef]
- Ahmad, Z.S.; Andersen, R.L.; Andersen, L.H.; O’Brien, E.C.; Kindt, I.; Shrader, P.; Vasandani, C.; Newman, C.B.; deGoma, E.M.; Baum, S.J.; et al. US Physician Practices for Diagnosing Familial Hypercholesterolemia: Data from the CASCADE-FH Registry. J. Clin. Lipidol. 2016, 10, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial Hypercholesterolaemia is Underdiagnosed and Undertreated in the General Population: Guidance for Clinicians to Prevent Coronary Heart Disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef]
- Ibrahim, S.; Nurmohamed, N.S.; Nierman, M.C.; De Goeij, J.N.; Zuurbier, L.; Van Rooij, J.; Schonck, W.A.M.; De Vries, J.; Hovingh, G.K.; Reeskamp, L.F.; et al. Enhanced Identification of Familial Hypercholesterolemia Using Central Laboratory Algorithms. Atherosclerosis 2024, 393, 117548. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| All Cohort (n = 245) | No Genetic Testing (n = 241) | Genetic Testing (n = 4) | p-Value | ||
|---|---|---|---|---|---|
| Clinical variables | |||||
| Age (years) | 62.19 ± 13.93 | 62.23 ± 14.03 | 59.51 ± 6.33 | 0.7 | |
| Male sex (%) | 200 (81.6) | 197 (81.7) | 3 (75) | 0.73 | |
| Hypercholesterolemia (%) | 219 (89.4) | 215 (89.2) | 4 (100) | 0.49 | |
| Hypertension (%) | 139 (56.7) | 137 (56.8) | 2 (50) | 0.78 | |
| Diabetes mellitus (%) | 58 (23.7) | 58 (24.1) | 0 (0) | 0.26 | |
| Smoking habit (%) | 121 (49.8) | ||||
| Killip class | I (%) | 173 (70.6) | 169 (70.1) | 4 (100) | 0.64 |
| II (%) | 55 (22.4) | 55 (22.8) | 0 (0) | ||
| III (%) | 7 (2.9) | 7 (2.9) | 0 (0) | ||
| IV (%) | 10 (4.1) | 10 (4.1) | 0 (0) | ||
| GRACE risk score | 119.09 ± 29.45 | 119.23 ± 29.61 | 110.25 ± 16.32 | 0.55 | |
| Infarct location | Anterior (%) | 105 (42.9) | 103 (42.7) | 2 (50) | 0.9 |
| Inferior (%) | 109 (44.5) | 107 (44.4) | 2 (50) | ||
| Lateral (%) | 15 (6.1) | 15 (6.2) | 0 (0) | ||
| OMI (%) | 16 (6.5) | 4 (6.6) | 0 (0) | ||
| LVEF (%) | 51.77 ± 10.58 | 51.72 ± 10.49 | 54.75 ± 17.21 | 0.57 | |
| LVEF <50% (%) | 93 (38) | 92 (38.2) | 1 (25) | 0.59 | |
| Previous chronic coronary syndrome (%) | 21 (8.6) | 21 (8.7) | 0 (0) | 0.54 | |
| Peripheral artery disease (%) | 19 (7.8) | 19 (7.9) | 0 (0) | 0.56 | |
| Previous cerebral vascular disease (%) | 10 (5.1) | 10 (5.2) | 0 (0) | 0.69 | |
| Lipid and metabolic profile before admission * | |||||
| Fasting blood glucose (mg/dL) | 100.95 ± 26.41 | 101.08 ± 26.63 | 93.75 ± 3.5 | 0.58 | |
| Total cholesterol (mg/dL) | 200.84 ± 55.19 | 199.54 ± 54.38 | 272 ± 60.16 | 0.009 | |
| Triglycerides (mg/dL) | 147.47 ± 83.74 | 147.55 ± 84.07 | 142.75 ± 72.68 | 0.91 | |
| HDL-C (mg/dL) | 46.66 ± 11.9 | 46.38 ± 11.73 | 61.75 ± 12.95 | 0.01 | |
| LDL-C (mg/dL) | 131.79 ± 45.34 | 130.84 ± 44.88 | 184.25 ± 44.91 | 0.02 | |
| HbA1c (%) | 6.32 ± 1.65 | 6.34 ± 1.65 | - | - | |
| Lipid and metabolic profile during admission # | |||||
| Fasting blood glucose (mg/dL) | 116.8 ± 39.6 | 117.08 ± 39.79 | 100.5 ± 24.24 | 0.41 | |
| Total cholesterol (mg/dL) | 162.38 ± 43.23 | 161.3 ± 42.17 | 225.25 ± 64.32 | 0.003 | |
| Triglycerides (mg/dL) | 134.38 ± 59.91 | 134.21 ± 59.84 | 143.75 ± 72.54 | 0.75 | |
| HDL-C (mg/dL) | 38.64 ± 10.26 | 38.39 ± 10.1 | 53 ± 10.68 | 0.005 | |
| LDL-C (mg/dL) | 101.68 ± 36.6 | 100.93 ± 35.97 | 145 ± 52.31 | 0.02 | |
| HbA1c (%) | 6.11 ± 0.99 | 6.12 ± 1 | 5.4 ± 0.41 | 0.15 | |
| Lipoprotein (a) (mg/dL) | 49.06 ± 47.16 | 48.82 ± 47 | 63 ± 61.56 | 0.55 | |
| Basal LDL-C levels (mg/dL) | 162.87 ± 44.17 | 161.71 ± 43.25 | 233 ± 49.09 | 0.001 | |
| Points in sDLCNS | Prevalence in the Cohort (%) | |
|---|---|---|
| Family history | ||
| First-degree relative with known premature * coronary disease | If any, 1 point | 17 (6.9) |
| First-degree relative with known premature * vascular disease | 2 (0.8) | |
| First-degree relative with known LDL-C level >95th percentile | 2 (0.8) | |
| Clinical history | ||
| Patient with premature * coronary artery disease | 2 points | 72 (29.4) |
| Patient with premature * cerebral vascular disease | If any, 1 point | 5 (2) |
| Patient with premature * peripheral vascular disease | 6 (2.4) | |
| Basal LDL-C levels mg/dL | ||
| LDL-C ≥ 330 mg/dL | 8 points | 0 (0) |
| LDL-C 250–329 mg/dL | 5 points | 9 (3.7) |
| LDL-C 190–249 mg/dL | 3 points | 52 (21.2) |
| LDL-C 155–189 mg/dL | 1 point | 67 (27.3) |
| LDL-C < 155 mg/dL | 0 points | 117 (47.8) |
| Genetic testing | ||
| Functional mutation in the LDL receptor, apoB or PCSK9 gene | 8 points | 0 (0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertolín-Boronat, C.; Marcos-Garcés, V.; Merenciano-González, H.; Martínez Mas, M.L.; Climent Alberola, J.I.; Perez, N.; López Bueno, L.; Esteban Argente, M.C.; Valls Reig, M.; Arizón Benito, A.; et al. Familial Hypercholesterolemia Screening in a Cardiac Rehabilitation Program After Myocardial Infarction. Cardiogenetics 2025, 15, 6. https://doi.org/10.3390/cardiogenetics15010006
Bertolín-Boronat C, Marcos-Garcés V, Merenciano-González H, Martínez Mas ML, Climent Alberola JI, Perez N, López Bueno L, Esteban Argente MC, Valls Reig M, Arizón Benito A, et al. Familial Hypercholesterolemia Screening in a Cardiac Rehabilitation Program After Myocardial Infarction. Cardiogenetics. 2025; 15(1):6. https://doi.org/10.3390/cardiogenetics15010006
Chicago/Turabian StyleBertolín-Boronat, Carlos, Víctor Marcos-Garcés, Héctor Merenciano-González, María Luz Martínez Mas, Josefa Inés Climent Alberola, Nerea Perez, Laura López Bueno, María Concepción Esteban Argente, María Valls Reig, Ana Arizón Benito, and et al. 2025. "Familial Hypercholesterolemia Screening in a Cardiac Rehabilitation Program After Myocardial Infarction" Cardiogenetics 15, no. 1: 6. https://doi.org/10.3390/cardiogenetics15010006
APA StyleBertolín-Boronat, C., Marcos-Garcés, V., Merenciano-González, H., Martínez Mas, M. L., Climent Alberola, J. I., Perez, N., López Bueno, L., Esteban Argente, M. C., Valls Reig, M., Arizón Benito, A., Payá Rubio, A., Ríos-Navarro, C., de Dios, E., Gavara, J., Jiménez-Navarro, M. F., Chorro, F. J., Sanchis, J., & Bodi, V. (2025). Familial Hypercholesterolemia Screening in a Cardiac Rehabilitation Program After Myocardial Infarction. Cardiogenetics, 15(1), 6. https://doi.org/10.3390/cardiogenetics15010006