



Cancers 2021, 13(6), 1360; https://doi.org/10.3390/cancers13061360 - 17 Mar 2021
Cited by 60 | Viewed by 8695
Abstract
►
Show Figures
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer, ranking third in cancer deaths worldwide. Over the last decade, several studies have emphasized the development of tyrosine kinase inhibitors (TKIs) to target the aberrant pathways in HCC. However, the outcomes
[...] Read more.
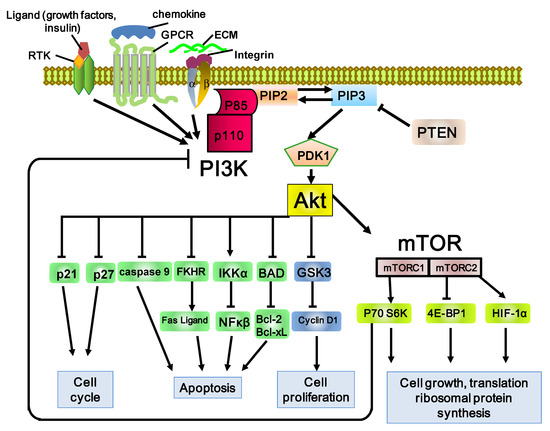
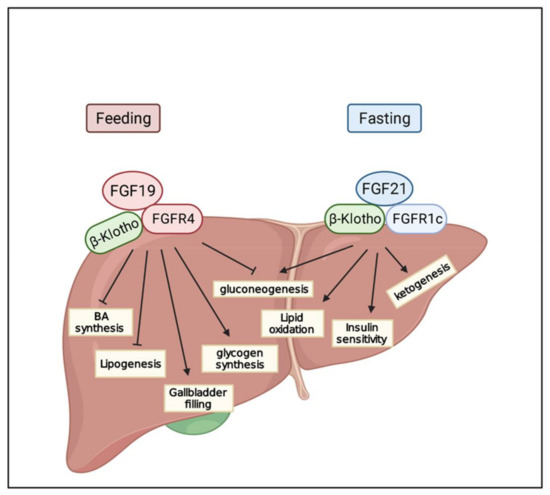
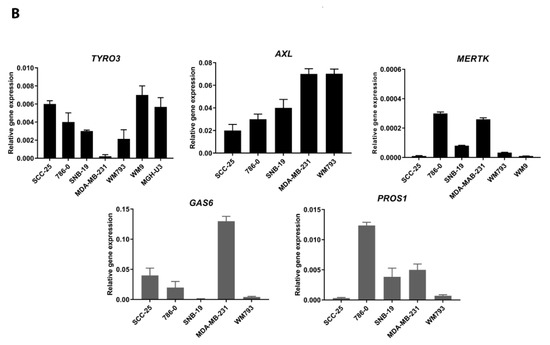
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer, ranking third in cancer deaths worldwide. Over the last decade, several studies have emphasized the development of tyrosine kinase inhibitors (TKIs) to target the aberrant pathways in HCC. However, the outcomes are far from satisfactory due to the increasing resistance and adverse effects. The family of fibroblast growth factor (FGF) and its receptors (FGFR) are involved in various biological processes, including embryogenesis, morphogenesis, wound repair, and cell growth. The aberrant FGF/FGFR signaling is also observed in multiple cancers, including HCC. Anti-FGF/FGFR provides delightful benefits for cancer patients, especially those with FGF signaling alteration. More and more multi-kinase inhibitors targeting FGF signaling, pan-FGFR inhibitors, and selective FGFR inhibitors are now under preclinical and clinical investigation. This review summarizes the aberrant FGF/FGFR signaling in HCC initiating, development and treatment status, and provide new insights into the treatment of HCC.
Full article
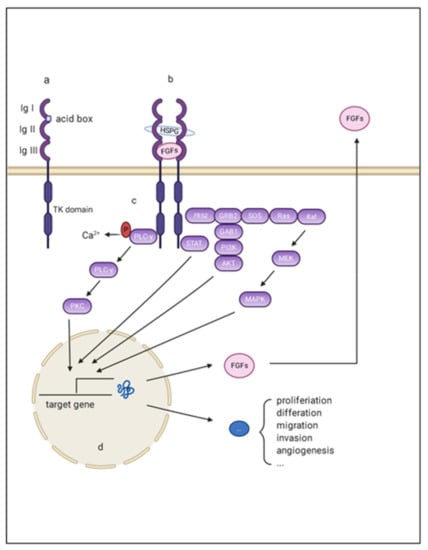
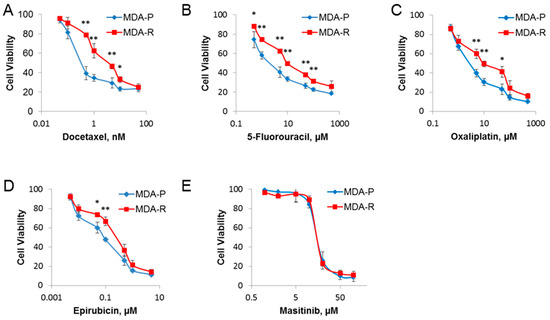
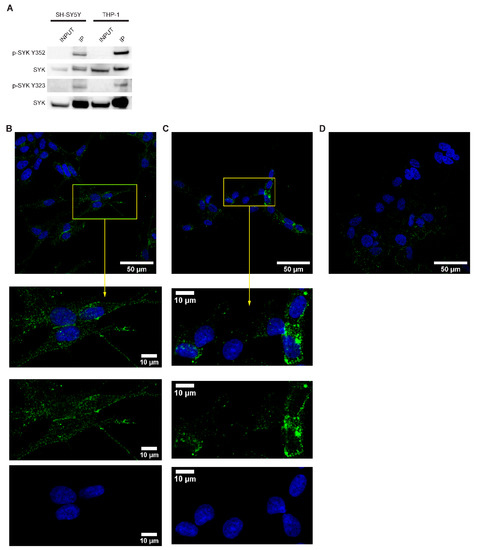
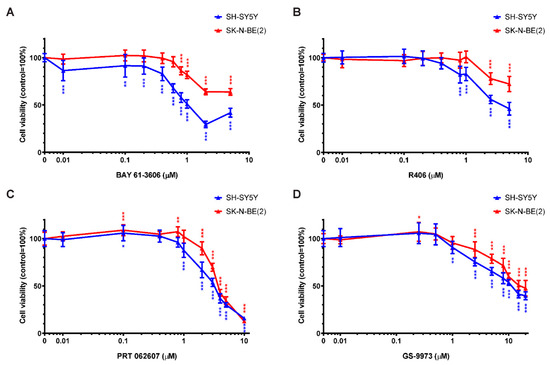
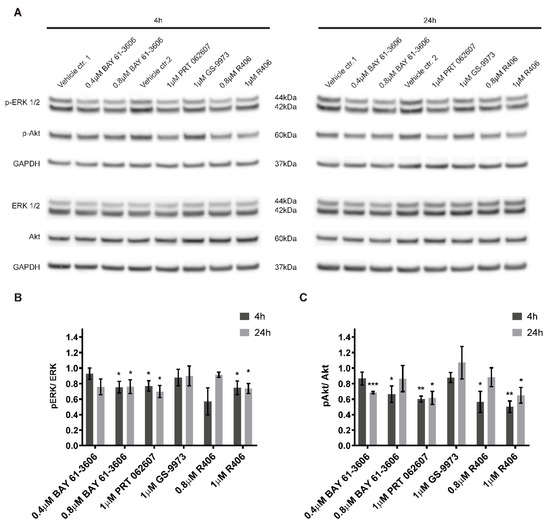
Figure 1







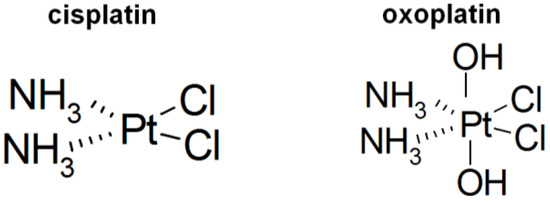


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}