Biomolecules In Silico: Contemporary Advances in Computational Approaches to Investigating the Molecular Dynamics of Biological Systems
Share This Topical Collection
Editors
 Dr. Thomas R. Caulfield
Dr. Thomas R. Caulfield
Dr. Thomas R. Caulfield
E-Mail
Website
Collection Editor
Department of Neuroscience, Division of QHS Computational Biology, Mayo Clinic, Jacksonville, FL 32224, USA
Interests: protein modeling and new drugs, such as structural studies of biomolecular targets, assessment of druggability, drug discovery (hit to lead through optimization) and de novo design
Special Issues, Collections and Topics in MDPI journals
 Dr. Manikandan Selvaraj
Dr. Manikandan Selvaraj
Dr. Manikandan Selvaraj
E-Mail
Collection Editor
Department of Clinical Genomics, Mayo Clinic, Rochester, MN 55902, USA
Interests: molecular docking; virtual screening; molecular modeling; docking studies; cheminformatics and computational chemistry; drug discovery; molecular dynamics simulation; protein modeling; computational chemistry; structural bioinformatics and structural biology
Topical Collection Information
Dear Colleagues,
Molecular dynamics simulations allow us to investigate physically realistic behaviors of biological systems in exquisite spatial and temporal resolution. This information is utilized to elucidate an array of biophysical characteristics, such as stability in different conditions, relative propensity to adopt relevant conformations, discovery of transient binding pockets, etc. Molecular dynamics simulations are employed to garner a fundamental understanding of macromolecules that can be applied from basic science to drug discovery. In this Topical Collection, we report current advancements in algorithms, software, and analytical techniques that are related to molecular dynamics simulations of biological systems.
Dr. Thomas R. Caulfield
Dr. Manikandan Selvaraj
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 250 words) can be sent to the Editorial Office for assessment.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Biomolecules is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- molecular dynamics simulation
- in silico design
- algorithms
- statistical mechanics
- biothermodynamics
- enhanced sampling
- conformational sampling
Published Papers (22 papers)
Open AccessArticle
Identification of Marine Compounds Inhibiting NF-κBInducing Kinase Through Molecular Docking and Molecular Dynamics Simulations
by
Muhammad Yasir, Jinyoung Park, Eun-Taek Han, Jin-Hee Han, Won Sun Park, Jongseon Choe and Wanjoo Chun
Cited by 7 | Viewed by 2906
Abstract
NF-κB-inducing kinase (NIK) plays a pivotal role in regulating both the canonical and non-canonical NF-κB signaling pathways, driving the expression of proteins involved in inflammation, immune responses, and cell survival. Overactivation of NIK is linked to various pathological conditions, including chronic inflammation, autoimmune
[...] Read more.
NF-κB-inducing kinase (NIK) plays a pivotal role in regulating both the canonical and non-canonical NF-κB signaling pathways, driving the expression of proteins involved in inflammation, immune responses, and cell survival. Overactivation of NIK is linked to various pathological conditions, including chronic inflammation, autoimmune diseases, metabolic disorders, and cancer progression. As such, NIK represents a compelling target for therapeutic intervention in these diseases. In this study, we explored the inhibitory potential of marine-derived compounds against NIK using integrated computational techniques, including molecular docking, molecular dynamics (MD) simulations, and free energy calculations. By screening a library of bioactive marine compounds, we identified several promising candidates with strong binding affinity to the NIK active site. By continuously narrowing down the library at each step, we found that the compounds santacruzamate A, xanthosine, and actinonine stand out at each step by demonstrating compact binding, highly stable interactions, and the most favorable free energy profiles, indicating their potential as effective NIK inhibitors. These findings not only advance our understanding of marine compounds as valuable resources for drug discovery but also highlight their potential for the development of natural anti-inflammatory therapies targeting NIK. This study opens new avenues for future research and therapeutic development aimed at combating inflammation and cancer through NIK inhibition.
Full article
►▼
Show Figures
Open AccessArticle
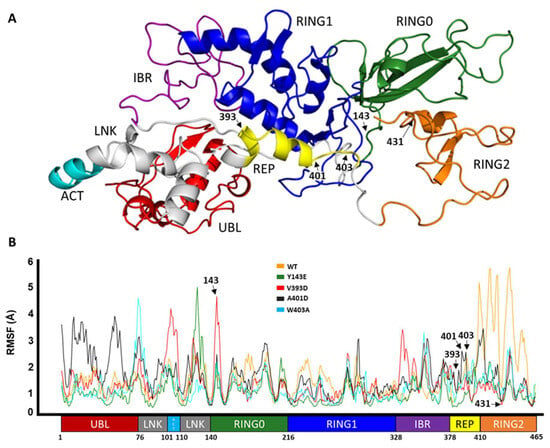
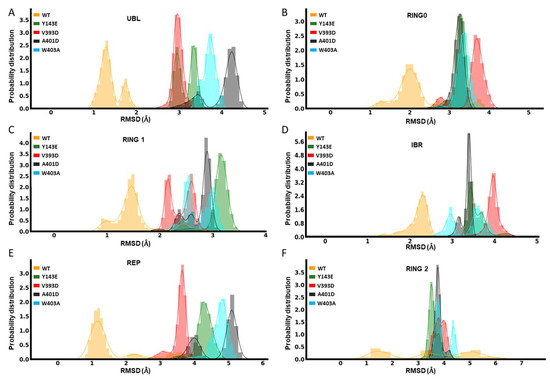
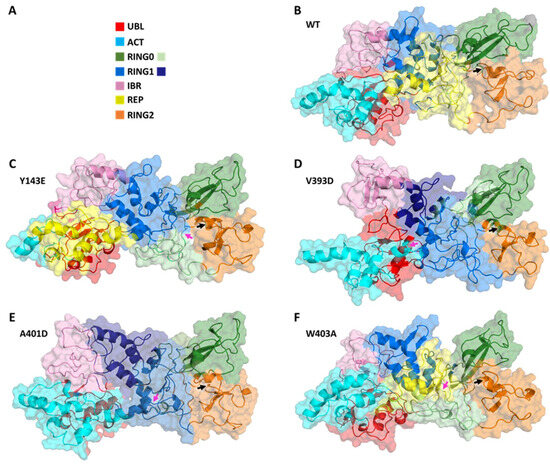
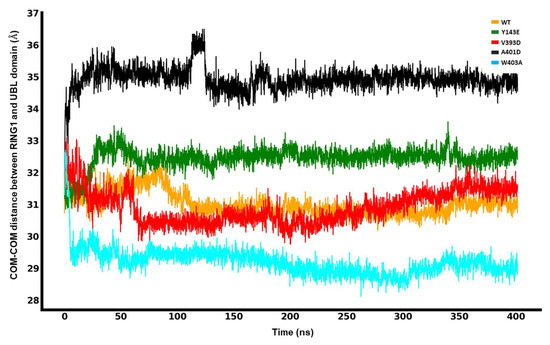
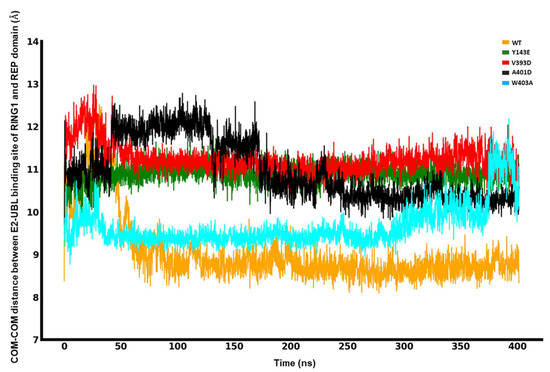
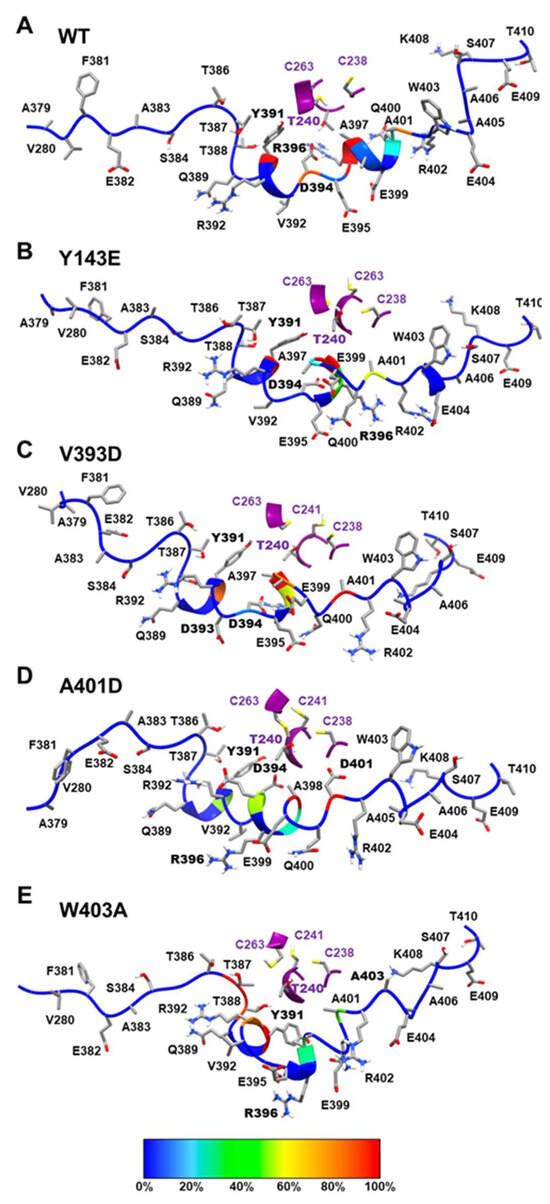
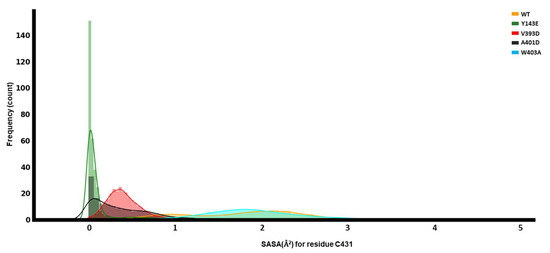
In Silico Investigation of Parkin-Activating Mutations Using Simulations and Network Modeling
by
Naeyma N. Islam, Caleb A. Weber, Matt Coban, Liam T. Cocker, Fabienne C. Fiesel, Wolfdieter Springer and Thomas R. Caulfield
Cited by 6 | Viewed by 3711
Abstract
Complete loss-of-function mutations in the PRKN gene are a major cause of early-onset Parkinson’s disease (PD). PRKN encodes the Parkin protein, an E3 ubiquitin ligase that works in conjunction with the ubiquitin kinase PINK1 in a distinct quality control pathway to tag damaged
[...] Read more.
Complete loss-of-function mutations in the PRKN gene are a major cause of early-onset Parkinson’s disease (PD). PRKN encodes the Parkin protein, an E3 ubiquitin ligase that works in conjunction with the ubiquitin kinase PINK1 in a distinct quality control pathway to tag damaged mitochondria for autophagic clearance, i.e., mitophagy. According to previous structural investigations, Parkin protein is typically kept in an inactive conformation via several intramolecular, auto-inhibitory interactions. Here, we performed molecular dynamics simulations (MDS) to provide insights into conformational changes occurring during the de-repression of Parkin and the gain of catalytic activity. We analyzed four different Parkin-activating mutations that are predicted to disrupt certain aspects of its auto-inhibition. All four variants showed greater conformational motions compared to wild-type protein, as well as differences in distances between domain interfaces and solvent-accessible surface area, which are thought to play critical roles as Parkin gains catalytic activity. Our findings reveal that the studied variants exert a notable influence on Parkin activation as they alter the opening of its closed inactive structure, a finding that is supported by recent structure- and cell-based studies. These findings not only helped further characterize the hyperactive variants but overall improved our understanding of Parkin’s catalytic activity and nominated targets within Parkin’s structure for potential therapeutic designs.
Full article
►▼
Show Figures
Open AccessArticle
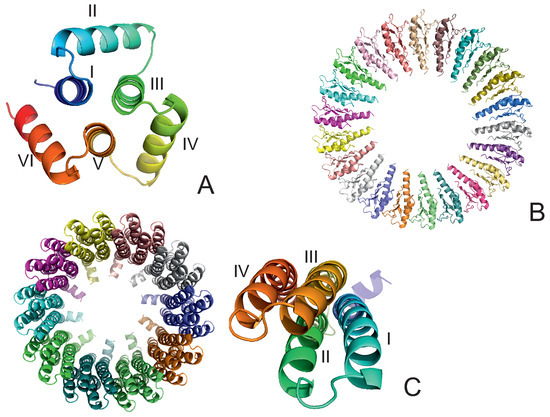
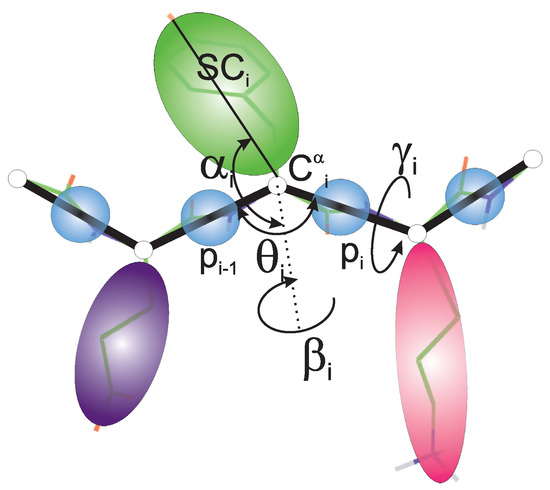
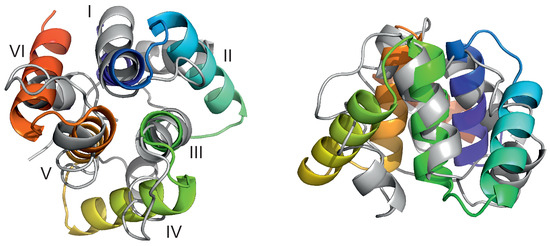
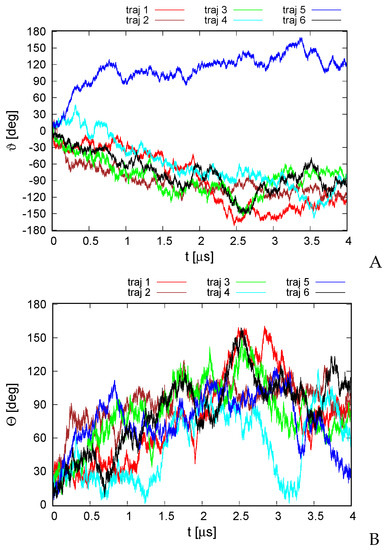
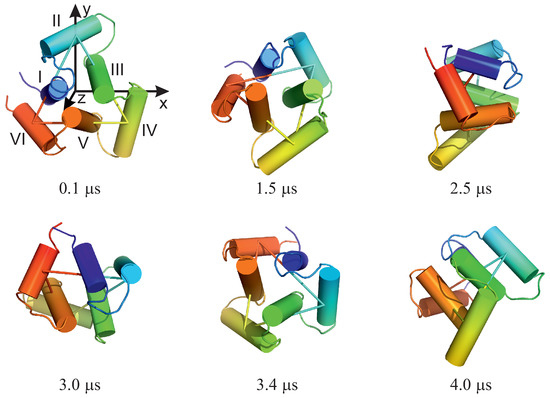
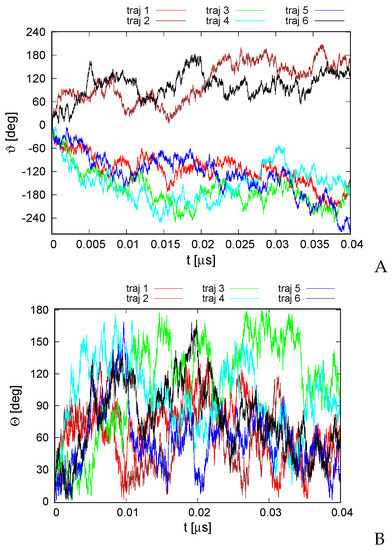
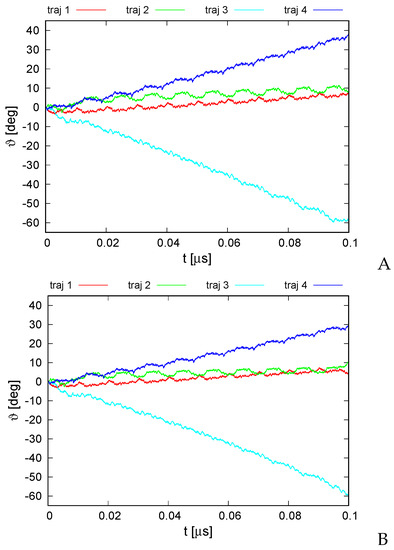
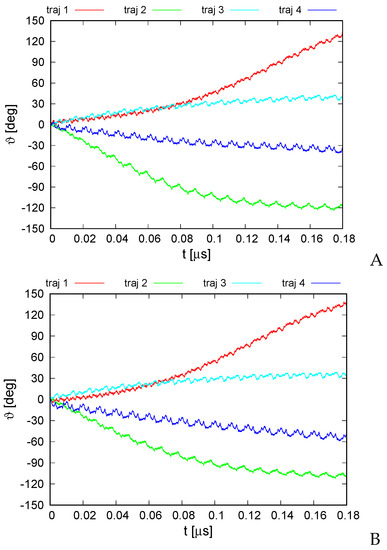
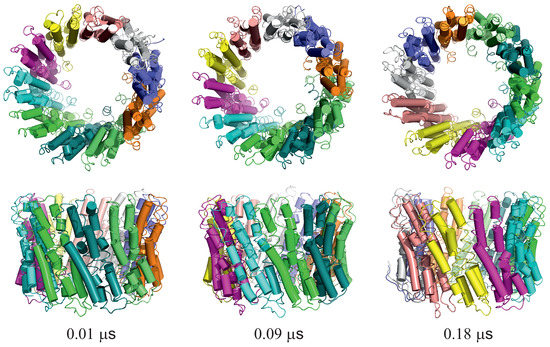
Long-Time Dynamics of Selected Molecular-Motor Components Using a Physics-Based Coarse-Grained Approach
by
Adam Liwo, Maciej Pyrka, Cezary Czaplewski, Xubiao Peng and Antti J. Niemi
Cited by 2 | Viewed by 3164
Abstract
Molecular motors are essential for the movement and transportation of macromolecules in living organisms. Among them, rotatory motors are particularly efficient. In this study, we investigated the long-term dynamics of the designed left-handed alpha/alpha toroid (PDB: 4YY2), the RBM2 flagellum protein ring from
[...] Read more.
Molecular motors are essential for the movement and transportation of macromolecules in living organisms. Among them, rotatory motors are particularly efficient. In this study, we investigated the long-term dynamics of the designed left-handed alpha/alpha toroid (PDB: 4YY2), the RBM2 flagellum protein ring from
Salmonella (PDB: 6SD5), and the V-type Na
-ATPase rotor in
Enterococcus hirae (PDB: 2BL2) using microcanonical and canonical molecular dynamics simulations with the coarse-grained UNRES force field, including a lipid-membrane model, on a millisecond laboratory time scale. Our results demonstrate that rotational motion can occur with zero total angular momentum in the microcanonical regime and that thermal motions can be converted into net rotation in the canonical regime, as previously observed in simulations of smaller cyclic molecules. For 6SD5 and 2BL2, net rotation (with a ratcheting pattern) occurring only about the pivot of the respective system was observed in canonical simulations. The extent and direction of the rotation depended on the initial conditions. This result suggests that rotatory molecular motors can convert thermal oscillations into net rotational motion. The energy from ATP hydrolysis is required probably to set the direction and extent of rotation. Our findings highlight the importance of molecular-motor structures in facilitating movement and transportation within living organisms.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
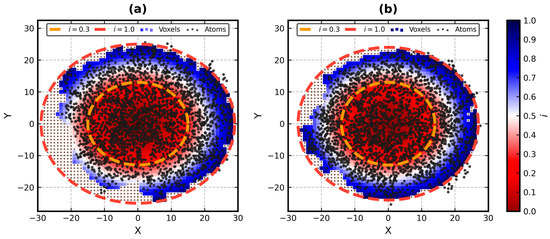
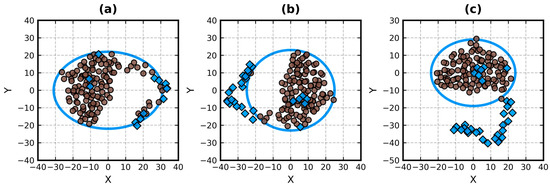
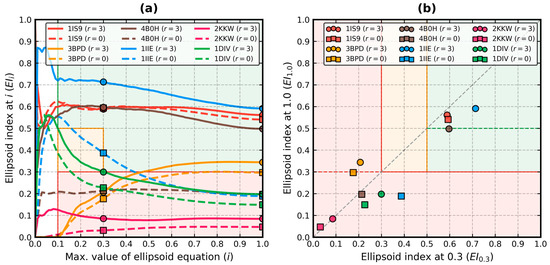
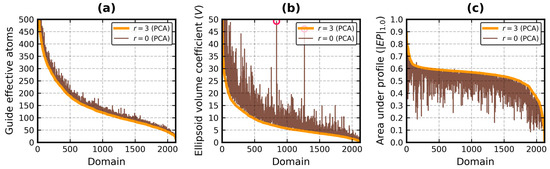
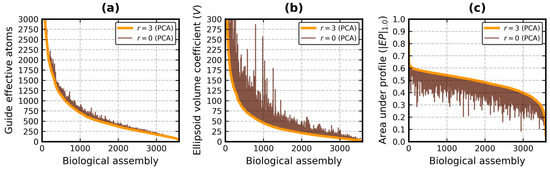
Improved Assessment of Globularity of Protein Structures and the Ellipsoid Profile of the Biological Assemblies from the PDB
by
Mateusz Banach
Cited by 3 | Viewed by 3204
Abstract
In this paper, we present an update to the ellipsoid profile algorithm (EP), a simple technique for the measurement of the globularity of protein structures without the calculation of molecular surfaces. The globularity property is understood in this context as the ability of
[...] Read more.
In this paper, we present an update to the ellipsoid profile algorithm (EP), a simple technique for the measurement of the globularity of protein structures without the calculation of molecular surfaces. The globularity property is understood in this context as the ability of the molecule to fill a minimum volume enclosing ellipsoid (MVEE) that approximates its assumed globular shape. The more of the interior of this ellipsoid is occupied by the atoms of the protein, the better are its globularity metrics. These metrics are derived from the comparison of the volume of the voxelized representation of the atoms and the volume of all voxels that can fit inside that ellipsoid (a uniform unit Å cube lattice). The so-called ellipsoid profile shows how the globularity changes with the distance from the center. Two of its values, the so-called ellipsoid indexes, are used to classify the structure as globular, semi-globular or non-globular. Here, we enhance the workflow of the EP algorithm via an improved outlier detection subroutine based on principal component analysis. It is capable of robust distinguishing between the dense parts of the molecules and, for example, disordered chain fragments fully exposed to the solvent. The PCA-based method replaces the current approach based on kernel density estimation. The improved EP algorithm was tested on 2124 representatives of domain superfamilies from SCOP 2.08. The second part of this work is dedicated to the survey of globularity of 3594 representatives of biological assemblies from molecules currently deposited in the PDB and analyzed by the 3DComplex database (monomers and complexes up to 60 chains).
Full article
►▼
Show Figures
Open AccessArticle
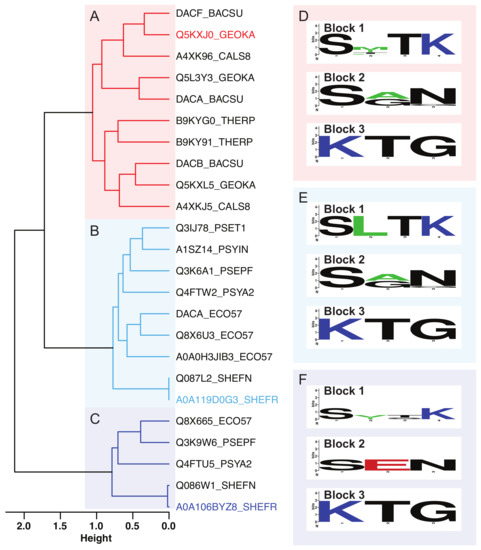
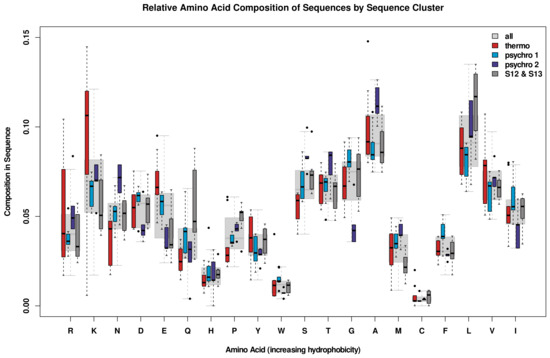
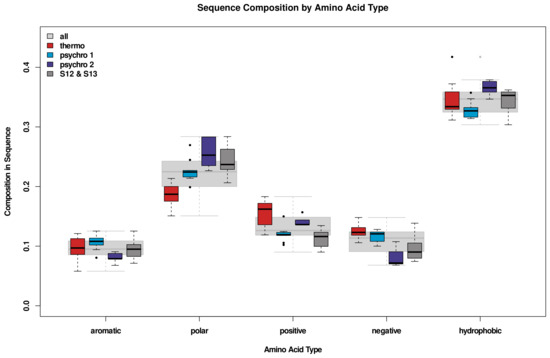
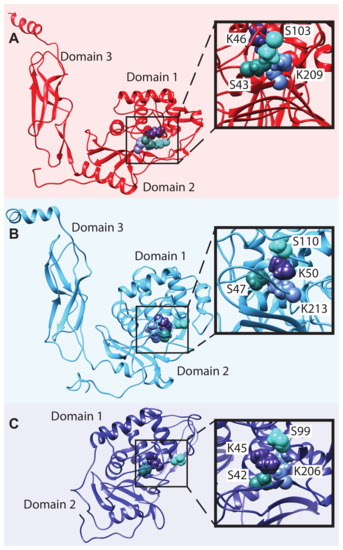
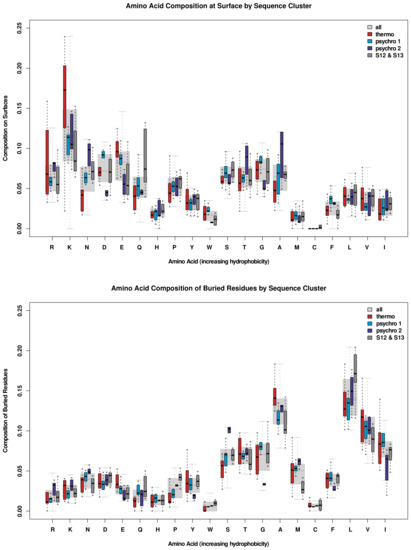
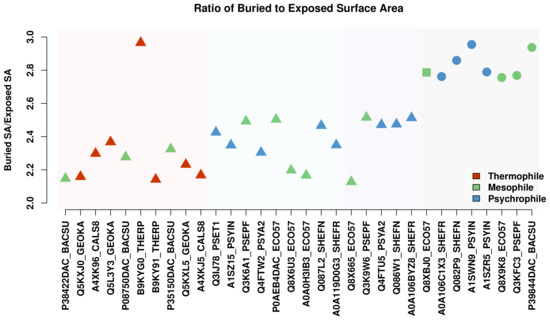
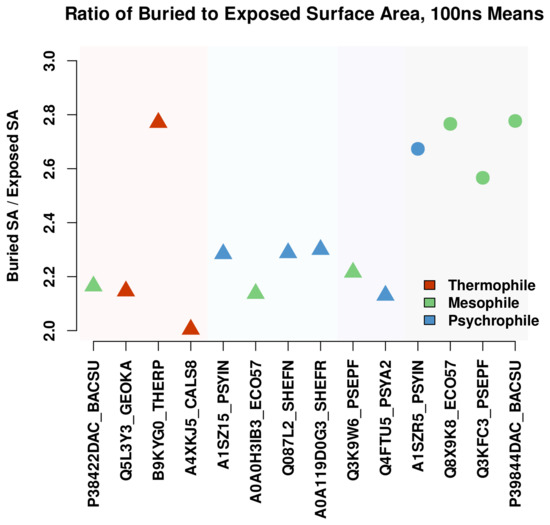
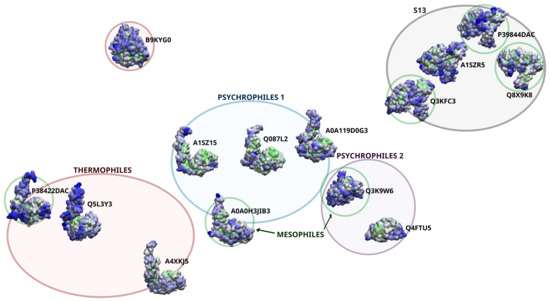
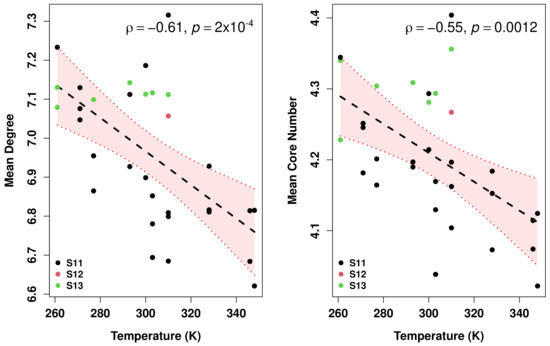
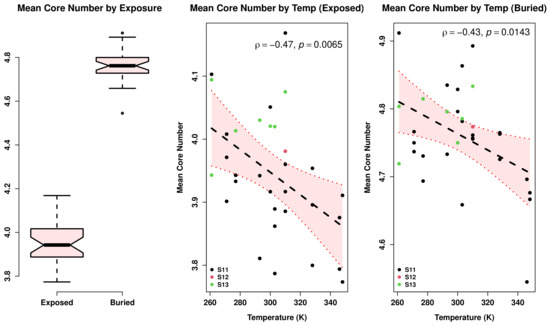
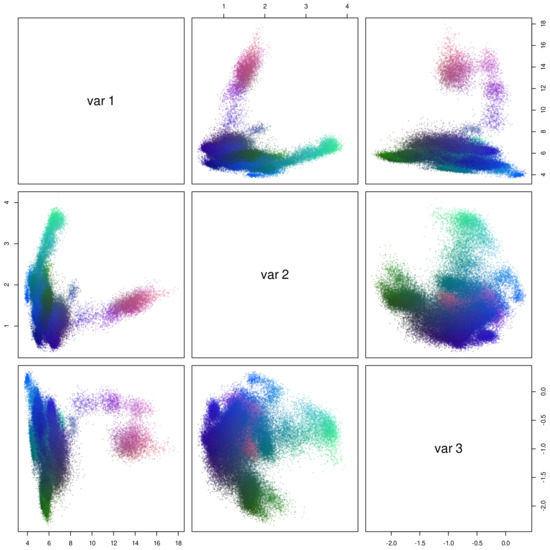
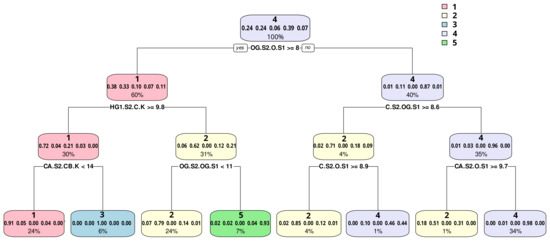
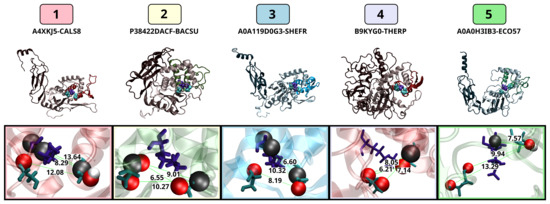
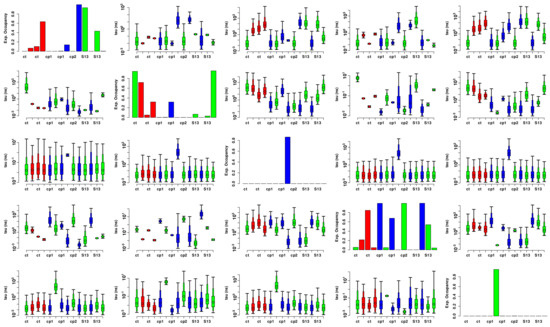
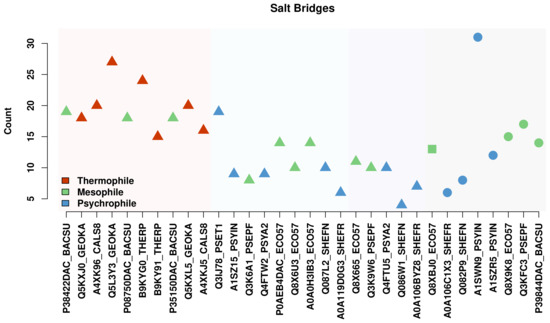
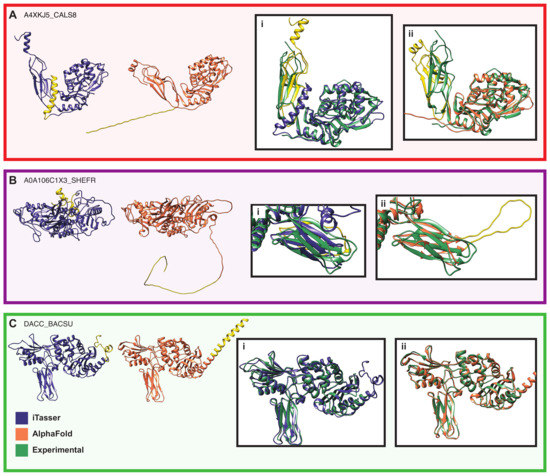
Comparative Modeling and Analysis of Extremophilic D-Ala-D-Ala Carboxypeptidases
by
Elizabeth M. Diessner, Gemma R. Takahashi, Rachel W. Martin and Carter T. Butts
Cited by 4 | Viewed by 3503
Abstract
Understanding the molecular adaptations of organisms to extreme environments requires a comparative analysis of protein structure, function, and dynamics across species found in different environmental conditions. Computational studies can be particularly useful in this pursuit, allowing exploratory studies of large numbers of proteins
[...] Read more.
Understanding the molecular adaptations of organisms to extreme environments requires a comparative analysis of protein structure, function, and dynamics across species found in different environmental conditions. Computational studies can be particularly useful in this pursuit, allowing exploratory studies of large numbers of proteins under different thermal and chemical conditions that would be infeasible to carry out experimentally. Here, we perform such a study of the MEROPS family S11, S12, and S13 proteases from psychophilic, mesophilic, and thermophilic bacteria. Using a combination of protein structure prediction, atomistic molecular dynamics, and trajectory analysis, we examine both conserved features and trends across thermal groups. Our findings suggest a number of hypotheses for experimental investigation.
Full article
►▼
Show Figures
Open AccessArticle
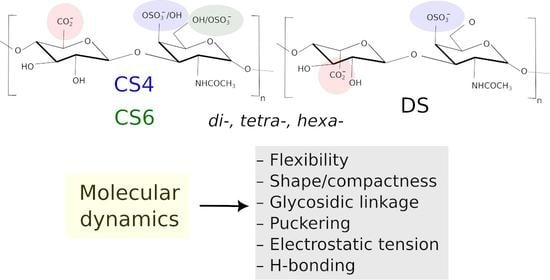
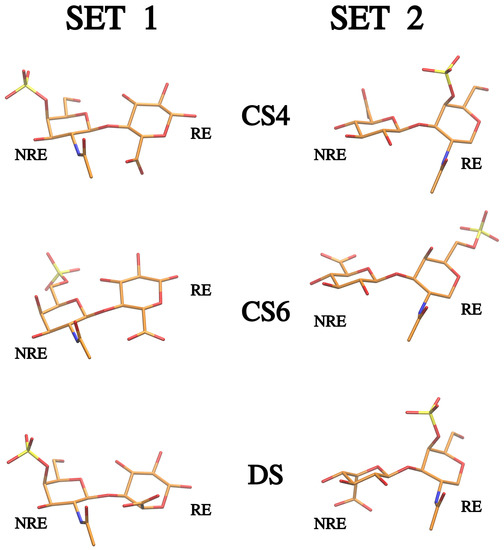
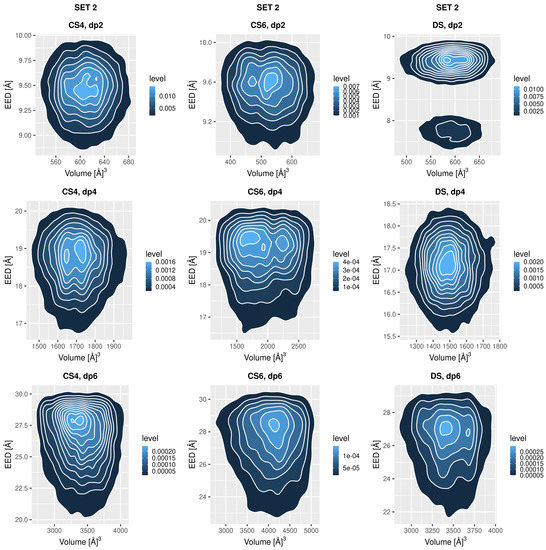
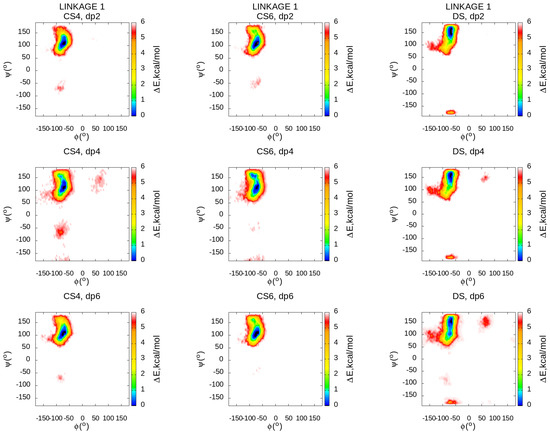
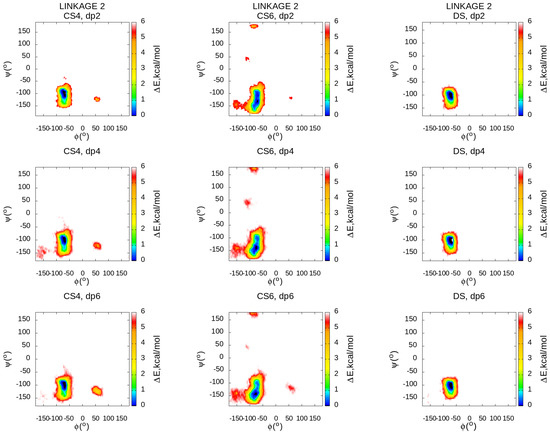
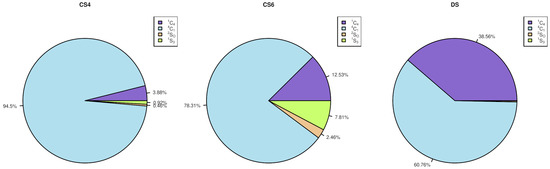
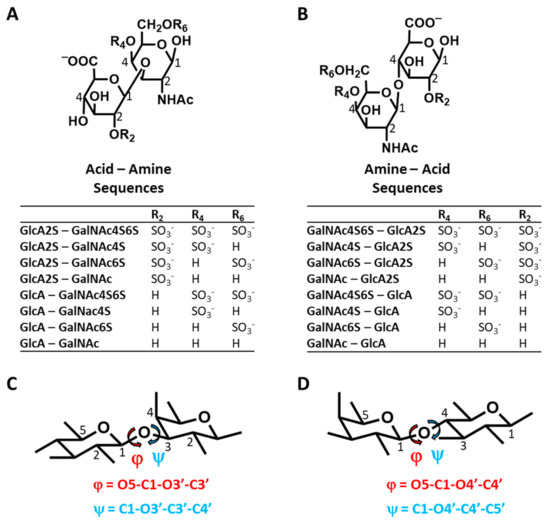
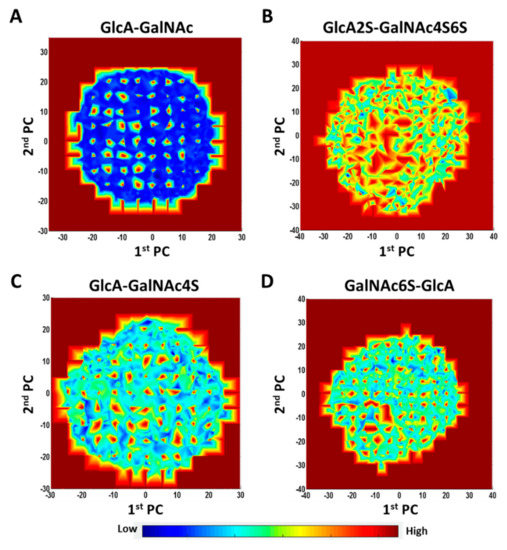
Molecular Dynamics-Based Comparative Analysis of Chondroitin and Dermatan Sulfates
by
Marta Pągielska and Sergey A. Samsonov
Cited by 5 | Viewed by 3231
Abstract
Glycosaminoglycans (GAGs) are a class of linear anionic periodic polysaccharides containing disaccharide repetitive units. These molecules interact with a variety of proteins in the extracellular matrix and so participate in biochemically crucial processes such as cell signalling affecting tissue regeneration as well as
[...] Read more.
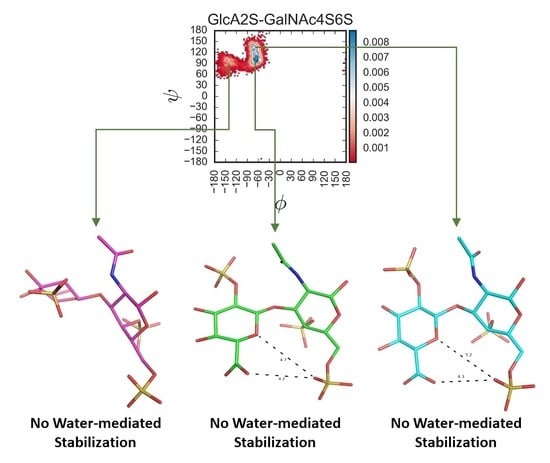
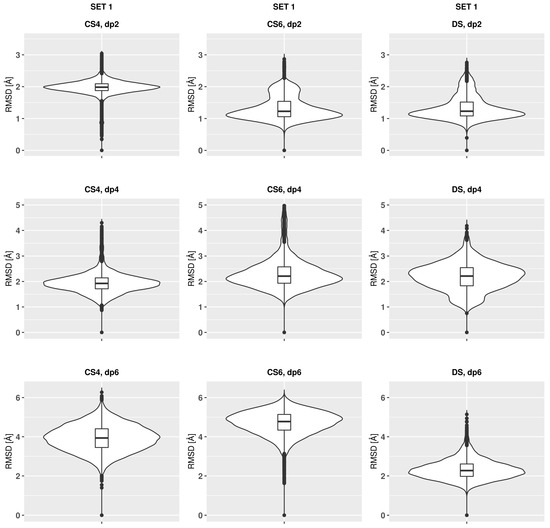
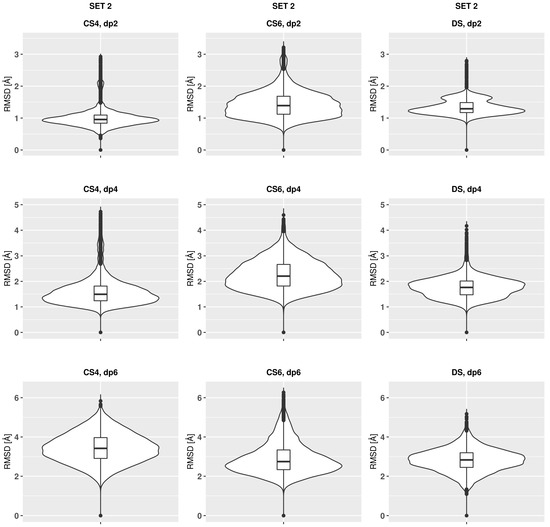
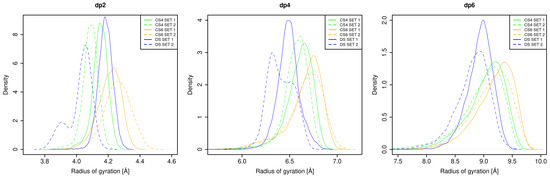
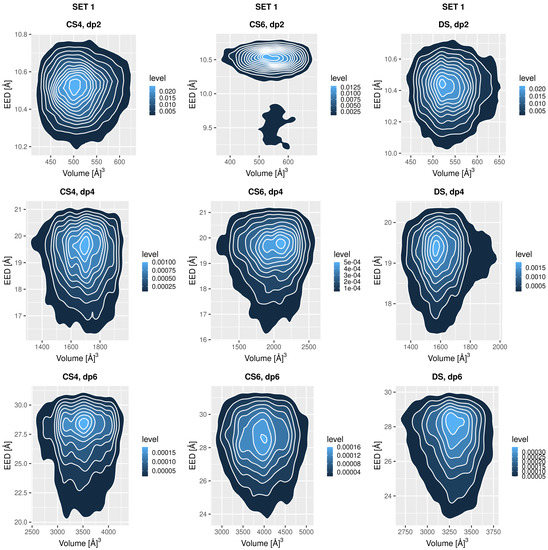
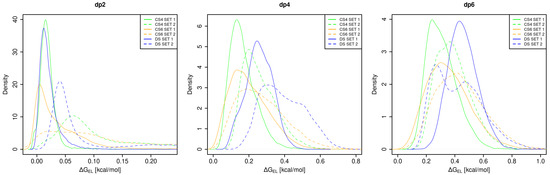
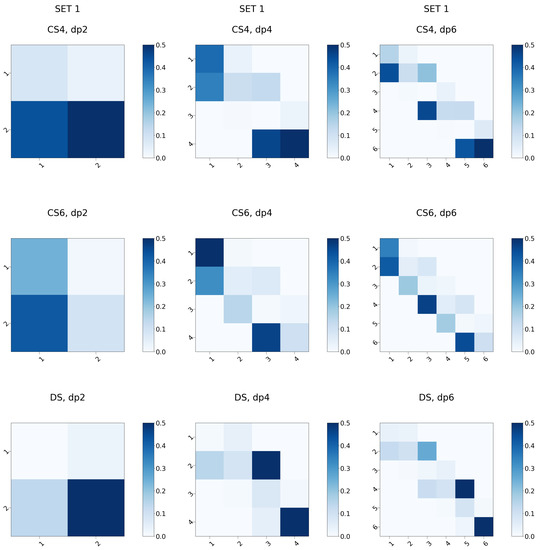
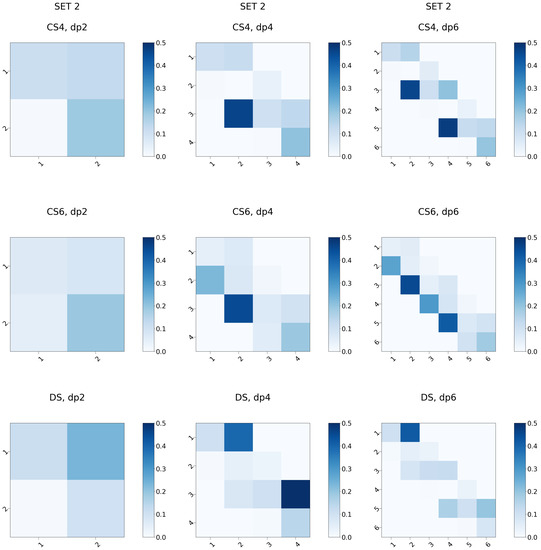
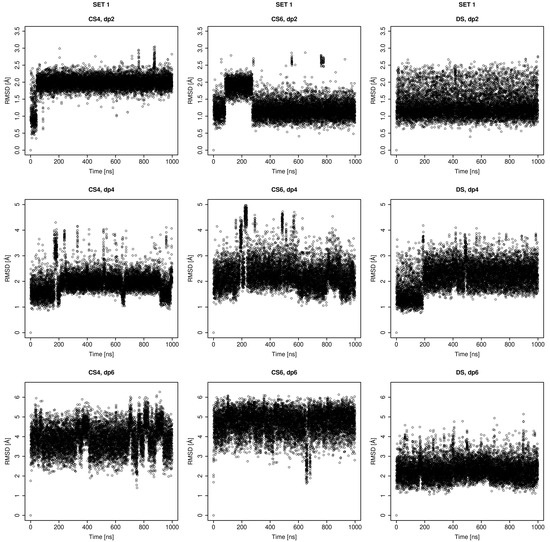
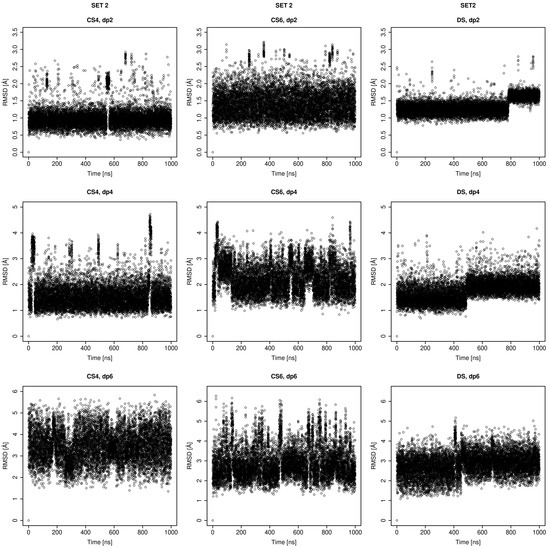
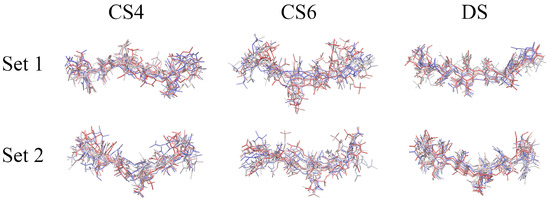
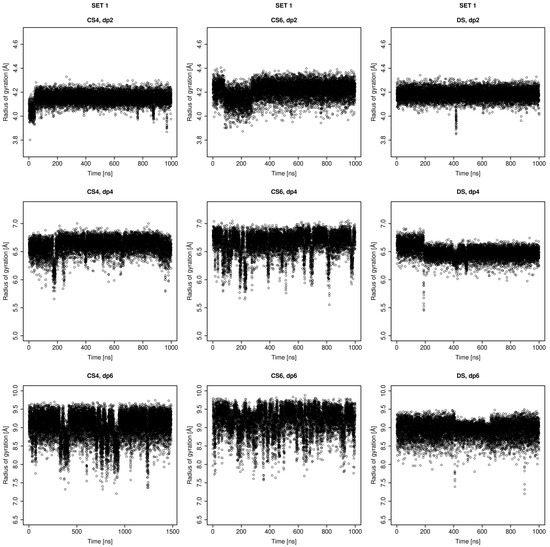
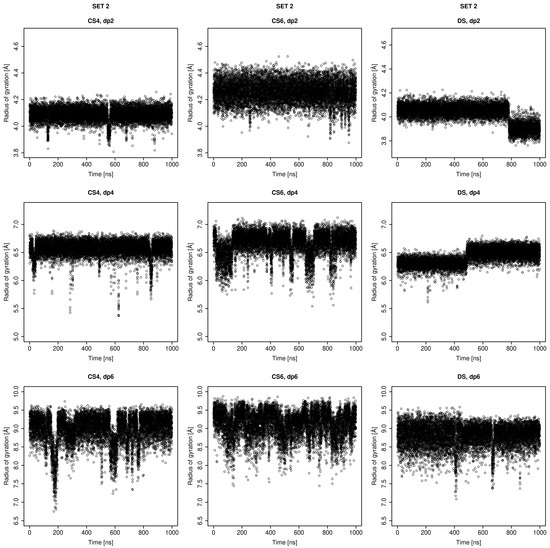
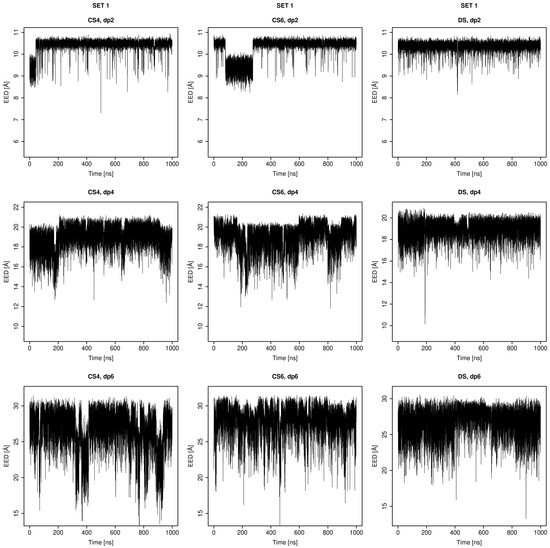
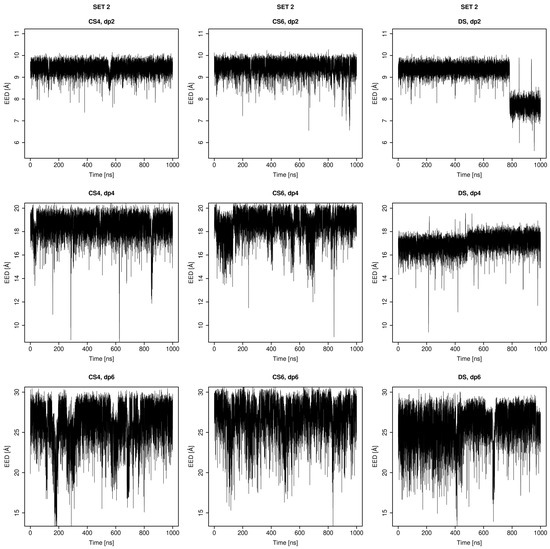
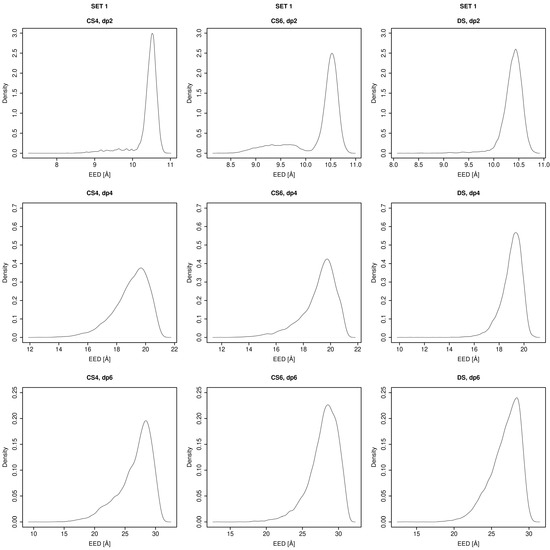
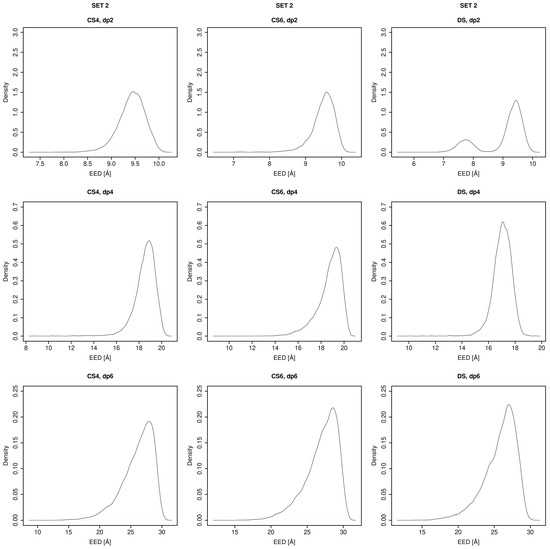
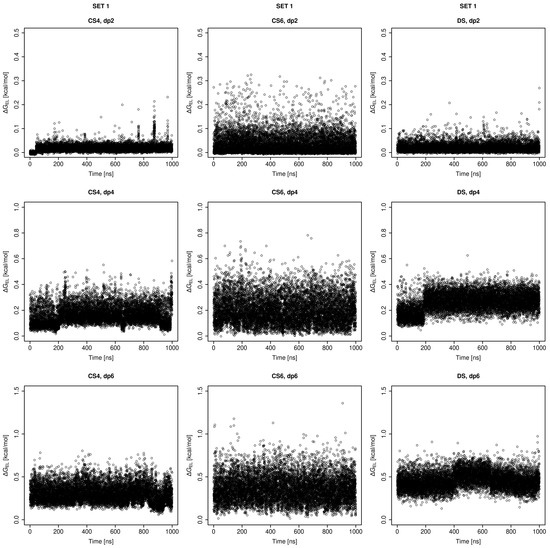
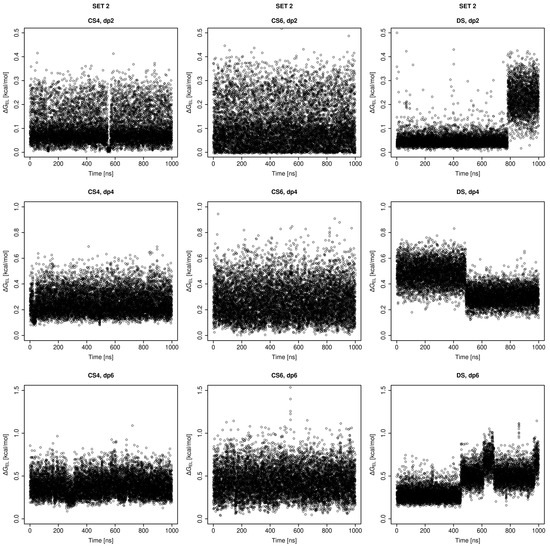
Glycosaminoglycans (GAGs) are a class of linear anionic periodic polysaccharides containing disaccharide repetitive units. These molecules interact with a variety of proteins in the extracellular matrix and so participate in biochemically crucial processes such as cell signalling affecting tissue regeneration as well as the onset of cancer, Alzheimer’s or Parkinson’s diseases. Due to their flexibility, periodicity and chemical heterogeneity, often termed “sulfation code”, GAGs are challenging molecules both for experiments and computation. One of the key questions in the GAG research is the specificity of their intermolecular interactions. In this study, we make a step forward to deciphering the “sulfation code” of chondroitin sulfates-4,6 (CS4, CS6, where the numbers correspond to the position of sulfation in NAcGal residue) and dermatan sulfate (DS), which is different from CSs by the presence of IdoA acid instead of GlcA. We rigorously investigate two sets of these GAGs in dimeric, tetrameric and hexameric forms with molecular dynamics-based descriptors. Our data clearly suggest that CS4, CS6 and DS are substantially different in terms of their structural, conformational and dynamic properties, which contributes to the understanding of how these molecules can be different when they bind proteins, which could have practical implications for the GAG-based drug design strategies in the regenerative medicine.
Full article
►▼
Show Figures
Open AccessArticle
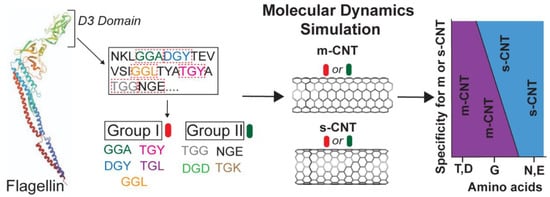
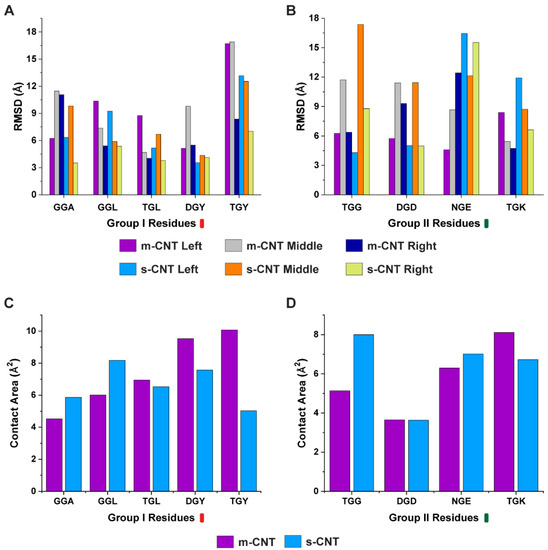
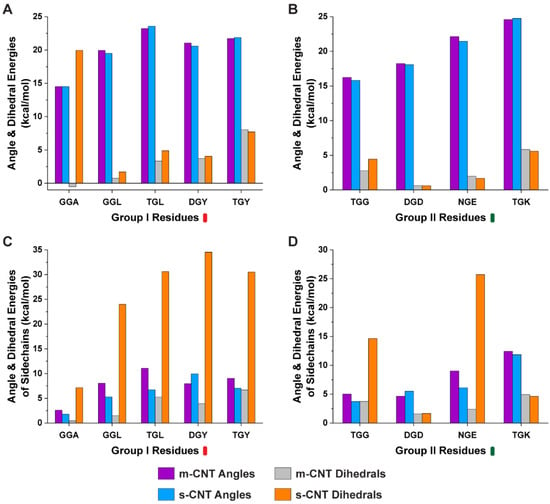
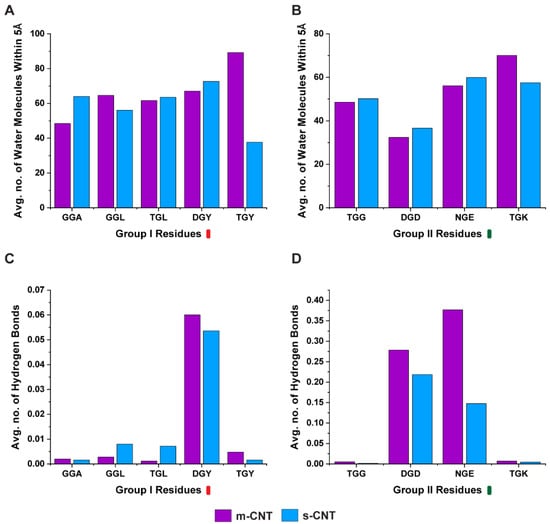
Computational Investigation of Chirality-Based Separation of Carbon Nanotubes Using Tripeptide Library
by
Shrishti Singh, Heena R. Divecha, Abimbola Ayoola, Marvin Xavierselvan, Jack Devlin and Isaac Macwan
Cited by 2 | Viewed by 3159
Abstract
Carbon nanotubes (CNT) have fascinating applications in flexible electronics, biosensors, and energy storage devices, and are classified as metallic or semiconducting based on their chirality. Semiconducting CNTs have been teased as a new material for building blocks in electronic devices, owing to their
[...] Read more.
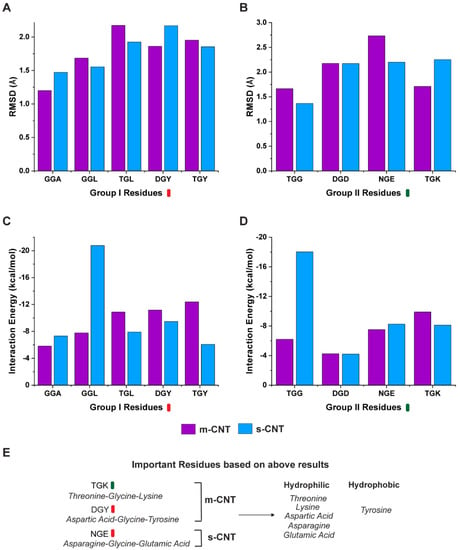
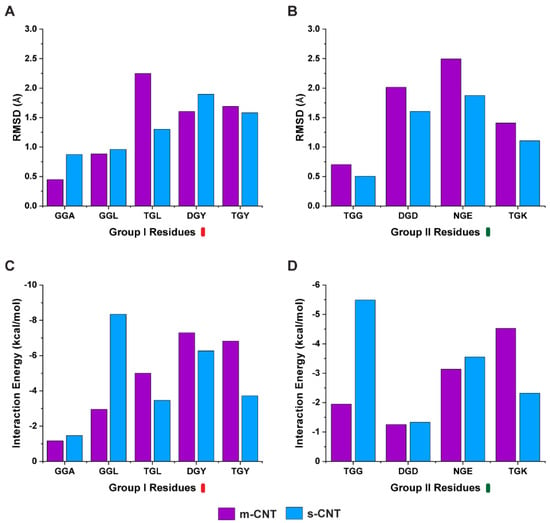
Carbon nanotubes (CNT) have fascinating applications in flexible electronics, biosensors, and energy storage devices, and are classified as metallic or semiconducting based on their chirality. Semiconducting CNTs have been teased as a new material for building blocks in electronic devices, owing to their band gap resembling silicon. However, CNTs must be sorted into metallic and semiconducting for such applications. Formerly, gel chromatography, ultracentrifugation, size exclusion chromatography, and phage display libraries were utilized for sorting CNTs. Nevertheless, these techniques are either expensive or have poor efficiency. In this study, we utilize a novel technique of using a library of nine tripeptides with glycine as a central residue to study the effect of flanking residues for large-scale separation of CNTs. Through molecular dynamics, we found that the tripeptide combinations with threonine as one of the flanking residues have a high affinity for metallic CNTs, whereas those with flanking residues having uncharged and negatively charged polar groups show selectivity towards semiconducting CNTs. Furthermore, the role of interfacial water molecules and the ability of the tripeptides to form hydrogen bonds play a crucial role in sorting the CNTs. It is envisaged that CNTs can be sorted based on their chirality-selective interaction affinity to tripeptides.
Full article
►▼
Show Figures
Open AccessArticle
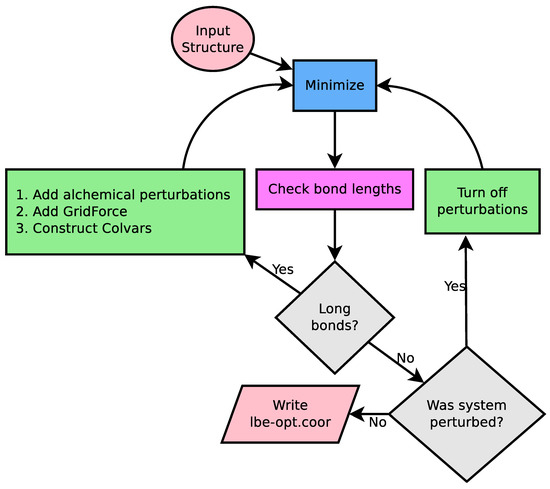
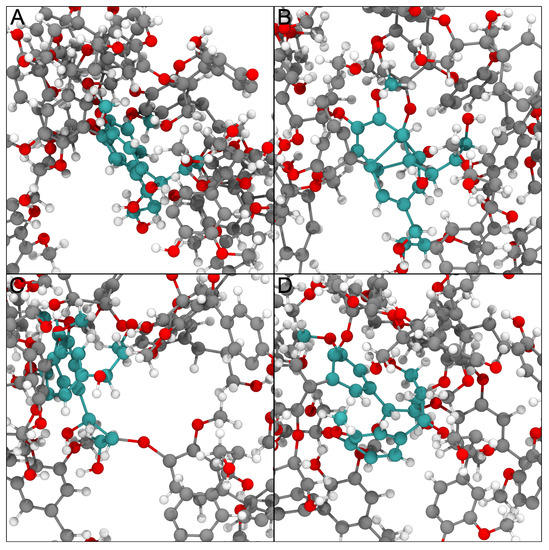
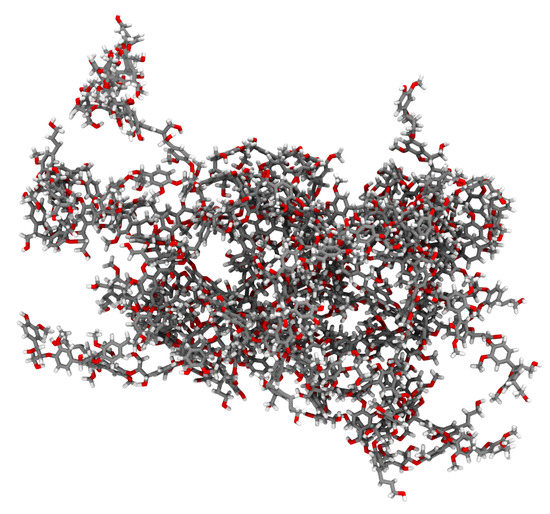
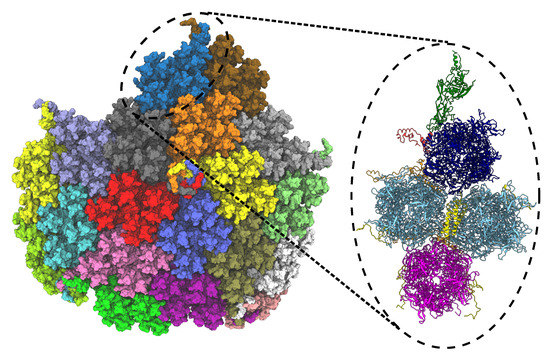
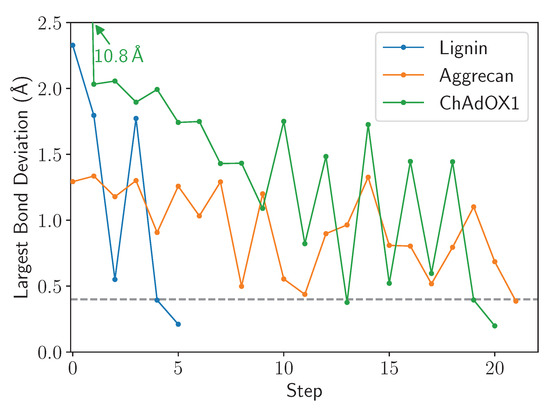
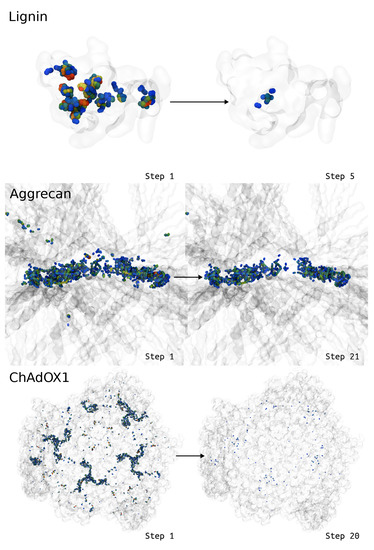
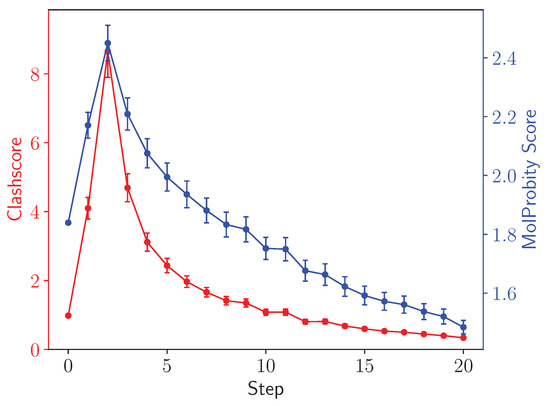
LongBondEliminator: A Molecular Simulation Tool to Remove Ring Penetrations in Biomolecular Simulation Systems
by
Daipayan Sarkar, Martin Kulke and Josh V. Vermaas
Cited by 5 | Viewed by 3478
Abstract
We develop a workflow, implemented as a plugin to the molecular visualization program VMD, that can fix ring penetrations with minimal user input. LongBondEliminator, detects ring piercing artifacts by the long, strained bonds that are the local minimum energy conformation during minimization for
[...] Read more.
We develop a workflow, implemented as a plugin to the molecular visualization program VMD, that can fix ring penetrations with minimal user input. LongBondEliminator, detects ring piercing artifacts by the long, strained bonds that are the local minimum energy conformation during minimization for some assembled simulation system. The LongBondEliminator tool then automatically treats regions near these long bonds using multiple biases applied through NAMD. By combining biases implemented through the collective variables module, density-based forces, and alchemical techniques in NAMD, LongBondEliminator will iteratively alleviate long bonds found within molecular simulation systems. Through three concrete examples with increasing complexity, a lignin polymer, an viral capsid assembly, and a large, highly glycosylated protein aggrecan, we demonstrate the utility for this method in eliminating ring penetrations from classical MD simulation systems. The tool is available via gitlab as a VMD plugin, and has been developed to be generically useful across a variety of biomolecular simulations.
Full article
►▼
Show Figures
Open AccessReview
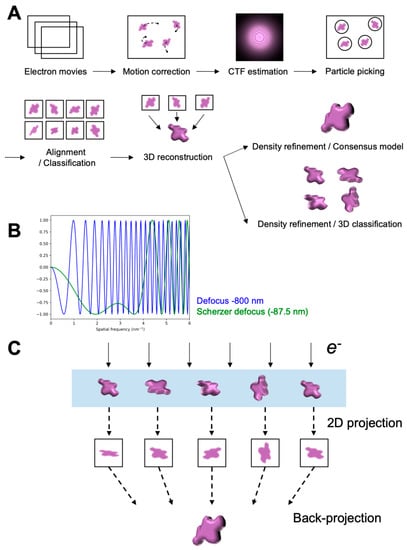
Probing Structural Perturbation of Biomolecules by Extracting Cryo-EM Data Heterogeneity
by
Kira DeVore and Po-Lin Chiu
Cited by 12 | Viewed by 5199
Abstract
Single-particle cryogenic electron microscopy (cryo-EM) has become an indispensable tool to probe high-resolution structural detail of biomolecules. It enables direct visualization of the biomolecules and opens a possibility for averaging molecular images to reconstruct a three-dimensional Coulomb potential density map. Newly developed algorithms
[...] Read more.
Single-particle cryogenic electron microscopy (cryo-EM) has become an indispensable tool to probe high-resolution structural detail of biomolecules. It enables direct visualization of the biomolecules and opens a possibility for averaging molecular images to reconstruct a three-dimensional Coulomb potential density map. Newly developed algorithms for data analysis allow for the extraction of structural heterogeneity from a massive and low signal-to-noise-ratio (SNR) cryo-EM dataset, expanding our understanding of multiple conformational states, or further implications in dynamics, of the target biomolecule. This review provides an overview that briefly describes the workflow of single-particle cryo-EM, including imaging and data processing, and new methods developed for analyzing the data heterogeneity to understand the structural variability of biomolecules.
Full article
►▼
Show Figures
Open AccessArticle
In-Depth Molecular Dynamics Study of All Possible Chondroitin Sulfate Disaccharides Reveals Key Insight into Structural Heterogeneity and Dynamism
by
Balaji Nagarajan, Nehru Viji Sankaranarayanan and Umesh R. Desai
Cited by 8 | Viewed by 3456
Abstract
GAGs exhibit a high level of conformational and configurational diversity, which remains untapped in terms of the recognition and modulation of proteins. Although GAGs are suggested to bind to more than 800 biologically important proteins, very few therapeutics have been designed or discovered
[...] Read more.
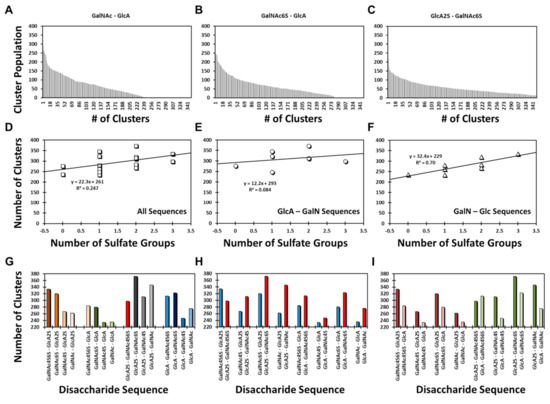
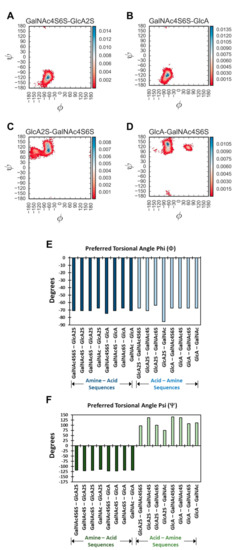
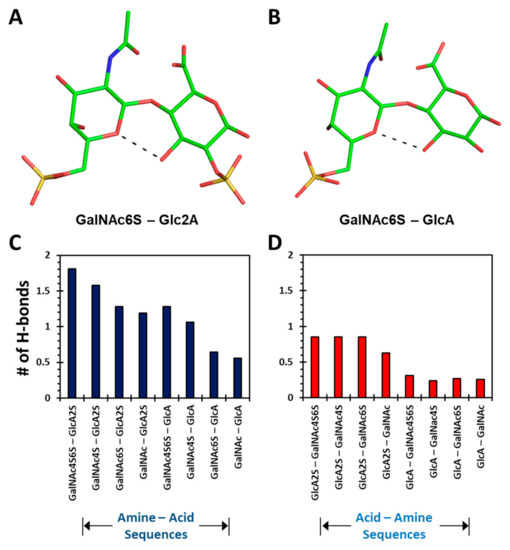
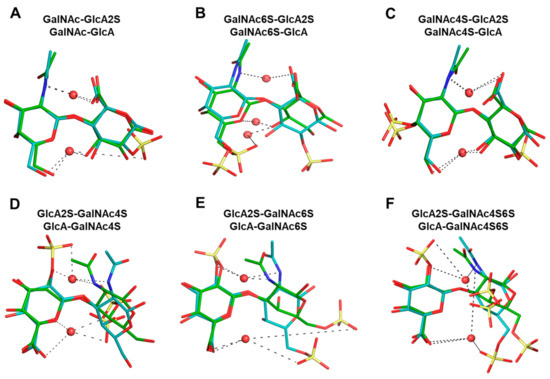
GAGs exhibit a high level of conformational and configurational diversity, which remains untapped in terms of the recognition and modulation of proteins. Although GAGs are suggested to bind to more than 800 biologically important proteins, very few therapeutics have been designed or discovered so far. A key challenge is the inability to identify, understand and predict distinct topologies accessed by GAGs, which may help design novel protein-binding GAG sequences. Recent studies on chondroitin sulfate (CS), a key member of the GAG family, pinpointing its role in multiple biological functions led us to study the conformational dynamism of CS building blocks using molecular dynamics (MD). In the present study, we used the all-atom GLYCAM06 force field for the first time to explore the conformational space of all possible CS building blocks. Each of the 16 disaccharides was solvated in a TIP3P water box with an appropriate number of counter ions followed by equilibration and a production run. We analyzed the MD trajectories for torsional space, inter- and intra-molecular H-bonding, bridging water, conformational spread and energy landscapes. An in-house phi and psi probability density analysis showed that 1→3-linked sequences were more flexible than 1→4-linked sequences. More specifically, phi and psi regions for 1→4-linked sequences were held within a narrower range because of intra-molecular H-bonding between the GalNAc O5 atom and GlcA O3 atom, irrespective of sulfation pattern. In contrast, no such intra-molecular interaction arose for 1→3-linked sequences. Further, the stability of 1→4-linked sequences also arose from inter-molecular interactions involving bridged water molecules. The energy landscape for both classes of CS disaccharides demonstrated increased ruggedness as the level of sulfation increased. The results show that CS building blocks present distinct conformational dynamism that offers the high possibility of unique electrostatic surfaces for protein recognition. The fundamental results presented here will support the development of algorithms that help to design longer CS chains for protein recognition.
Full article
►▼
Show Figures
Open AccessArticle
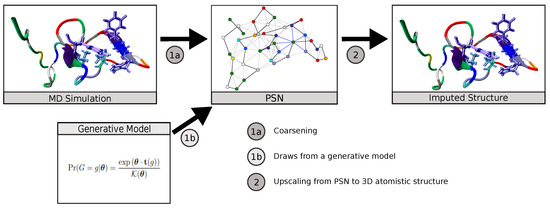
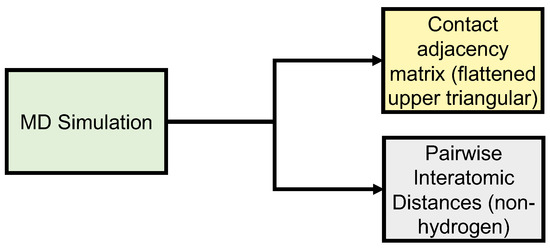
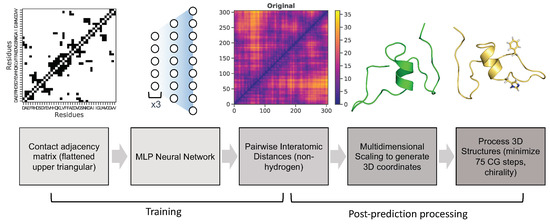
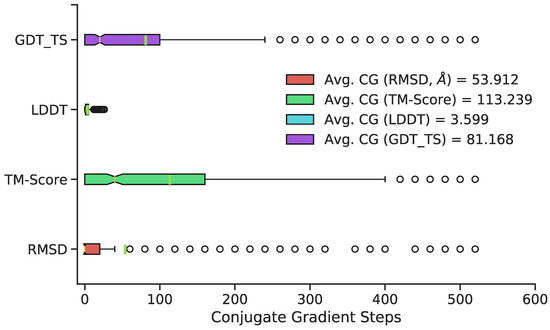
Neural Upscaling from Residue-Level Protein Structure Networks to Atomistic Structures
by
Vy T. Duong, Elizabeth M. Diessner, Gianmarc Grazioli, Rachel W. Martin and Carter T. Butts
Cited by 6 | Viewed by 4185
Abstract
Coarse-graining is a powerful tool for extending the reach of dynamic models of proteins and other biological macromolecules. Topological coarse-graining, in which biomolecules or sets thereof are represented via graph structures, is a particularly useful way of obtaining highly compressed representations of molecular
[...] Read more.
Coarse-graining is a powerful tool for extending the reach of dynamic models of proteins and other biological macromolecules. Topological coarse-graining, in which biomolecules or sets thereof are represented via graph structures, is a particularly useful way of obtaining highly compressed representations of molecular structures, and simulations operating via such representations can achieve substantial computational savings. A drawback of coarse-graining, however, is the loss of atomistic detail—an effect that is especially acute for topological representations such as protein structure networks (PSNs). Here, we introduce an approach based on a combination of machine learning and physically-guided refinement for inferring atomic coordinates from PSNs. This “neural upscaling” procedure exploits the constraints implied by PSNs on possible configurations, as well as differences in the likelihood of observing different configurations with the same PSN. Using a 1 μs atomistic molecular dynamics trajectory of A
, we show that neural upscaling is able to effectively recapitulate detailed structural information for intrinsically disordered proteins, being particularly successful in recovering features such as transient secondary structure. These results suggest that scalable network-based models for protein structure and dynamics may be used in settings where atomistic detail is desired, with upscaling employed to impute atomic coordinates from PSNs.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
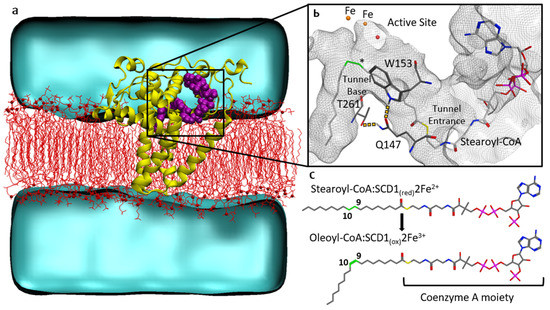
Sequential Dynamics of Stearoyl-CoA Desaturase-1(SCD1)/Ligand Binding and Unbinding Mechanism: A Computational Study
by
Anna B. Petroff, Rebecca L. Weir, Charles R. Yates, Joseph D. Ng and Jerome Baudry
Cited by 3 | Viewed by 4410
Abstract
Stearoyl-CoA desaturase-1 (SCD1 or delta-9 desaturase, D9D) is a key metabolic protein that modulates cellular inflammation and stress, but overactivity of SCD1 is associated with diseases, including cancer and metabolic syndrome. This transmembrane endoplasmic reticulum protein converts saturated fatty acids into monounsaturated fatty
[...] Read more.
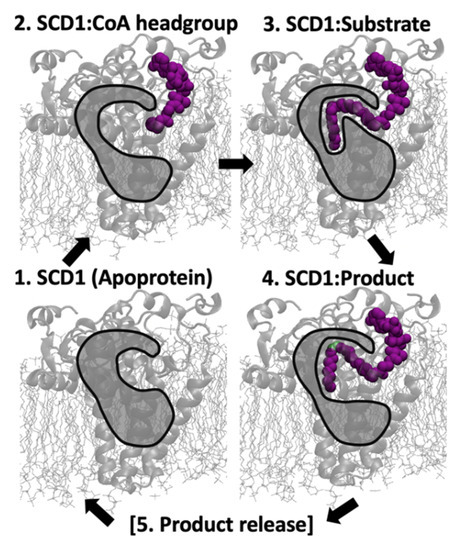
Stearoyl-CoA desaturase-1 (SCD1 or delta-9 desaturase, D9D) is a key metabolic protein that modulates cellular inflammation and stress, but overactivity of SCD1 is associated with diseases, including cancer and metabolic syndrome. This transmembrane endoplasmic reticulum protein converts saturated fatty acids into monounsaturated fatty acids, primarily stearoyl-CoA into oleoyl-CoA, which are critical products for energy metabolism and membrane composition. The present computational molecular dynamics study characterizes the molecular dynamics of SCD1 with substrate, product, and as an apoprotein. The modeling of SCD1:fatty acid interactions suggests that: (1) SCD1:CoA moiety interactions open the substrate-binding tunnel, (2) SCD1 stabilizes a substrate conformation favorable for desaturation, and (3) SCD1:product interactions result in an opening of the tunnel, possibly allowing product exit into the surrounding membrane. Together, these results describe a highly dynamic series of SCD1 conformations resulting from the enzyme:cofactor:substrate interplay that inform drug-discovery efforts.
Full article
►▼
Show Figures
Open AccessReview
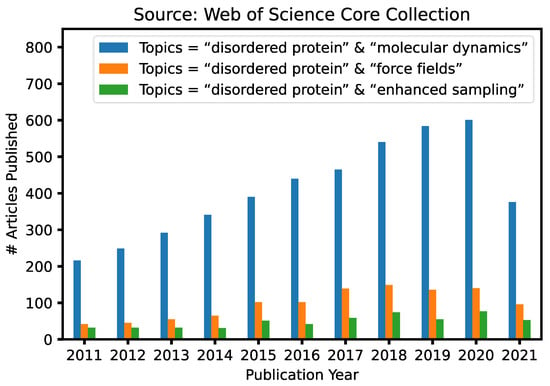
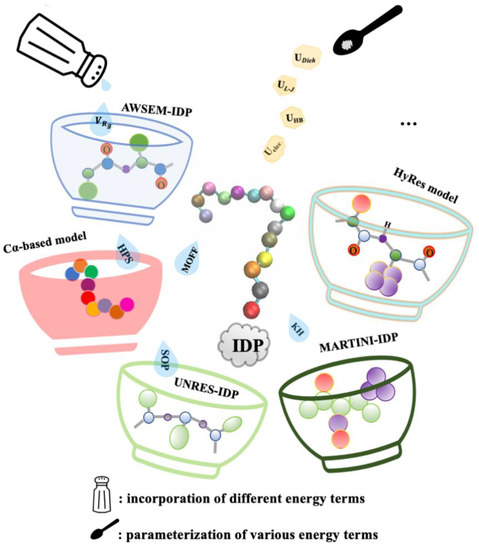
Advanced Sampling Methods for Multiscale Simulation of Disordered Proteins and Dynamic Interactions
by
Xiping Gong, Yumeng Zhang and Jianhan Chen
Cited by 33 | Viewed by 5838
Abstract
Intrinsically disordered proteins (IDPs) are highly prevalent and play important roles in biology and human diseases. It is now also recognized that many IDPs remain dynamic even in specific complexes and functional assemblies. Computer simulations are essential for deriving a molecular description of
[...] Read more.
Intrinsically disordered proteins (IDPs) are highly prevalent and play important roles in biology and human diseases. It is now also recognized that many IDPs remain dynamic even in specific complexes and functional assemblies. Computer simulations are essential for deriving a molecular description of the disordered protein ensembles and dynamic interactions for a mechanistic understanding of IDPs in biology, diseases, and therapeutics. Here, we provide an in-depth review of recent advances in the multi-scale simulation of disordered protein states, with a particular emphasis on the development and application of advanced sampling techniques for studying IDPs. These techniques are critical for adequate sampling of the manifold functionally relevant conformational spaces of IDPs. Together with dramatically improved protein force fields, these advanced simulation approaches have achieved substantial success and demonstrated significant promise towards the quantitative and predictive modeling of IDPs and their dynamic interactions. We will also discuss important challenges remaining in the atomistic simulation of larger systems and how various coarse-grained approaches may help to bridge the remaining gaps in the accessible time- and length-scales of IDP simulations.
Full article
►▼
Show Figures
Open AccessFeature PaperReview
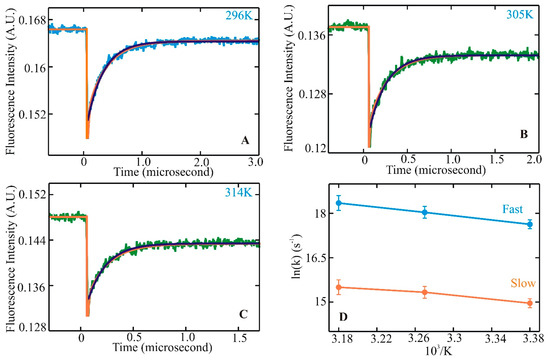
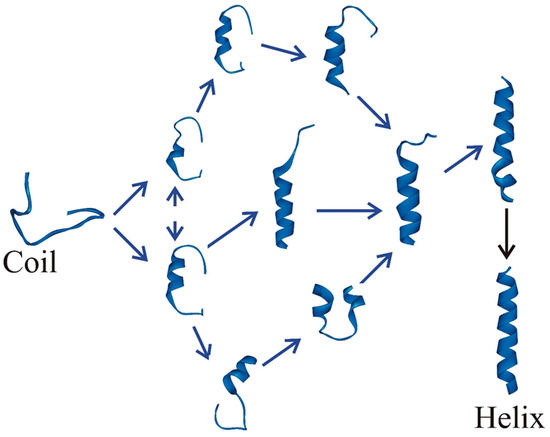
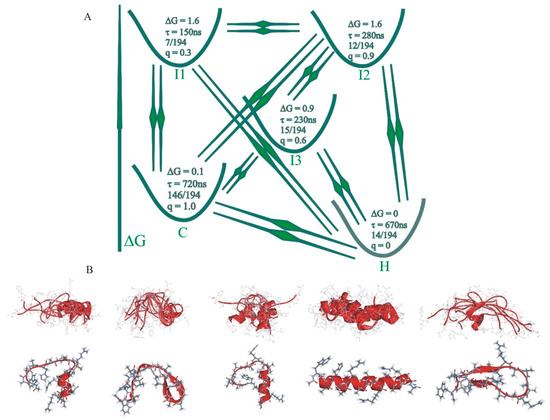
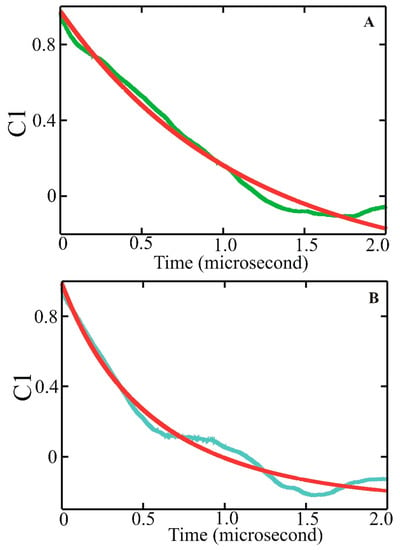
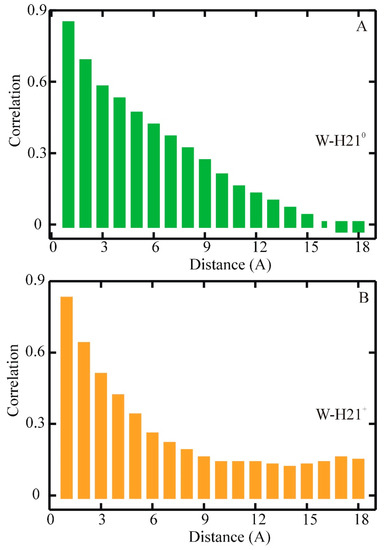
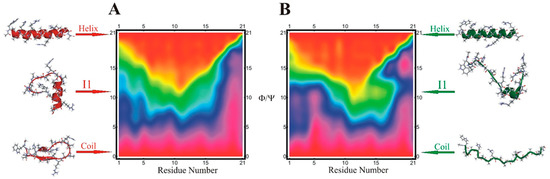
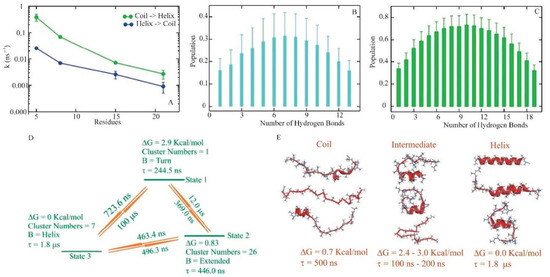
Dissecting Multiple Pathways in the Relaxation Dynamics of Helix <==> Coil Transitions with Optimum Dimensionality Reduction
by
Gouri S. Jas, Ed W. Childs, C. Russell Middaugh and Krzysztof Kuczera
Cited by 2 | Viewed by 3480
Abstract
Fast kinetic experiments with dramatically improved time resolution have contributed significantly to understanding the fundamental processes in protein folding pathways involving the formation of a-helices and b-hairpin, contact formation, and overall collapse of the peptide chain. Interpretation of experimental results through application of
[...] Read more.
Fast kinetic experiments with dramatically improved time resolution have contributed significantly to understanding the fundamental processes in protein folding pathways involving the formation of a-helices and b-hairpin, contact formation, and overall collapse of the peptide chain. Interpretation of experimental results through application of a simple statistical mechanical model was key to this understanding. Atomistic description of all events observed in the experimental findings was challenging. Recent advancements in theory, more sophisticated algorithms, and a true long-term trajectory made way for an atomically detailed description of kinetics, examining folding pathways, validating experimental results, and reporting new findings for a wide range of molecular processes in biophysical chemistry. This review describes how optimum dimensionality reduction theory can construct a simplified coarse-grained model with low dimensionality involving a kinetic matrix that captures novel insights into folding pathways. A set of metastable states derived from molecular dynamics analysis generate an optimally reduced dimensionality rate matrix following transition pathway analysis. Analysis of the actual long-term simulation trajectory extracts a relaxation time directly comparable to the experimental results and confirms the validity of the combined approach. The application of the theory is discussed and illustrated using several examples of helix <==> coil transition pathways. This paper focuses primarily on a combined approach of time-resolved experiments and long-term molecular dynamics simulation from our ongoing work.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
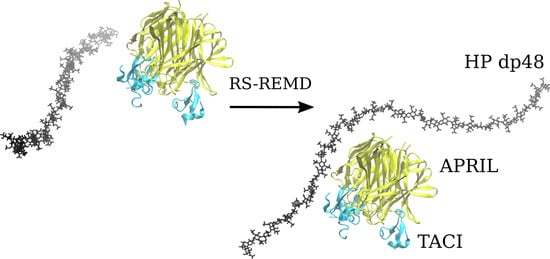
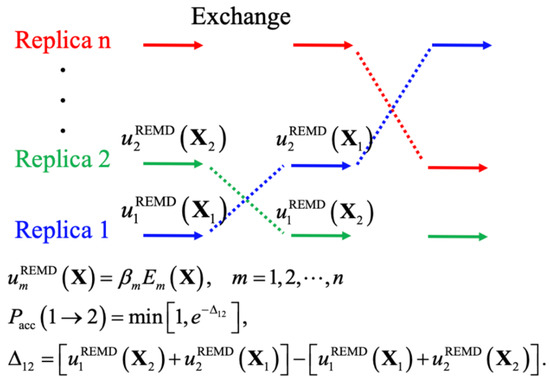
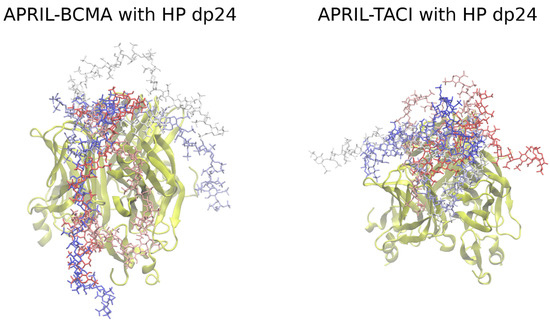
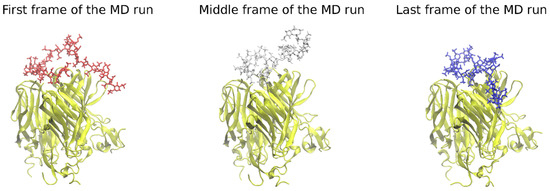
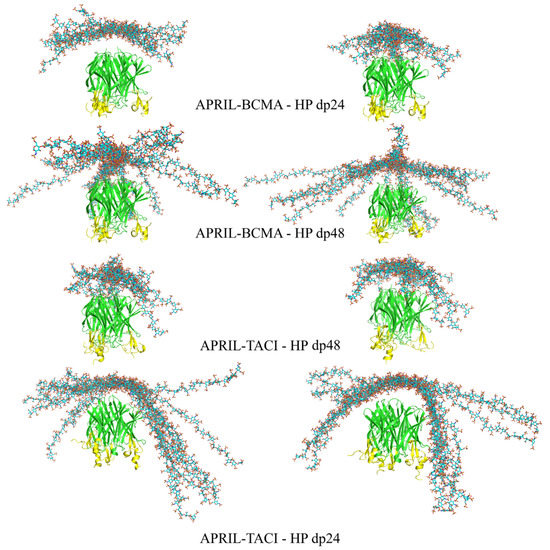
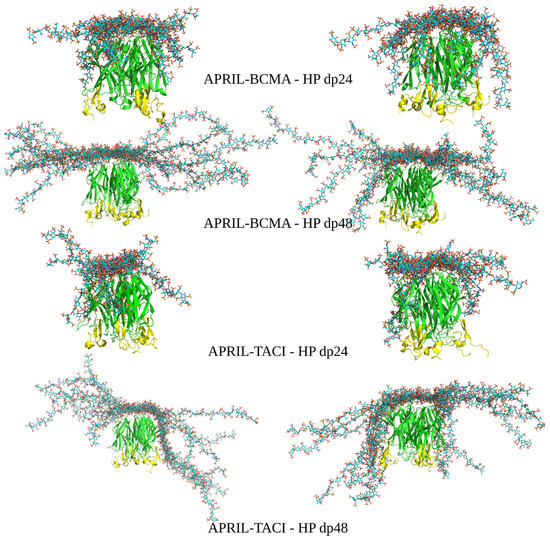
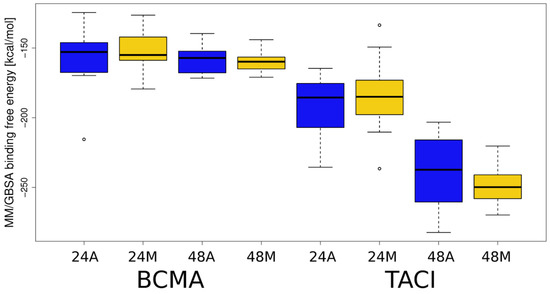
Advanced Molecular Dynamics Approaches to Model a Tertiary Complex APRIL/TACI with Long Glycosaminoglycans
by
Mateusz Marcisz, Martyna Maszota-Zieleniak, Bertrand Huard and Sergey A. Samsonov
Cited by 8 | Viewed by 3275
Abstract
Glycosaminoglycans (GAGs) are linear anionic periodic polysaccharides participating in a number of biologically relevant processes in the extracellular matrix via interactions with their protein targets. Due to their periodicity, conformational flexibility, pseudo-symmetry of the sulfation pattern, and the key role of electrostatics, these
[...] Read more.
Glycosaminoglycans (GAGs) are linear anionic periodic polysaccharides participating in a number of biologically relevant processes in the extracellular matrix via interactions with their protein targets. Due to their periodicity, conformational flexibility, pseudo-symmetry of the sulfation pattern, and the key role of electrostatics, these molecules are challenging for both experimental and theoretical approaches. In particular, conventional molecular docking applied for GAGs longer than 10-mer experiences severe difficulties. In this work, for the first time, 24- and 48-meric GAGs were docked using all-atomic repulsive-scaling Hamiltonian replica exchange molecular dynamics (RS-REMD), a novel methodology based on replicas with van der Waals radii of interacting molecules being scaled. This approach performed well for proteins complexed with oligomeric GAGs and is independent of their length, which distinguishes it from other molecular docking approaches. We built a model of long GAGs in complex with a proliferation-inducing ligand (APRIL) prebound to its receptors, the B cell maturation antigen and the transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI). Furthermore, the prediction power of the RS-REMD for this tertiary complex was evaluated. We conclude that the TACI–GAG interaction could be potentially amplified by TACI’s binding to APRIL. RS-REMD outperformed Autodock3, the docking program previously proven the best for short GAGs.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceArticle
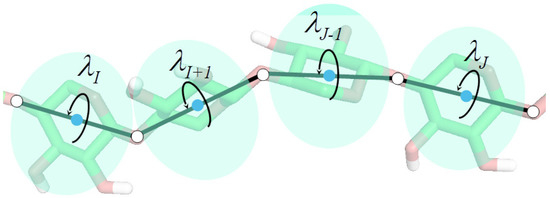
Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems
by
Adam Liwo, Cezary Czaplewski, Adam K. Sieradzan, Agnieszka G. Lipska, Sergey A. Samsonov and Rajesh K. Murarka
Cited by 56 | Viewed by 9789
Abstract
Molecular dynamics with coarse-grained models is nowadays extensively used to simulate biomolecular systems at large time and size scales, compared to those accessible to all-atom molecular dynamics. In this review article, we describe the physical basis of coarse-grained molecular dynamics, the coarse-grained force
[...] Read more.
Molecular dynamics with coarse-grained models is nowadays extensively used to simulate biomolecular systems at large time and size scales, compared to those accessible to all-atom molecular dynamics. In this review article, we describe the physical basis of coarse-grained molecular dynamics, the coarse-grained force fields, the equations of motion and the respective numerical integration algorithms, and selected practical applications of coarse-grained molecular dynamics. We demonstrate that the motion of coarse-grained sites is governed by the potential of mean force and the friction and stochastic forces, resulting from integrating out the secondary degrees of freedom. Consequently, Langevin dynamics is a natural means of describing the motion of a system at the coarse-grained level and the potential of mean force is the physical basis of the coarse-grained force fields. Moreover, the choice of coarse-grained variables and the fact that coarse-grained sites often do not have spherical symmetry implies a non-diagonal inertia tensor. We describe selected coarse-grained models used in molecular dynamics simulations, including the most popular MARTINI model developed by Marrink’s group and the UNICORN model of biological macromolecules developed in our laboratory. We conclude by discussing examples of the application of coarse-grained molecular dynamics to study biologically important processes.
Full article
►▼
Show Figures
Open AccessArticle
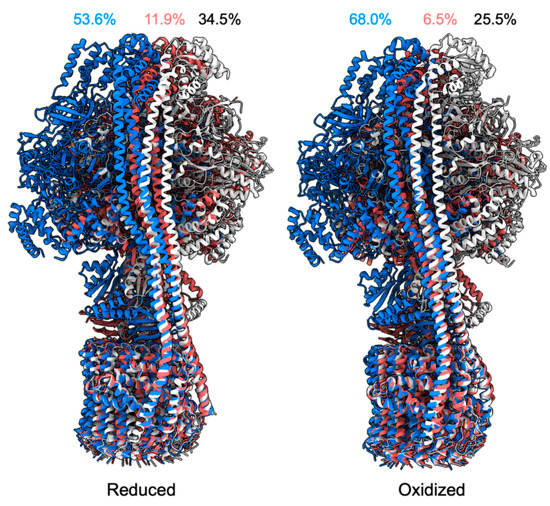
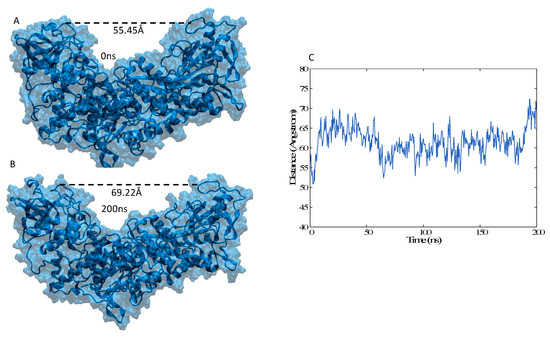
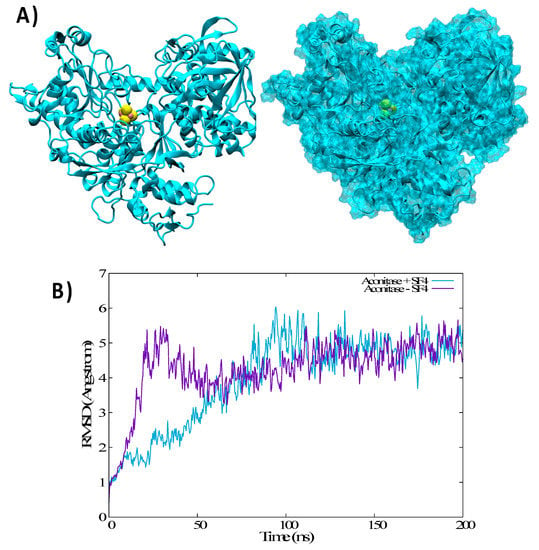
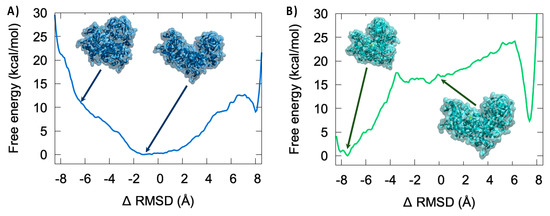
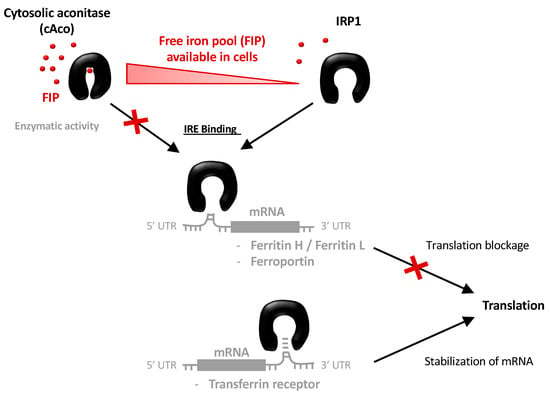
The Iron Maiden. Cytosolic Aconitase/IRP1 Conformational Transition in the Regulation of Ferritin Translation and Iron Hemostasis
by
Cécilia Hognon, Emmanuelle Bignon, Guillaume Harle, Nadège Touche, Stéphanie Grandemange and Antonio Monari
Cited by 7 | Viewed by 4410
Abstract
Maintaining iron homeostasis is fundamental for almost all living beings, and its deregulation correlates with severe and debilitating pathologies. The process is made more complicated by the omnipresence of iron and by its role as a fundamental component of a number of crucial
[...] Read more.
Maintaining iron homeostasis is fundamental for almost all living beings, and its deregulation correlates with severe and debilitating pathologies. The process is made more complicated by the omnipresence of iron and by its role as a fundamental component of a number of crucial metallo proteins. The response to modifications in the amount of the free-iron pool is performed via the inhibition of ferritin translation by sequestering consensus messenger RNA (mRNA) sequences. In turn, this is regulated by the iron-sensitive conformational equilibrium between cytosolic aconitase and IRP1, mediated by the presence of an iron–sulfur cluster. In this contribution, we analyze by full-atom molecular dynamics simulation, the factors leading to both the interaction with mRNA and the conformational transition. Furthermore, the role of the iron–sulfur cluster in driving the conformational transition is assessed by obtaining the related free energy profile via enhanced sampling molecular dynamics simulations.
Full article
►▼
Show Figures
Open AccessArticle
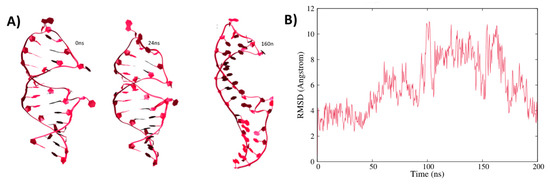
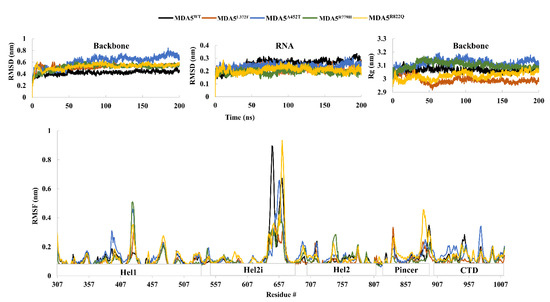
Computational Insights into the Structural Dynamics of MDA5 Variants Associated with Aicardi–Goutières Syndrome and Singleton–Merten Syndrome
by
Vijayakumar Gosu, Santanu Sasidharan, Prakash Saudagar, Hak-Kyo Lee and Donghyun Shin
Cited by 17 | Viewed by 4193
Abstract
Melanoma differentiation-associated protein 5 (MDA5) is a crucial RIG-I-like receptor RNA helicase enzyme encoded by
IFIH1 in humans. Single nucleotide polymorphisms in the
IFIH1 results in fatal genetic disorders such as Aicardi–Goutières syndrome and Singleton–Merten syndrome, and in increased risk of type I
[...] Read more.
Melanoma differentiation-associated protein 5 (MDA5) is a crucial RIG-I-like receptor RNA helicase enzyme encoded by
IFIH1 in humans. Single nucleotide polymorphisms in the
IFIH1 results in fatal genetic disorders such as Aicardi–Goutières syndrome and Singleton–Merten syndrome, and in increased risk of type I diabetes in humans. In this study, we chose four different amino acid substitutions of the MDA5 protein responsible for genetic disorders: MDA5
L372F, MDA5
A452T, MDA5
R779H, and MDA5
R822Q and analyzed their structural and functional relationships using molecular dynamic simulations. Our results suggest that the mutated complexes are relatively more stable than the wild-type MDA5. The radius of gyration, interaction energies, and intra-hydrogen bond analysis indicated the stability of mutated complexes over the wild type, especially MDA5
L372F and MDA5
R822Q. The dominant motions exhibited by the wild-type and mutant complexes varied significantly. Moreover, the betweenness centrality of the wild-type and mutant complexes showed shared residues for intra-signal propagation. The observed results indicate that the mutations lead to a gain of function, as reported in previous studies, due to increased interaction energies and stability between RNA and MDA5 in mutated complexes. These findings are expected to deepen our understanding of MDA5 variants and may assist in the development of relevant therapeutics against the disorders.
Full article
►▼
Show Figures
Open AccessArticle
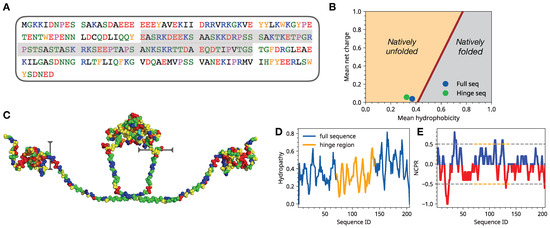
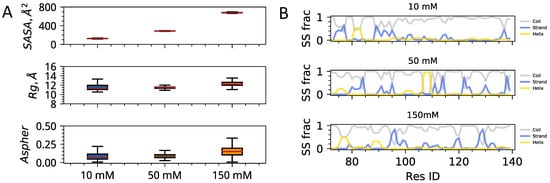
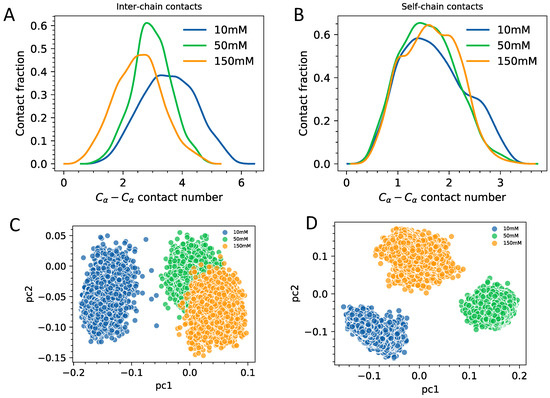
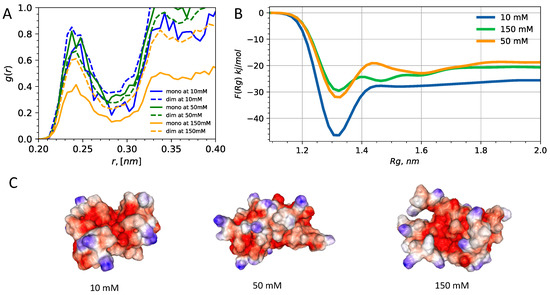
Solvent Exposure and Ionic Condensation Drive Fuzzy Dimerization of Disordered Heterochromatin Protein Sequence
by
Jazelli Mueterthies and Davit A. Potoyan
Cited by 5 | Viewed by 3687
Abstract
Proteins with low complexity, disordered sequences are receiving increasing attention due to their central roles in the biogenesis and regulation of membraneless organelles. In eukaryotic organisms, a substantial fraction of disordered proteins reside in the nucleus, thereby facilitating the formation of nuclear bodies,
[...] Read more.
Proteins with low complexity, disordered sequences are receiving increasing attention due to their central roles in the biogenesis and regulation of membraneless organelles. In eukaryotic organisms, a substantial fraction of disordered proteins reside in the nucleus, thereby facilitating the formation of nuclear bodies, nucleolus, and chromatin compartmentalization. The heterochromatin family of proteins (HP1) is an important player in driving the formation of gene silenced mesoscopic heterochromatin B compartments and pericentric regions. Recent experiments have shown that the HP1a sequence of
Drosophila melanogaster can undergo liquid-liquid phase separation under both in vitro and in vivo conditions, induced by changes of the monovalent salt concentration. While the phase separation of HP1a is thought to be the mechanism underlying chromatin compartmentalization, the molecular level mechanistic picture of salt-driven phase separation of HP1a has remained poorly understood. The disordered hinge region of HP1a is seen as the driver of salt-induced condensation because of its charge enriched sequence and post-translational modifications. Here, we set out to decipher the mechanisms of salt-induced condensation of HP1a through a systematic study of salt-dependent conformations of single chains and fuzzy dimers of disordered HP1a hinge sequences. Using multiple independent all-atom simulations with and without enhanced sampling, we carry out detailed characterization of conformational ensembles of disordered HP1a chains under different ionic conditions using various polymeric and structural measures. We show that the mobile ion release, enhancement of local transient secondary structural elements, and side-chain exposure to solvent are robust trends that accompany fuzzy dimer formation. Furthermore, we find that salt-induced changes in the ensemble of conformations of HP1a disordered hinge sequence fine-tune the inter-chain vs. self-chain interactions in ways that favor fuzzy dimer formation under low salt conditions in the agreement with condensation trends seen in experiments.
Full article
►▼
Show Figures
Open AccessArticle
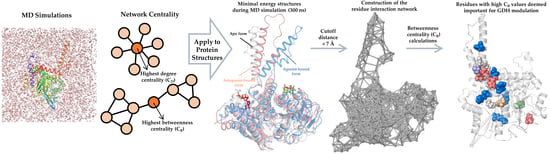
Mapping the Intramolecular Communications among Different Glutamate Dehydrogenase States Using Molecular Dynamics
by
Shaherin Basith, Balachandran Manavalan, Tae Hwan Shin and Gwang Lee
Cited by 14 | Viewed by 4227
Abstract
Glutamate dehydrogenase (GDH) is a ubiquitous enzyme that catalyzes the reversible oxidative deamination of glutamate to α-ketoglutarate. It acts as an important branch-point enzyme between carbon and nitrogen metabolisms. Due to the multifaceted roles of GDH in cancer, hyperinsulinism/hyperammonemia, and central nervous system
[...] Read more.
Glutamate dehydrogenase (GDH) is a ubiquitous enzyme that catalyzes the reversible oxidative deamination of glutamate to α-ketoglutarate. It acts as an important branch-point enzyme between carbon and nitrogen metabolisms. Due to the multifaceted roles of GDH in cancer, hyperinsulinism/hyperammonemia, and central nervous system development and pathologies, tight control of its activity is necessitated. To date, several GDH structures have been solved in its closed form; however, intrinsic structural information in its open and apo forms are still deficient. Moreover, the allosteric communications and conformational changes taking place in the three different GDH states are not well studied. To mitigate these drawbacks, we applied unbiased molecular dynamic simulations (MD) and network analysis to three different GDH states i.e., apo, active, and inactive forms, for investigating their modulatory mechanisms. In this paper, based on MD and network analysis, crucial residues important for signal transduction, conformational changes, and maps of information flow among the different GDH states were elucidated. Moreover, with the recent findings of allosteric modulators, an allosteric wiring illustration of GDH intramolecular signal transductions would be of paramount importance to obtain the process of this enzyme regulation. The structural insights gained from this study will pave way for large-scale screening of GDH regulators and could support researchers in the design and development of new and potent GDH ligands.
Full article
►▼
Show Figures
Open AccessArticle
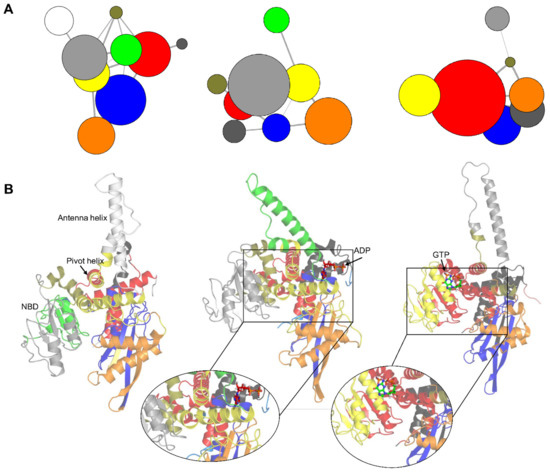
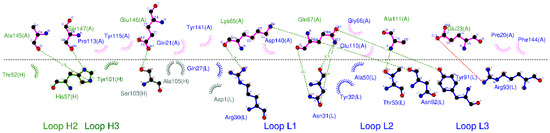
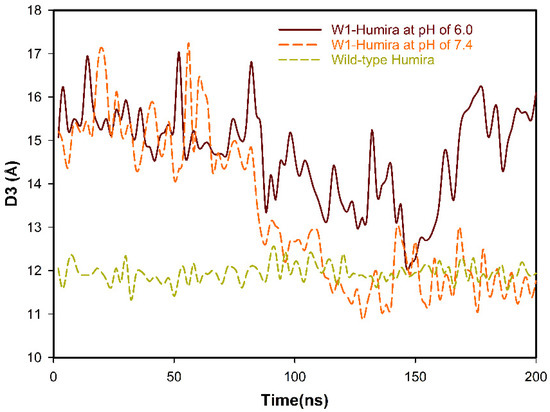
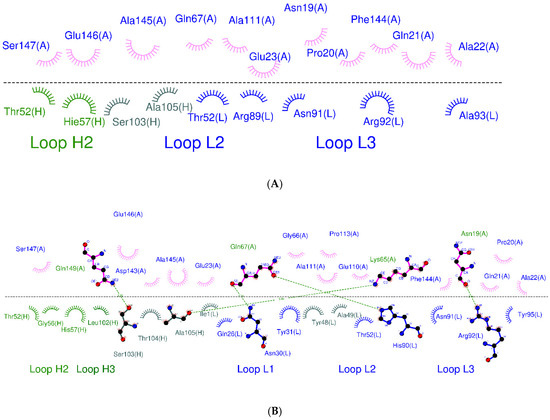
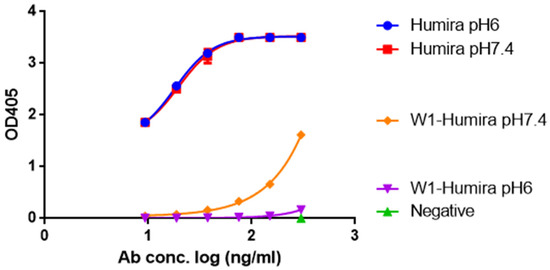
Anti-TNF Alpha Antibody Humira with pH-dependent Binding Characteristics: A constant-pH Molecular Dynamics, Gaussian Accelerated Molecular Dynamics, and In Vitro Study
by
Shih-Ting Hong, Yu-Cheng Su, Yu-Jen Wang, Tian-Lu Cheng and Yeng-Tseng Wang
Cited by 6 | Viewed by 6268
Abstract
Humira is a monoclonal antibody that binds to TNF alpha, inactivates TNF alpha receptors, and inhibits inflammation. Neonatal Fc receptors can mediate the transcytosis of Humira–TNF alpha complex structures and process them toward degradation pathways, which reduces the therapeutic effect of Humira. Allowing
[...] Read more.
Humira is a monoclonal antibody that binds to TNF alpha, inactivates TNF alpha receptors, and inhibits inflammation. Neonatal Fc receptors can mediate the transcytosis of Humira–TNF alpha complex structures and process them toward degradation pathways, which reduces the therapeutic effect of Humira. Allowing the Humira–TNF alpha complex structures to dissociate to Humira and soluble TNF alpha in the early endosome to enable Humira recycling is crucial. We used the cytoplasmic pH (7.4), the early endosomal pH (6.0), and pK
a of histidine side chains (6.0–6.4) to mutate the residues of complementarity-determining regions with histidine. Our engineered Humira (W1-Humira) can bind to TNF alpha in plasma at neutral pH and dissociate from the TNF alpha in the endosome at acidic pH. We used the constant-pH molecular dynamics, Gaussian accelerated molecular dynamics, two-dimensional potential mean force profiles, and in vitro methods to investigate the characteristics of W1-Humira. Our results revealed that the proposed Humira can bind TNF alpha with pH-dependent affinity in vitro. The W1-Humira was weaker than wild-type Humira at neutral pH in vitro, and our prediction results were close to the in vitro results. Furthermore, our approach displayed a high accuracy in antibody pH-dependent binding characteristics prediction, which may facilitate antibody drug design. Advancements in computational methods and computing power may further aid in addressing the challenges in antibody drug design.
Full article
►▼
Show Figures
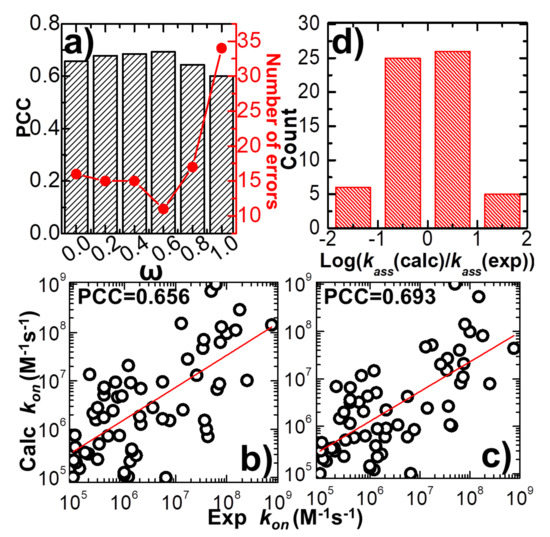
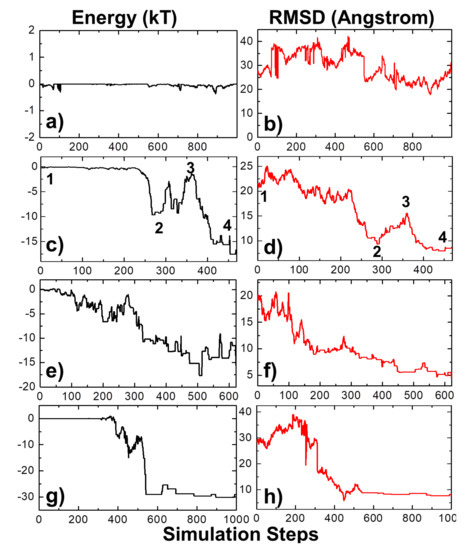
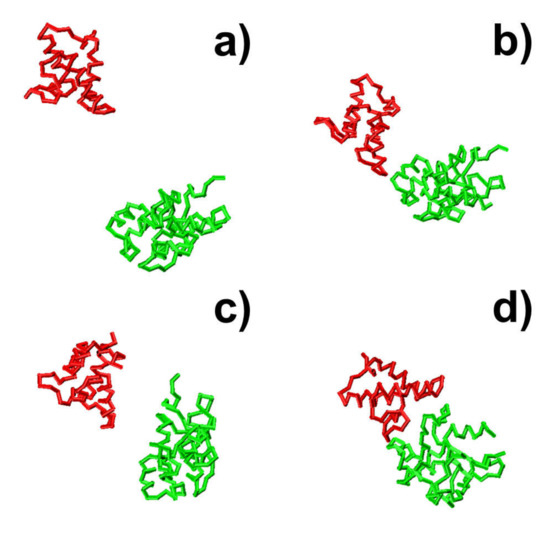
Open AccessArticle
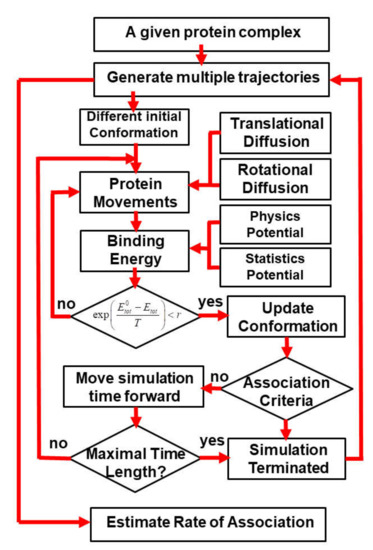
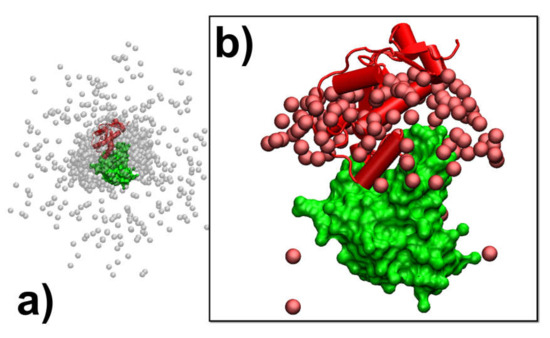
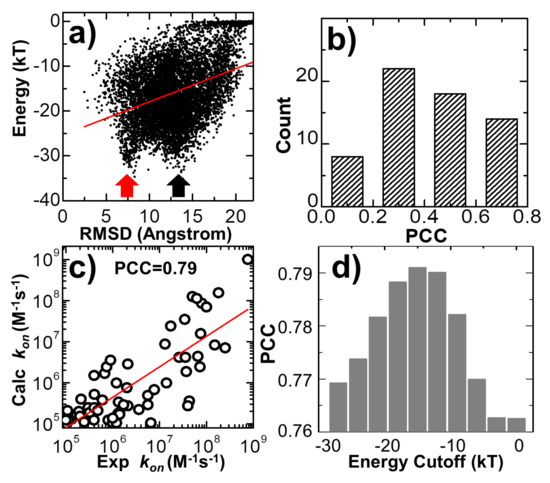
Using Coarse-Grained Simulations to Characterize the Mechanisms of Protein–Protein Association
by
Kalyani Dhusia, Zhaoqian Su and Yinghao Wu
Cited by 7 | Viewed by 4438
Abstract
The formation of functionally versatile protein complexes underlies almost every biological process. The estimation of how fast these complexes can be formed has broad implications for unravelling the mechanism of biomolecular recognition. This kinetic property is traditionally quantified by association rates, which can
[...] Read more.
The formation of functionally versatile protein complexes underlies almost every biological process. The estimation of how fast these complexes can be formed has broad implications for unravelling the mechanism of biomolecular recognition. This kinetic property is traditionally quantified by association rates, which can be measured through various experimental techniques. To complement these time-consuming and labor-intensive approaches, we developed a coarse-grained simulation approach to study the physical processes of protein–protein association. We systematically calibrated our simulation method against a large-scale benchmark set. By combining a physics-based force field with a statistically-derived potential in the simulation, we found that the association rates of more than 80% of protein complexes can be correctly predicted within one order of magnitude relative to their experimental measurements. We further showed that a mixture of force fields derived from complementary sources was able to describe the process of protein–protein association with mechanistic details. For instance, we show that association of a protein complex contains multiple steps in which proteins continuously search their local binding orientations and form non-native-like intermediates through repeated dissociation and re-association. Moreover, with an ensemble of loosely bound encounter complexes observed around their native conformation, we suggest that the transition states of protein–protein association could be highly diverse on the structural level. Our study also supports the idea in which the association of a protein complex is driven by a “funnel-like” energy landscape. In summary, these results shed light on our understanding of how protein–protein recognition is kinetically modulated, and our coarse-grained simulation approach can serve as a useful addition to the existing experimental approaches that measure protein–protein association rates.
Full article
►▼
Show Figures







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}