1. Introduction
Nearly all biological processes in living cells are achieved by the interactions among functionally different proteins [
1,
2,
3,
4,
5,
6]—for instance, the assembly of transient signaling complexes [
7,
8] or more stable biomolecular machines [
9,
10,
11]. The dynamic properties of protein interactions are not only characterized by the dissociation constants (
Kd), which determines the thermodynamic stability of a protein complex [
12], but also the association rate (
kon), which measures its kinetics, i.e., how fast this complex can be formed [
13,
14]. In a real cellular environment, interactions between proteins are often under kinetic, rather than thermodynamic, control [
15,
16]. For instance, proteins in cell signaling networks often have more than one binding partner that compete with each other. The difference in the speed of binding between these interactions, no matter how strong they are, regulates the dynamics of signal flows in the network. One example is the natural-killer (NK) cell receptor NKG2D (natural-killer group 2, member D) [
17]. The receptor recognizes both cellular and viral ligands with the same binding interface, indicating that these ligands must compete with each other for receptor binding when they coexist in the system. As a result, the difference in association rates of receptor binding between cellular and viral ligands directly regulates the NK cytolytic activity [
18]. Additionally, mutations at the binding interfaces of protein complexes often alter the rates of their associations and thereby result in severe pathological outcomes [
19]. Using the same example of NKG2D, individuals who carry specific mutants that lower the binding specificity of the receptor to the viral ligands are more likely to get infected by the virus [
20]. Therefore, studies of the protein–protein association mechanism on a quantitative level are significant for us to understand the dynamics of many cellular activities; furthermore, they have broad impacts on many other fields, such as drug discovery and protein design.
Relative to the current experimental techniques for measuring the rates of protein–protein interactions, such as surface plasma resonance (SPR) [
21] and spectroscopic inhibition assay (IASP) [
22], computational modeling approaches are not only less time-consuming and labor-intensive, but can also provide mechanistic details that are inaccessible in the laboratory. As a result, a large variety of computational methods have already been developed to study protein–protein association. Some methods can directly predict association rate constants by feeding the structural and chemical features collected from the binding interfaces of protein complexes into machine-learning-based models [
23,
24]. These models, however, are not able to provide any insights along the pathway of protein–protein association. Methods using physics-based principles, on the contrary, are used to simulate the detailed process of association. Among these methods, all-atom molecular dynamic (MD) simulations based on the explicit solvent model can reveal the complete protein–protein association kinetics [
25,
26,
27,
28,
29,
30,
31,
32]. Through MD simulation, it was found that the native protein complexes can be associated through a very structurally diverse transition state ensemble in which no more than 20% of native contacts remained [
33]. These all-atom simulations, however, are extremely demanding for computational resources and have so far only been successfully applied to a limited number of cases [
33,
34]. In comparison, the Brownian dynamic (BD) simulations, based on the implicit solvent model, are more computationally efficient and thus are widely used to study protein–protein association [
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57]. In particular, a recent method based on BD simulation and a “transient-complex” theory was proven to be able to successfully predict protein association rates and provide mechanistic insights into the association processes [
58,
59,
60,
61].
We have previously also developed a coarse-grained simulation approach to study protein–protein association [
62]. Positive correlations have been observed between the experimental measurements and our calculated values of association rates. However, the method has not been used to explore the detailed mechanism of association. In this work, we discussed this possibility by systematically calibrating the simulations against a comprehensive benchmark set. For each complex in the benchmark, a large number of simulation trajectories was carried out. Based on the statistical distributions of these trajectories, we showed that an ensemble of loosely bound encounter complexes were formed around their native conformation, suggesting that the transition states of protein–protein association could be highly diverse on the structural level. The analysis of each individual trajectory further suggested that association could be a dynamic process for searching local binding configurations by repeated dissociation and re-association. We found a correlation between the binding energy used in the simulations and the structural similarity of encounter complexes to their native conformation, implying a “funnel-like” landscape of protein–protein interactions along the pathways of their association. The correlation between experimental and our computationally simulated rates of association became stronger after we introduced a statistical potential into the simulations. Finally, we explored the combination of different criteria for encounter complex formation. In summary, our results provided insights into the common features underlying the general process of protein–protein association. This approach therefore offers quantitative characterizations of the dynamics of protein complex formation and can improve our understanding of the mechanisms of these important biological processes.
3. Results
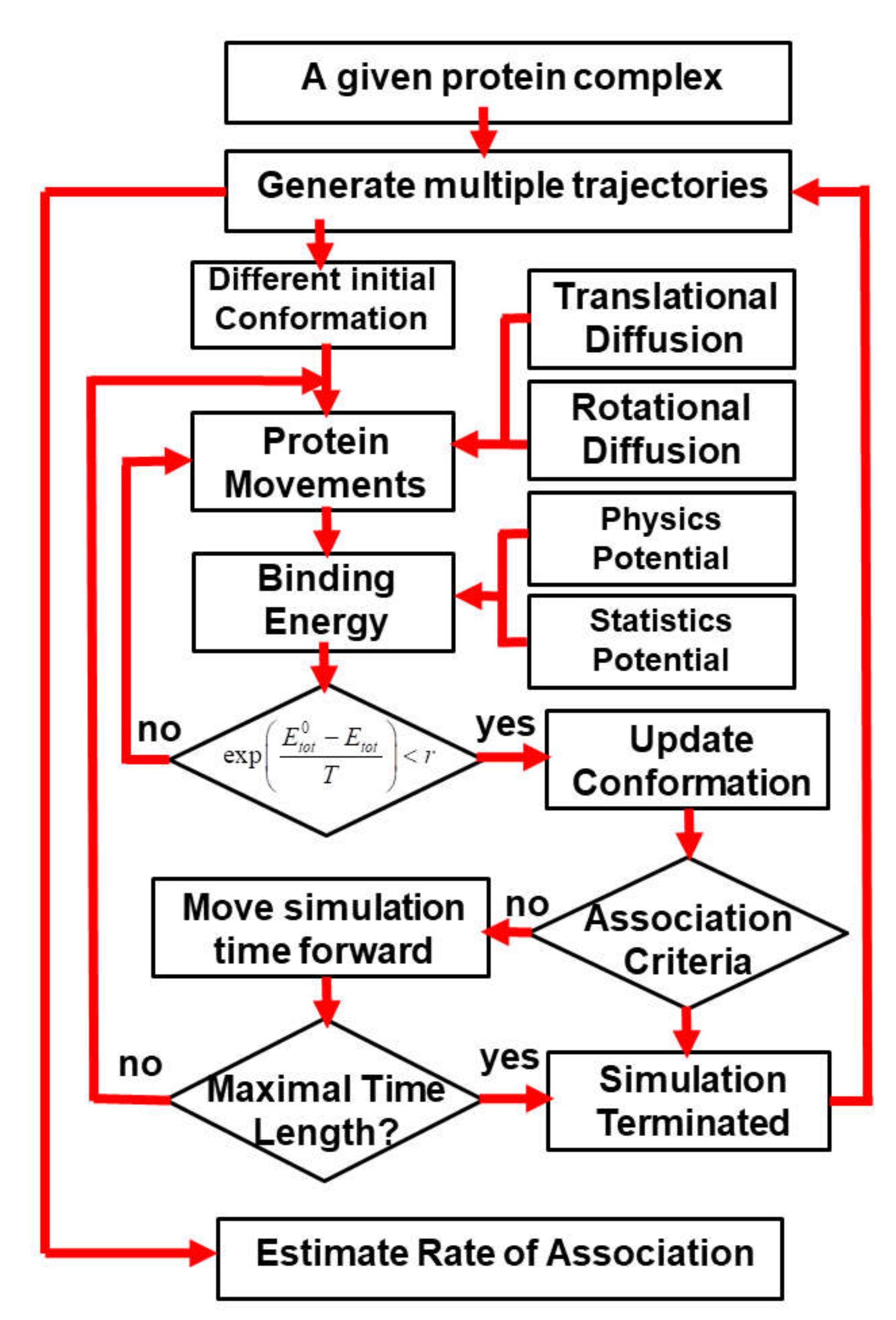
In order to systematically test the generality of our kinetic Monte Carlo (kMC) simulation in estimating the association rates of different protein complexes, we collected 62 protein complexes from previous literature as a benchmark set. The criteria of benchmark construction are described in the Methods. Detailed information about the benchmark set, including the PDB IDs, the index of two binding partners and their corresponding experimental values of association rates, is listed in
Table S1 in the Supplementary Materials. For all these protein complexes, 10
4 trajectories were carried out by the kinetic Monte Carlo simulation algorithm (a detailed description of the algorithm can be found in the Methods and in
Figure 1). Each trajectory started from a different initial random configuration. After the initial conformation, diffusions of each binding are guided by the intermolecular energies. At the end of each trajectory, two binding partners either successfully or unsuccessfully form an encounter complex through the pre-defined association criteria. Based on counting how many encounter complexes are observed from all the trajectories, the association probability can be derived for each complex in the benchmark set. The calculated association probability
Pi for complex
i will be further converted into a rate constant
by the following equation:
The parameters Pmin and Pmax in Equation (5) stand for the minimal and maximal values of association probabilities that were obtained from all the protein complexes in the benchmark. The parameter equals 1.03 × 105, which stands for the lowest value of experimentally measured association rates observed in the benchmark. The parameter N indicates how many orders of magnitudes for experimental association rates are considered in our benchmark. As described in the Methods, because we only considered protein complexes whose rate constants are within the range of 1.0 × 105 and 1.0 × 109 M−1s−1, the value of N equals 4 in Equation (5).
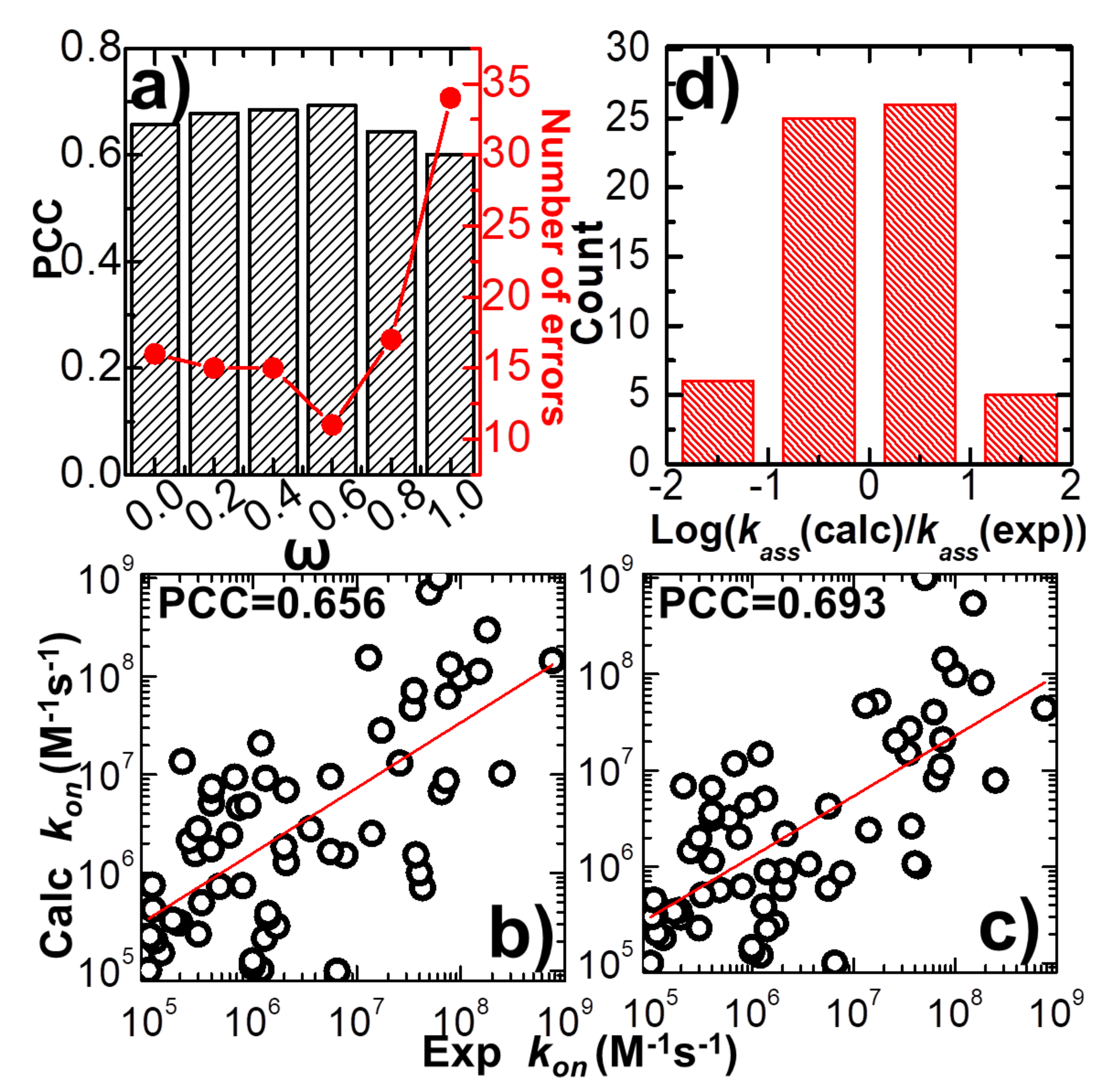
The force field which was developed to delineate the intermolecular interaction in our kinetic Monte Carlo simulations combines a previously constructed physics-based potential with a newly added statistics-based potential. A weight constant, ω, is used to define the relative contributions of these two potentials, as shown in Equation (1). In order to explore the capability of this new hybrid force field in estimating the association rates of different protein complexes, we adjusted the value of this weight constant from 0 to 1. Only the physics-based potential is used in the simulations when the weight constant equals 0, while only the statistics-based potential is used when the weight equals 1. Given a specific value for the weight constant, the association rate was calculated for each protein complex in the benchmark based on the statistical analysis from its 104 simulation trajectories, as illustrated in the previous paragraph. We compared the calculated association rates of all protein complexes with their corresponding experimental measurements. Consequently, the Pearson correlation coefficient (PCC) between these two datasets was obtained under different values of weight constant.
In detail, the variation between weight constant and calculated PCC is plotted in
Figure 2a. The figure shows that without the statistics-based potential (ω = 0), we can still achieve a positive correlation between simulations and experiments. A log 10 base plot between our calculated values of association rates and their experimental data for all 62 complexes is displayed in
Figure 2b under this circumstance, with a Pearson correlation coefficient of 0.656. When the statistics-based potential started to be added into the force field,
Figure 2a suggests that a higher correlation was obtained. The value of PCC peaks when the weight constant equals 0.6. The comparison between simulated and experimental association rates for all 62 complexes is displayed in
Figure 2c under this circumstance, with a Pearson correlation coefficient of 0.693. However, the correlation between simulations and experiments will drop if the weight of the statistics-based potential increases further. Finally, the lowest correlation (PCC = 0.6) was attained without the physics-based potential (ω = 1). Taken together, our simulation results indicate that our kinetic Monte Carlo simulation can distinguish the kinetics of protein–protein interactions within a wide range of association rates. Moreover, the optimal combination between the physics-based and the statistics-based potentials was able to improve the simulation’s accuracy.
The common logarithm of the ratio between simulated and experimental association rates
was further calculated for all complexes in the benchmark, under the optimal value of weight constant (ω = 0.6). If this value was larger than 1 for a protein complex, its association rate was overestimated over an order of magnitude relative to the experimental data. On the other hand, a protein complex with a value smaller than -1 indicates that its association rate was underestimated over an order of magnitude relative to the experimental data.
Figure 2d shows the distribution of our calculations. The histogram in the figure indicates that among all 62 complexes, only six were underestimated over an order of magnitude, and five were overestimated over an order of magnitude. The association rates of the remaining 51 protein complexes were reproduced within one order of magnitude. We assume that the association rate of a protein complex can be correctly predicted if the difference between our calculated value and its corresponding experimental data is below one order of magnitude. This is a reasonable estimation given the fact that the experimentally observed association rates span an extremely wide range, with over ten orders of magnitudes. Based on this criterion, we show that our simulation to predict the protein–protein association rates can reach an overall accuracy of 80%. The calculated association probabilities and rates for all protein complexes in the benchmark under the weight constant 0.6 are listed in
Table S1.
Based on the definition of
, we further counted the number of errors in the prediction of association rates for all protein complexes in the benchmark. The error of prediction for a specific protein complex is marked as its absolute value of calculated
, which is larger than 1, indicating that the difference between experimental and predicted association rates is higher than one order of magnitude. The total numbers of errors from the prediction were derived under all different values of the weight constant ω. The values are plotted in
Figure 2a as red dots and lines. The figure shows that the change in prediction errors as a function of weight constant ω correlates well to the variation in PCC. The highest value of PCC corresponds to the lowest number of errors when ω equals 0.6. This result confirms that the accuracy in predicting the rates of protein–protein association can be marginally improved when an original version of the physics-based potential is complemented with a new statistics-based potential with an optimal weight.
We selected a protein complex from the benchmark as a specific example to characterize the dynamic mechanisms of association. In detail, the complex formed between the E9 DNase domain of colicin endonucleases and immunity protein Im9 (PDB id 2VLN) was selected as a test system [
78]. Among a total number of 10
4 simulation trajectories, with a maximal duration of 1000 ns for each individual trajectory, we found that encounter complexes were successfully formed in 1819 of them. This indicates that the value of the association probability equals 0.1819. Using Equation (5), we further calculated the rate of association between proteins E9 DNase domain and Im9. The association rate of the complex equals 9.86 × 10
7 M
−1s
−1, which is close to the experimental measurement (1.0 × 10
8 M
−1s
−1) [
78]. The physical processes along four representative simulation trajectories are illustrated in
Figure 3. The changes in the total intermolecular interactions between two binding partners in these four trajectories are plotted by black curves in
Figure 3a,c,e,f as a function of simulation steps, while their changes in the root-mean-square difference (RMSD) from the native complex are plotted by red curves in
Figure 3b,d,f,g, respectively. The RMSD is large at the beginning of all four trajectories, resulting from their initial random configurations. As a result, the intermolecular interactions at the initial stage of association in all four systems are very close to 0, indicating that two binding partners were separated from each other and diffused independently in the simulation box.
In the first system, small fluctuations in total energy were observed at around 0 throughout the simulation trajectory, as shown in
Figure 3a. In comparison, a high level of RMSD was sustained along the trajectory, with large fluctuations (
Figure 3b), indicating that the proteins cannot associate into complexes by the end of the maximal time duration in this system. On the contrary, complexes were successfully formed in the next three trajectories. This is reflected by the fact that the intermolecular interactions dropped to negative values in these systems, while their RMSD values also decreased to levels below 10 Å by the end of the simulations. In these cases, the proteins have spatially approached each other and found their actual binding sites after they diffused throughout the simulation box. Additionally,
Figure 3 shows that the formation of the protein complex is faster in some trajectories than others. For instance, the complex in the second trajectory was formed at 470 ns, whereas the complex in the third trajectory was formed at 630 ns. Moreover, in most of these trajectories, we found that local energy minimal states were formed between proteins along the pathways to their final associations. Similar to the process of folding [
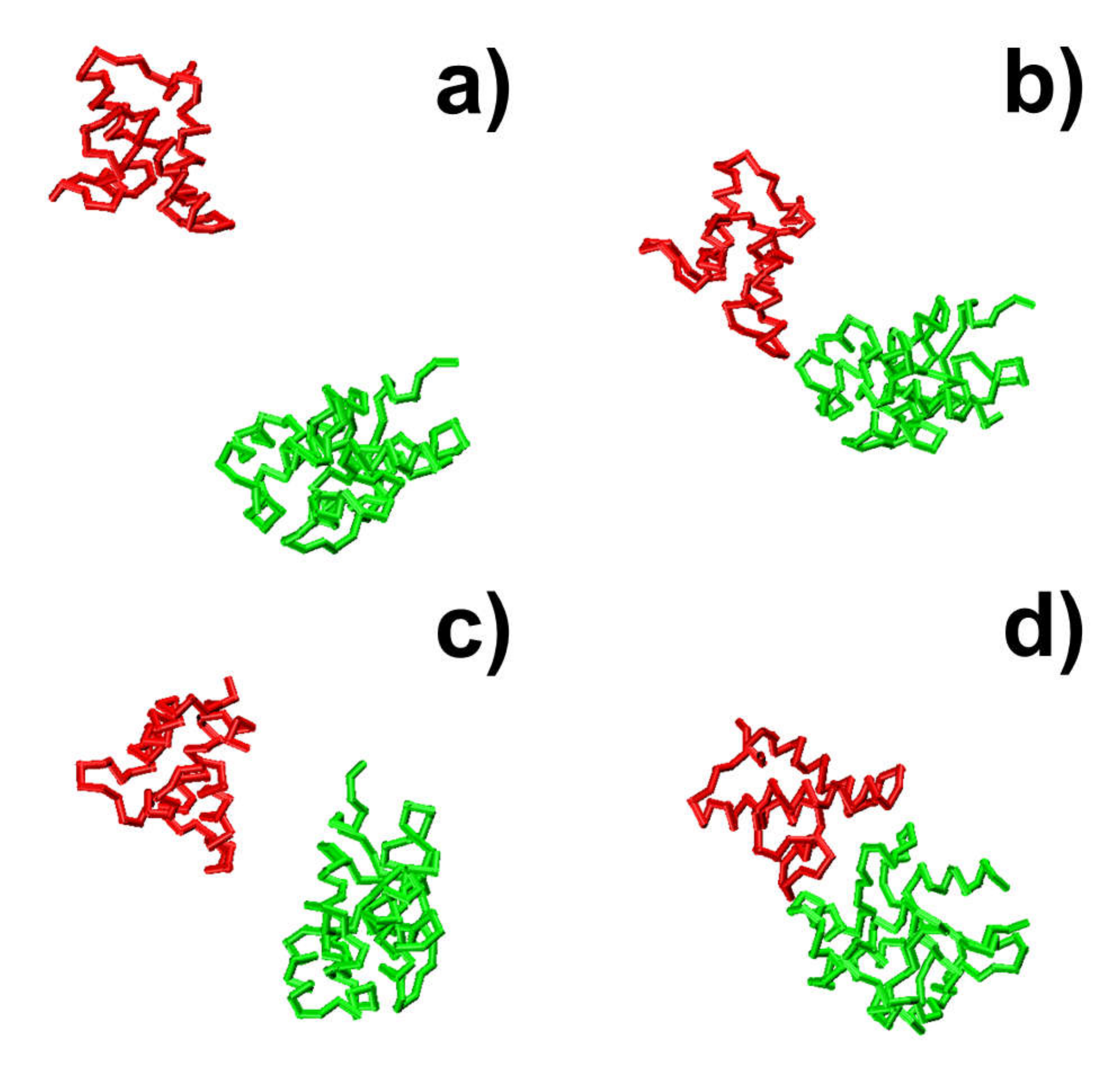
79], protein complexes are able to overcome the energy barrels between these local minimal states and reach the final native-like configuration. For instance, in the second trajectory, following the initial configuration, which has an energy level of zero and a large RMSD value, we found that the system formed a relatively stable structure, in which both energy and RMSD dropped to relatively low levels. The initial configuration is denoted by the number “1” in
Figure 3c,d, and its corresponding snapshot from the trajectory is shown in
Figure 4a. The local energy minimal state is denoted by the number “2” in the figures, and its corresponding snapshot from the trajectory is shown in
Figure 4b. A tentative binding interface is formed between two proteins, as observed in the figure. After the formation of this tentative complex, both the energy in the system and the RMSD of the protein complex started to increase and peaked at the point denoted by the number “3” in
Figure 3c,d and the corresponding snapshot shown in
Figure 4c. The comparison between structures in
Figure 4c suggests that the tentative complex was dissociated, and two proteins were reorganized into a different orientation. Finally, both the energy in the system and the RMSD of the two proteins started to drop again until they formed the final encounter complex, which is denoted by the number “4” in
Figure 3c,d and the corresponding structure shown in
Figure 4d. The comparison between structures in
Figure 4b,d suggests that a more compact complex was formed in the final configuration compared to the tentative complex.
Taken together, the analysis of simulation details reveals the dynamic mechanism of association. We showed that the individual association pathway is a multistep process which involves continuously searching local binding configurations and repeatedly forming kinetic intermediates with dissociation and re-association. A similar phenomenon has also been observed in a previous study using all-atom molecular dynamic simulations [
33,
34].
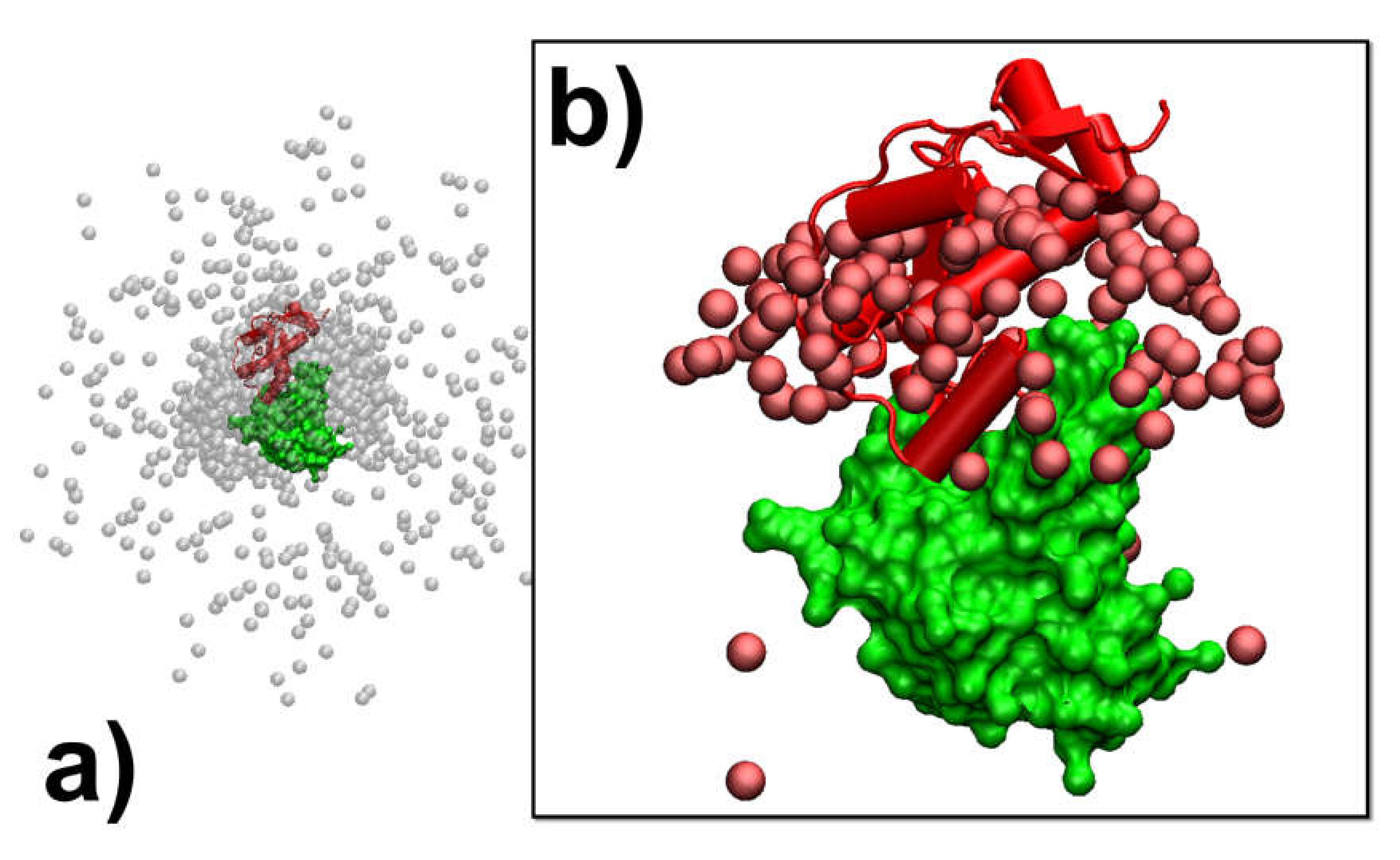
In addition to the analysis of the individual association pathways, it is also helpful to compare results between different simulation trajectories. As a result, protein complex 2VLN was still used as a test system. We plotted the spatial distribution of the final configurations that were generated from all the simulation trajectories in order to obtain an overview of the structural similarity among all the successfully formed encounter complexes. This distribution is plotted in
Figure 5a. In the figure, the native structure of the protein complex is placed in the center of the box, while the protein E9 DNase domain is shown in green with its surface profile and Im9 is shown in red in the cartoon representation. In order to attain a better visual effect, 10
3 final configurations were randomly selected from the 10
4 trajectories to avoid spatial crowding in the figure. The structures of protein E9 DNase domain in these configurations were aligned with the native configuration, while the centers of mass for the protein Im9 are represented by the grey points.
Figure 5a shows that there are a number of grey points uniformly distributed in the simulation box. These points represent the situation in which protein Im9 still diffuses around its binding partner. With the exception of these configurations, the majority of grey points are distributed around the center. These points represent the situation in which Im9 has formed a physical interface with protein E9 DNase domain.
According to the association probability calculated for protein complex 2VLN, encounter complexes were successfully formed in about 200 final configurations out of 10
3 selected trajectories. We further plotted the spatial distribution of these configurations in
Figure 5b. As in
Figure 5a, the native structure of the protein complex is placed in the center of the box, with the protein E9 DNase domain in a green surface profile and Im9 in a red cartoon representation, while the centers of mass for the protein Im9 are represented by the red points. The figure shows that the centers of mass for almost all Im9 proteins in the encounter complexes are distributed relatively closer to the native conformation than the distribution observed in
Figure 5a, although a highly noticeable range of spatial deviation exists. This indicates that encounter complexes constitute an ensemble of loosely bound structures formed around their native conformation. This ensemble of encounter complex thus suggests that the transition states of protein–protein association could be highly diverse on the structural level. Furthermore, the combination of
Figure 5a,b with the previous two figures enables the following mechanistic insights into the physical process of protein association. There are overall three outcomes observed in the simulations of association. In particular, two binding partners either 1) freely diffuse by the end of the maximal time duration, 2) form a physical interface that is different from the native conformation and remain kinetically trapped in this nonnative state, or 3) form an initial tentative contact, that is different from the native conformation, but later successfully transit into the correct binding interface through the process of dissociation and re-association. Moreover, for all trajectories in which encounter complexes were formed through the third pathway, their structures displayed a wide variety of relative protein–protein orientations around the native conformation.
In summary, the analysis of the overall simulation trajectories reveals, firstly, that association between proteins could be kinetically trapped into nonnative states, secondly, that the pathway towards the final formation of the encounter complex is a dynamic process consisting of local structural reorganization and, finally, that even the structures in the final ensemble of encounter complexes are highly diverse. These features are consistent with the observations in another previous study using all-atom molecular dynamic simulations [
33,
34].
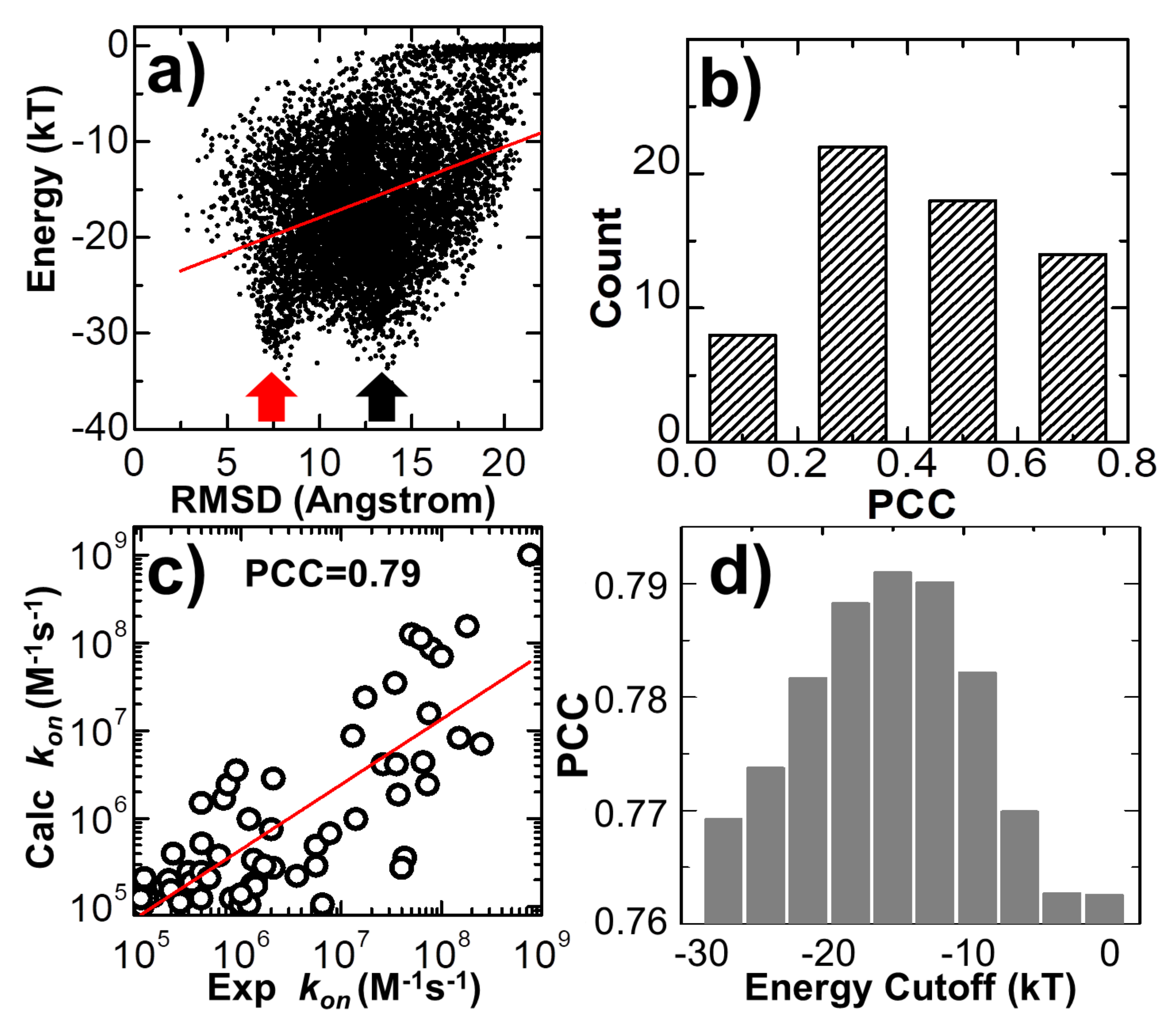
In the last three trajectories plotted in
Figure 3, we found that the decreases in the intermolecular energy in the systems co-variated with the RMSD between the conformation of the protein complex and its native structure. This suggests that the simulations of association were driven by the intermolecular energy. In order to further test whether this energetic variable could be used as an indicator to capture the features shared among encounter complexes, we compared the values of RMSD with intermolecular energy for all the final configurations taken from the end of 10
4 trajectories. The correlation is shown in
Figure 6a for the complex 2VLN. Each of the ten thousand points in the figure stands for the final configuration from one trajectory, while its
y-axis equals the intermolecular energy calculated based on this configuration, and the
x-axis is its RMSD from the native structure of the protein complex. The group of trajectories in which two binding partners failed to form a contact are reflected by the tip in the top-right corner of the figure. The distribution below the tip, on the other hand, indicates a large variety of complexes formed between these two binding partners in the remaining trajectories. We found a strong positive correlation in this distribution. The calculated Pearson correlation coefficient equals 0.7. In particular, a large group of encounter complexes formed through the lowest intermolecular energy, as indicated by the red arrow in
Figure 6a, show small values of RMSD of around 7 Å. We further calculated the Pearson correlation coefficient between RMSD and intermolecular energy from the end of the 10
4 simulation trajectories for all 62 protein complexes in the benchmark. The distribution of Pearson correlation coefficient values is plotted in
Figure 6b. The figure shows that the correlations for all protein complexes are positive, while more than half of them have strong correlations that are higher than 0.5. This positive correlation between binding energy and RMSD from the native structures suggests that protein complexes formed with native-like protein interfaces tend to have lower binding energies than complexes that are trapped in the non-native states. As a result, the structural similarity embedded in the ensemble of encounter complexes can be characterized by the intermolecular energy. In other words, the positive correlation between energetic and structural features in protein complex formation indicates that protein–protein association is a kinetic process similar to protein folding [
79], in which diffusions of binding partners are driven by a “funnel-like” energy landscape.
In the last section of the Results, we explored the combination of different criteria for encounter complex formation. As described in the Methods, the number of intermolecular contacts recovered from the native structure was used as a single criterion to determine whether an encounter complex can be successfully formed along simulations. Here, in addition to the number of native contacts, we further considered four other criteria. The first criterion is that the percentage of native contacts must be above a cutoff value. The percentage is calculated by the ratio of the number of recovered native contacts to the total number of intermolecular contacts in the native complex. The second criterion is that the RMSD between the encounter complex and the native complex needs to be below a cutoff value. The third is the distance cutoff between the interfaces of two binding partners, while the last criterion is the energy cutoff calculated by Equation (1). The number of encounter complexes observed in all simulation trajectories strongly depends on the combinations and values of these different binding criteria. We enumerated all different combinations for these criteria. For each combination, a wide range of discretized values was adopted for different criteria. In detail, the range of native contact numbers was initially 0 to 10, with an interval of 1; the range of the native contact percentage was initially 0% to 10%, with an interval of 1%; the range of RMSD was initially 0 to 30Å, with an interval of 3Å; the range of interface distance cutoff was initially 0 to 10Å, with an interval of 1Å; and the range of binding energy cutoff was initially 0 to -30kT, with an interval of 3kT. For all possible combinations and values, we calculated association probabilities for each protein complex in the benchmark. Consequently, we found that by selecting the optimal combinations and values of these binding criteria, we were able to achieve a higher correlation between experimental and simulated association rates than using individual binding criterion. One example is shown in
Figure 6c. The specific combination of binding criteria adopted in the figure is as follows: the native contact percentage should be higher than 6%; the native contact number should be more than 5; the binding energy should be lower than -15kT; finally, the RMSD and interface distance should be lower than 12 and 10 Å, respectively. Under this combination of binding criteria, a high PCC value of 0.79 was achieved.
We further found that this correlation also depends on the values of the adopted binding criteria. For instance, in
Figure 6d, we found that a high correlation between simulation results and experimental data can only be attained when the value of energy cutoff was adopted within a small range. For instance, the best PCC of 0.79 could only be attained with an energy cutoff of -15.0kT, as shown at the end of the last paragraph. Under higher energy cutoff values, e.g., -3.0kT, the highest PCC found in the system is 0.76, if the native contact percentage is higher than 4% or the native contact number is more than 5, with the RMSD and interface distance cutoffs equal 15 and 1 Å, respectively. Similarly, under lower energy cutoff values, e.g., -27.0kT, the highest PCC found in the test is also lower than 0.77, if the native contact percentage is higher than 3%, or the native contact number is more than five, with the RMSD and interface distance cutoffs equal to 12 and 10 Å, respectively. This can be explained as follows: if the energy cutoff is too low, the criterion for association will not be strict enough and systems will contain encounter complexes that are not correctly bound. As a result, the association probability will be overestimated. On the other hand, if the energy cutoff is too high, the criterion for association will be too strict and systems will not contain enough encounter complexes that are correctly bound. As a result, the association probability will be underestimated. In general, the range of energy cutoff that results in a high correlation with experimental association rates corresponds to the scope of average binding energy that is found in the transition state ensemble of all protein complexes. Taken together, our statistical results imply that although there is strong diversity in the structures and energetics of different protein complexes, the common mechanisms underlying their association can be characterized by a combination of different binding criteria.
4. Concluding and Discussions
The versatile functions of protein complexes in cells strongly depend on how quickly the building blocks in these complexes can be assembled together [
80,
81,
82]. This kinetic property of protein–protein interactions is quantified by the association rate, which can be traditionally measured through various experimental methods. Compared with these methods, computational modeling approaches are much less time-consuming and labor-intensive. Moreover, they can provide a mechanistic understanding of biophysical problems on a spatial-temporal level, which is currently inaccessible in the laboratory. Relative to other all-atom simulation techniques that are highly demanding in terms of computational resources, we study the physics process of protein–protein association using a coarse-grained model and a new hybrid force field which contains both physics-based and statistics-based potentials. Our physics-based potential consists of two terms that describe the electrostatic interactions and hydrophobic effect. The parameter used to balance the weights between these two factors has been tuned in order to achieve the best performance, as in our previous study [
62]. However, there are various types of molecular interactions missing in this simplified potential, such as the side-chain hydrogen bonds, the dipole–dipole interactions and the π-stacking of aromatic rings between different side-chains. We assume that the contributions of these complicated energy contributions can be implicitly included in the statistics-based potential, thereby complementing the original physics-based potential. This assumption was verified by our results, in which we showed that a mixed form of these two different potential functions can improve the correlation between simulated and experimental association rates, although this improvement is not substantial. The best correlation was obtained when the weight constant between the contributions of the physics-based and statistics-based potentials was equal to 0.6. However, it is worth mentioning that the value of this weight depends on the protein complexes tested in the benchmark. In other words, when a various selection or a subset of benchmarks is used, it is likely that the best correlation will be derived under a different value of weight constant. Nevertheless, it is beyond the scope of this study to offer an optimal value of weight constant that can provide the best prediction of the association rate of any protein complex. The purpose here is to explore the idea that a mixture of force fields derived from complementary sources is better able to describe the process of protein–protein association.
Previous works showed that the association rate constant of forming transient complexes purely via unbiased diffusions in which the diffusion coefficients of individual proteins can be calculated by methods such as Hydropro [
83] is ~10
5M
−1s
−1 [
50,
84,
85]. The real values of association rates higher than this “basal” rate constant are the result of the intermolecular interactions between proteins, such as the long-range electrostatic attraction, which biases diffusions toward the formation of transient complexes. As a result, the association rates calculated from simulation models that incorporate both diffusions and intermolecular interactions, such as the method developed in this study, can differ by several orders of magnitude and thus are closer to the experimentally observed values. We systematically calibrated our simulation method against a large-scale benchmark set. For each complex in the benchmark, a large number of simulation trajectories were carried out. Based on the statistical analysis of these trajectories, we found that common mechanisms underlie the association of structurally diverse protein complexes. In particular, we revealed that the association of a protein complex contains multiple steps, in which proteins continuously search their local binding orientations and form non-native-like intermediates through repeated dissociation and re-association. Moreover, we showed that encounter complexes constitute an ensemble of loosely bound structures formed around their native conformation, suggesting that the transition states of protein–protein association could be highly diverse on the structural level. Finally, a positive correlation between the binding energy and structural similarity of encounter complexes with their native conformation was found, which indicates that protein–protein association is a kinetic process in which the diffusions of binding partners are driven by a “funnel-like” energy landscape. We also noticed that encounter complexes formed in a number of trajectories have a low binding energy but a large RMSD from the native structure, as shown by the black arrow in
Figure 6a, indicating that the binding partners of the protein complexes in these trajectories associate into non-native-like encounter complexes. These low-energy, non-native-like encounter complexes cannot be discerned by other binding criteria either, including the number or the percentage of native contacts and the distance between the binding interfaces. The formation of these complexes is either due to the fact that they are kinetically trapped in the intermediate states during the repeated dissociation and re-association process along their association pathways or due to the large energy frustration in the simplified force field used in this study. In summary, these results shed light on our understanding of how protein–protein recognition is kinetically modulated, and the computational method developed in this study can therefore complement existing experimental approaches that measure protein–protein association rates.
In our previous study, the association rates were derived directly from the calculated association probability and related simulation parameters, such as volume and length of association time. However, the potential used to describe the intermolecular interactions in the simulation is simplified and thus not accurate enough, so the energy landscape for guiding association is not funnel-like. As a result, one problem in estimating the association rates in our previous study is that the association with high experimental rate constants tends to be underestimated, while the association with low experimental rate constants tends to be overestimated. To tackle this issue, in this study, we attempt to use an empirical formula (as shown in Equation (5)) to minimize the effect of the simplification of force field. It is worth mentioning that this equation is only applied to rescaled simulation results. There is no free parameter which can be adjusted to fit against the experimental data. The parameters and N are experimental constants observed in the benchmark. The association probabilities Pi were derived from the first-principle simulation, while their upper and lower limits Pmin and Pmax were fixed variables, as long as all the association probabilities were derived. Therefore, there is no issue of overfitting when using Equation (5) to compare our simulation results with experimental data. However, it is possible that this empirical formula could bring some artifacts. For instance, the probability of association is time dependent. It will approach a value of 1 when given an extremely long simulation time, but it will approach 0 with an extremely short simulation time. This effect, fortunately, can be potentially canceled out by calculating the relative difference of association probability from its maximal and minimal values, as shown in the equation. Moreover, the length of simulation duration in our study is carefully determined to obtain an appropriate range of association probability, while the same value was applied to all protein complexes in the benchmark. Additionally, there is another limitation in Equation (5). It only takes into account the association rates across four orders of magnitude, as the range considered in the benchmark set. Although we adopted this limited range of rate constant values in the equation, it can still be performed to study protein associations whose rates are beyond this range. For instance, if the simulated probability for one protein complex is smaller than the minimal probability in Equation (5), this will lead to the result that the calculated association rate is lower than 105M−1s−1. On the other hand, if the simulated probability for one protein complex is larger than the maximal probability in Equation (5), this will lead to the result that the calculated association rate is higher than 109M−1s−1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





