
Mapping the Intramolecular Communications among Different Glutamate Dehydrogenase States Using Molecular Dynamics


Abstract
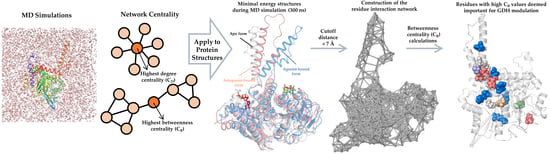
1. Introduction
2. Materials and Methods
2.1. Molecular Dynamic Simulations of GDH States
2.2. Structural Analysis
2.3. Essential Dynamics
2.4. Construction of the Residue Interaction Network
2.5. Calculation of Network Centralities
2.6. Correlation Network Analysis
3. Results
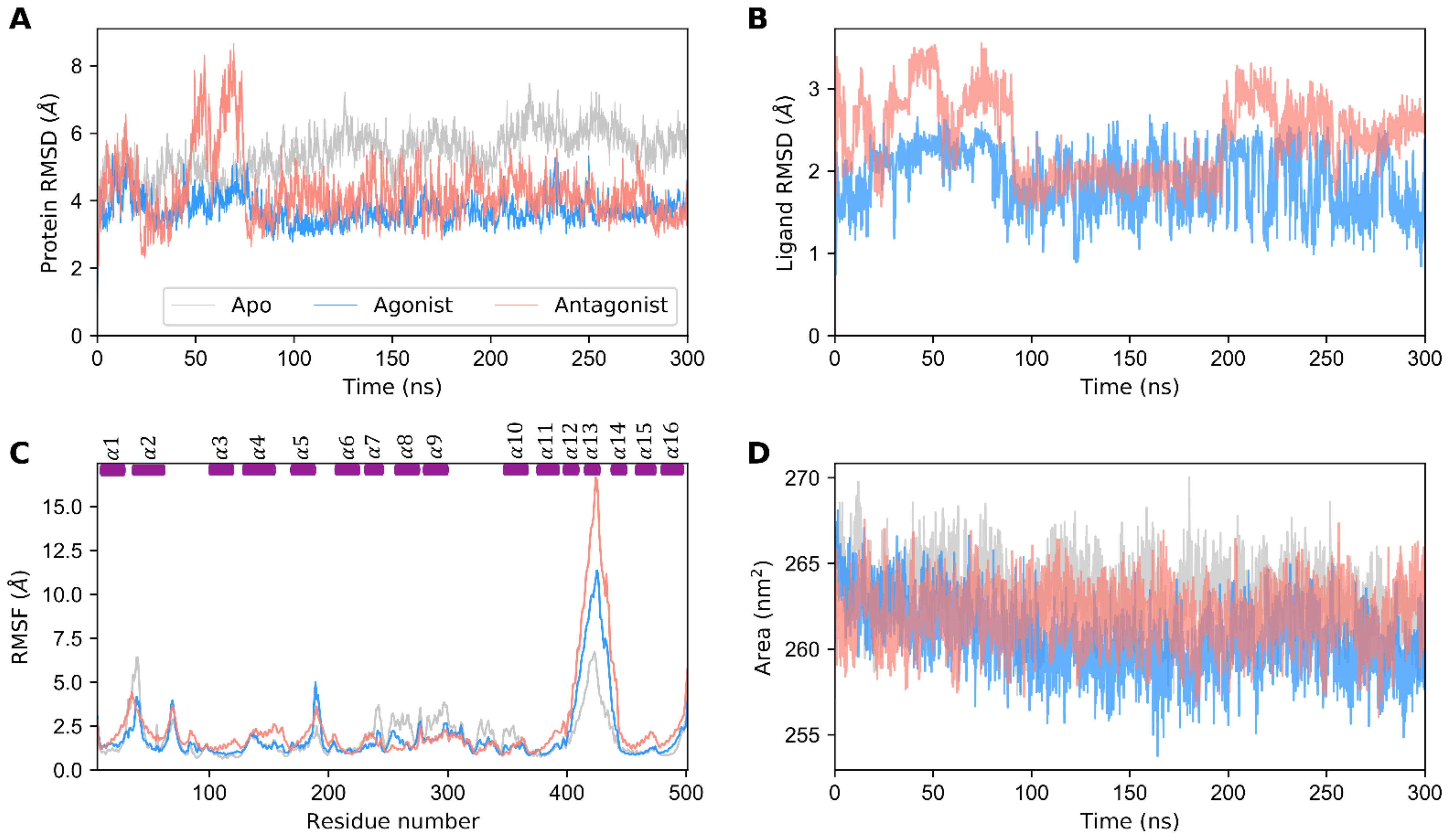
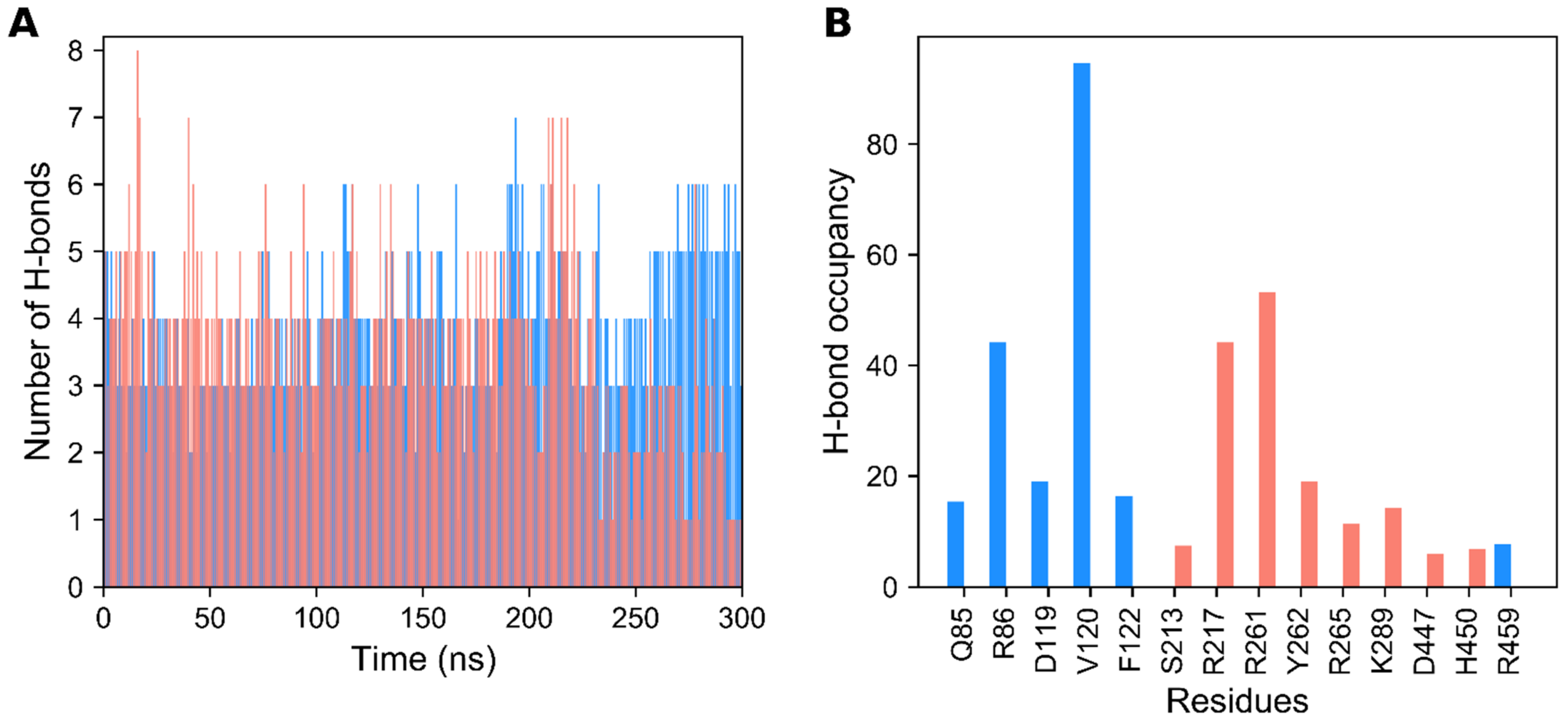
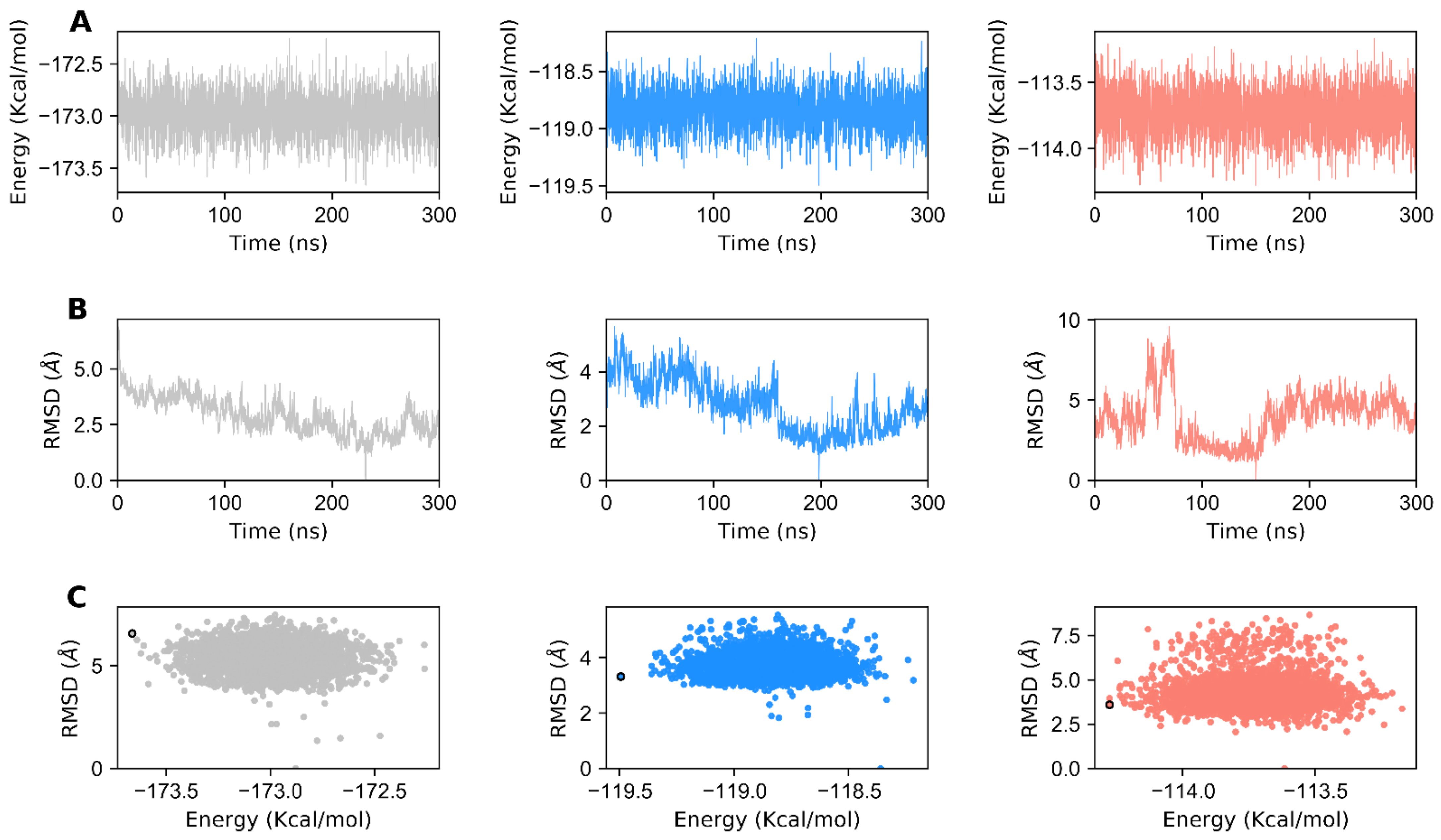
3.1. Structural Stability of GDH States
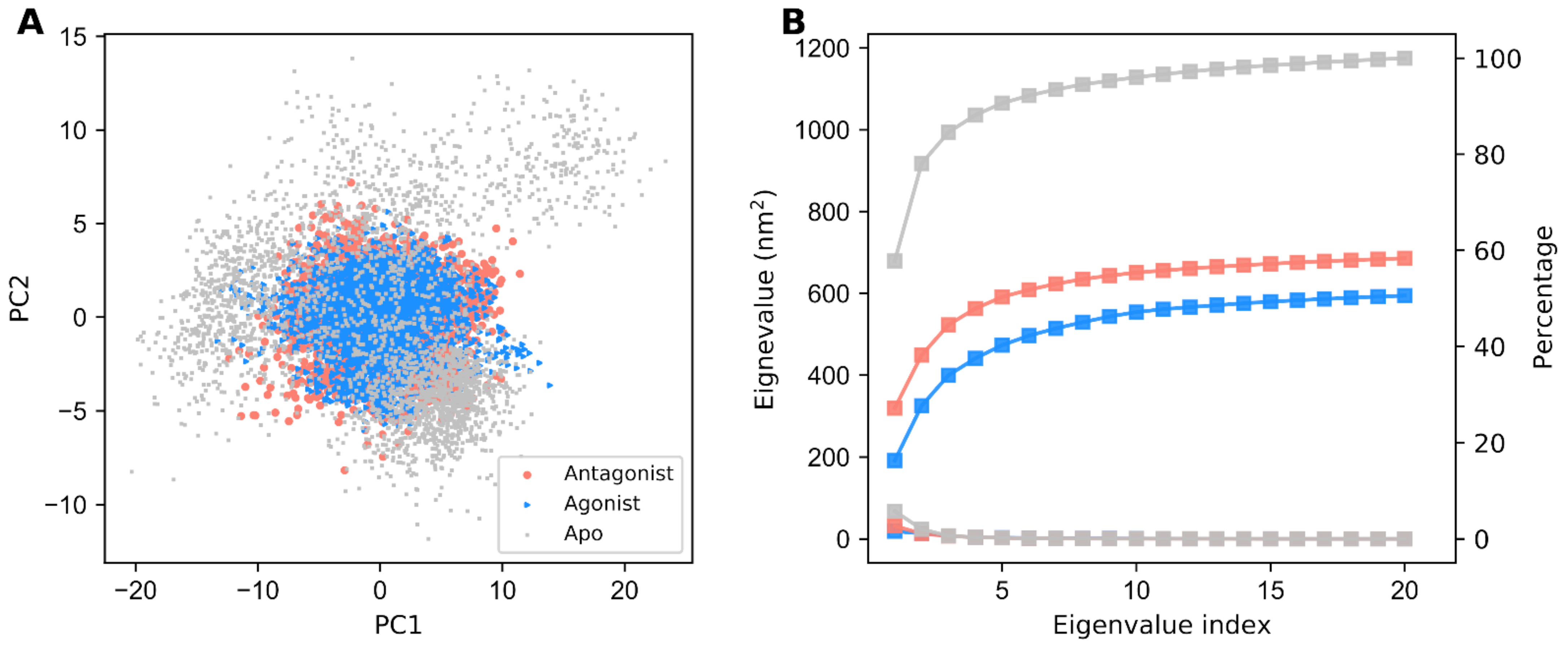
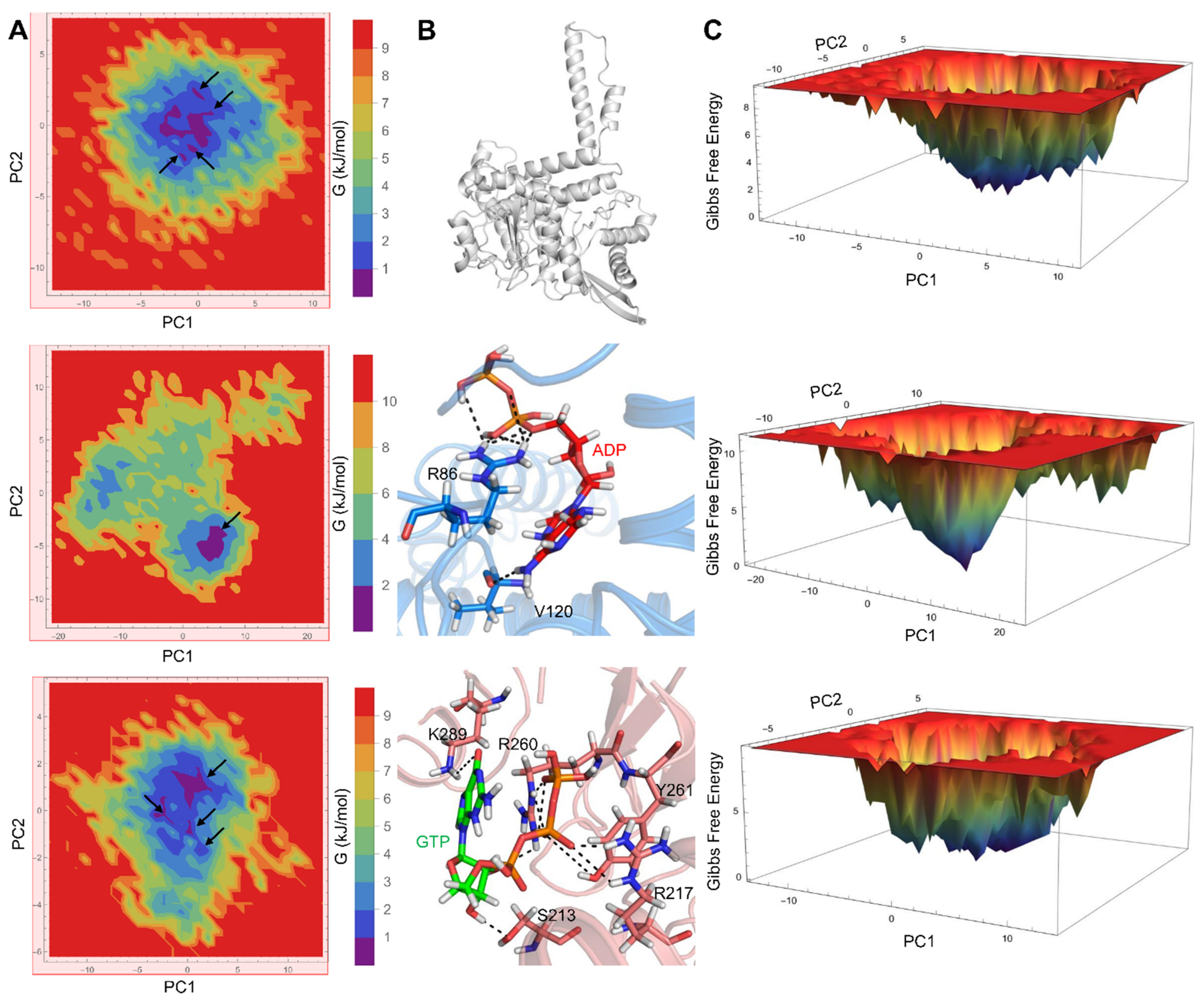
3.2. PCA and FEL Analysis
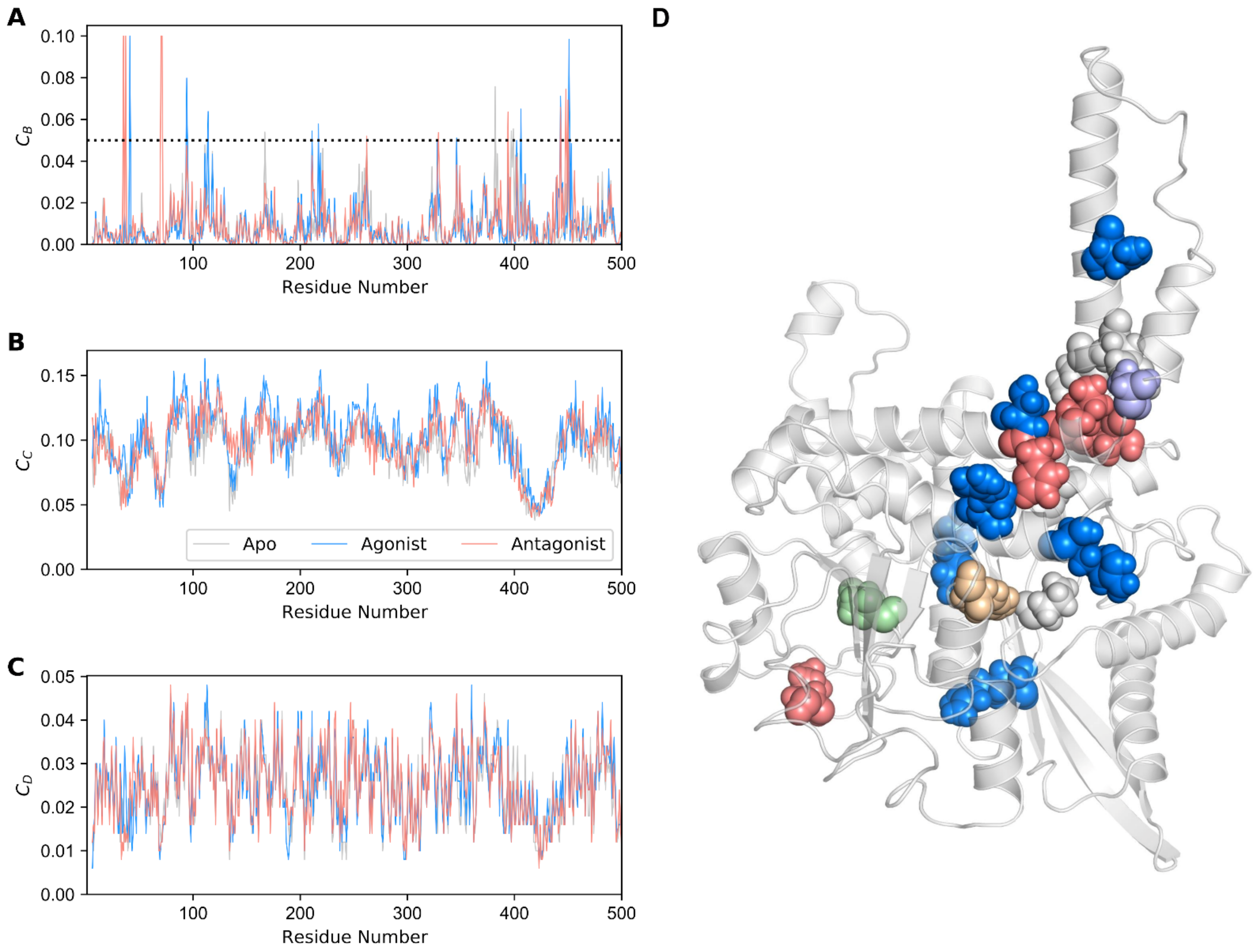
3.3. Identification of GDH Hot Spot Residues Using Network Analysis
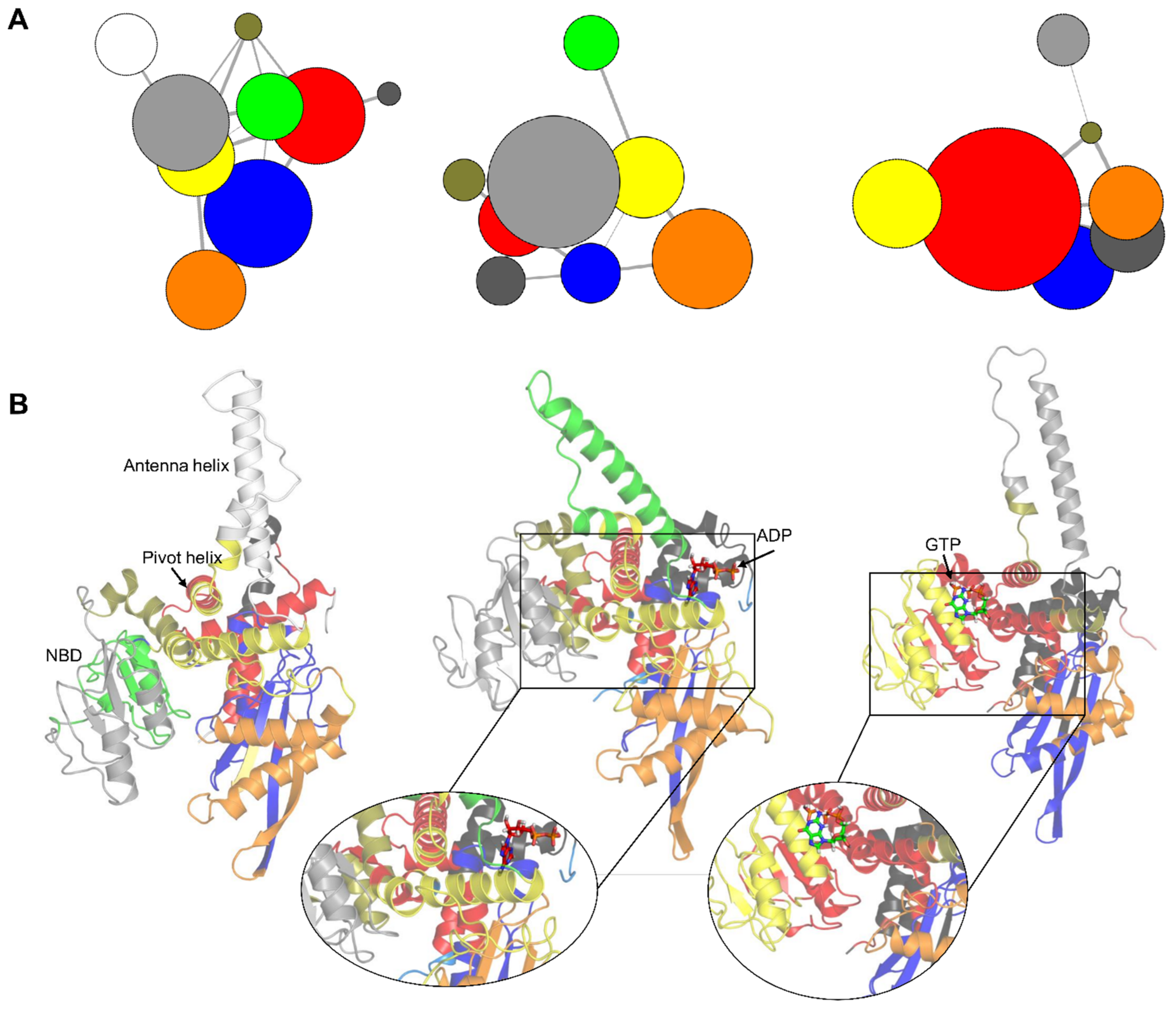
3.4. Correlation Network Analysis of GDH States
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Frieden, C. The dissociation of glutamic dehydrogenase by reduced diphosphopyridine nucleotide (DPNH). Biochim. Biophys. Acta 1958, 27, 431–432. [Google Scholar] [CrossRef]
- Frieden, C. Glutamic dehydrogenase. I. The effect of coenzyme on the sedimentation velocity and kinetic behavior. J. Biol. Chem. 1959, 234, 809–814. [Google Scholar] [CrossRef]
- Olson, J.A.; Anfinsen, C.B. The crystallization and characterization of L-glutamic acid dehydrogenase. J. Biol. Chem. 1952, 197, 67–79. [Google Scholar] [CrossRef]
- Strecker, H.J. Glutamic dehydrogenase. Arch. Biochem. Biophys. 1953, 46, 128–140. [Google Scholar] [CrossRef]
- Hudson, R.C.; Daniel, R.M. L-glutamate dehydrogenases: Distribution, properties and mechanism. Comp. Biochem. Physiol. B 1993, 106, 767–792. [Google Scholar] [CrossRef]
- Rife, J.E.; Cleland, W.W. Determination of the chemical mechanism of glutamate dehydrogenase from pH studies. Biochemistry 1980, 19, 2328–2333. [Google Scholar] [CrossRef]
- Rife, J.E.; Cleland, W.W. Kinetic mechanism of glutamate dehydrogenase. Biochemistry 1980, 19, 2321–2328. [Google Scholar] [CrossRef]
- Britton, K.L.; Baker, P.J.; Rice, D.W.; Stillman, T.J. Structural relationship between the hexameric and tetrameric family of glutamate dehydrogenases. Eur. J. Biochem. 1992, 209, 851–859. [Google Scholar] [CrossRef]
- Grzechowiak, M.; Sliwiak, J.; Jaskolski, M.; Ruszkowski, M. Structural Studies of Glutamate Dehydrogenase (Isoform 1) From Arabidopsis thaliana, an Important Enzyme at the Branch-Point Between Carbon and Nitrogen Metabolism. Front. Plant. Sci. 2020, 11, 754. [Google Scholar] [CrossRef]
- Banerjee, S.; Schmidt, T.; Fang, J.; Stanley, C.A.; Smith, T.J. Structural studies on ADP activation of mammalian glutamate dehydrogenase and the evolution of regulation. Biochemistry 2003, 42, 3446–3456. [Google Scholar] [CrossRef]
- Li, M.; Li, C.; Allen, A.; Stanley, C.A.; Smith, T.J. Glutamate dehydrogenase: Structure, allosteric regulation, and role in insulin homeostasis. Neurochem. Res. 2014, 39, 433–445. [Google Scholar] [CrossRef]
- Stillman, T.J.; Migueis, A.M.; Wang, X.G.; Baker, P.J.; Britton, K.L.; Engel, P.C.; Rice, D.W. Insights into the mechanism of domain closure and substrate specificity of glutamate dehydrogenase from Clostridium symbiosum. J. Mol. Biol. 1999, 285, 875–885. [Google Scholar] [CrossRef]
- Knapp, S.; de Vos, W.M.; Rice, D.; Ladenstein, R. Crystal structure of glutamate dehydrogenase from the hyperthermophilic eubacterium Thermotoga maritima at 3.0 A resolution. J. Mol. Biol. 1997, 267, 916–932. [Google Scholar] [CrossRef]
- Britton, K.L.; Yip, K.S.; Sedelnikova, S.E.; Stillman, T.J.; Adams, M.W.; Ma, K.; Maeder, D.L.; Robb, F.T.; Tolliday, N.; Vetriani, C.; et al. Structure determination of the glutamate dehydrogenase from the hyperthermophile Thermococcus litoralis and its comparison with that from Pyrococcus furiosus. J. Mol. Biol. 1999, 293, 1121–1132. [Google Scholar] [CrossRef]
- Werner, C.; Stubbs, M.T.; Krauth-Siegel, R.L.; Klebe, G. The crystal structure of Plasmodium falciparum glutamate dehydrogenase, a putative target for novel antimalarial drugs. J. Mol. Biol. 2005, 349, 597–607. [Google Scholar] [CrossRef]
- Oliveira, T.; Panjikar, S.; Carrigan, J.B.; Hamza, M.; Sharkey, M.A.; Engel, P.C.; Khan, A.R. Crystal structure of NAD+-dependent Peptoniphilus asaccharolyticus glutamate dehydrogenase reveals determinants of cofactor specificity. J. Struct. Biol. 2012, 177, 543–552. [Google Scholar] [CrossRef]
- Smith, T.J.; Stanley, C.A. Untangling the glutamate dehydrogenase allosteric nightmare. Trends Biochem. Sci. 2008, 33, 557–564. [Google Scholar] [CrossRef]
- Mathioudakis, L.; Bourbouli, M.; Daklada, E.; Kargatzi, S.; Michaelidou, K.; Zaganas, I. Localization of Human Glutamate Dehydrogenases Provides Insights into Their Metabolic Role and Their Involvement in Disease Processes. Neurochem. Res. 2019, 44, 170–187. [Google Scholar] [CrossRef]
- Bera, S.; Rashid, M.; Medvinsky, A.B.; Sun, G.Q.; Li, B.L.; Acquisti, C.; Sljoka, A.; Chakraborty, A. Allosteric regulation of glutamate dehydrogenase deamination activity. Sci. Rep. 2020, 10, 16523. [Google Scholar] [CrossRef] [PubMed]
- Yielding, K.L.; Tomkins, G.M. An effect of L-leucine and other essential amino acids on the structure and activity of glutamic dehydrogenase. Proc. Natl. Acad. Sci. USA 1961, 47, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Couee, I.; Tipton, K.F. Inhibition of ox brain glutamate dehydrogenase by perphenazine. Biochem. Pharmacol. 1990, 39, 1167–1173. [Google Scholar] [CrossRef]
- George, A.; Bell, J.E. Effects of adenosine 5′-diphosphate on bovine glutamate dehydrogenase: Diethyl pyrocarbonate modification. Biochemistry 1980, 19, 6057–6061. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, M.; Chen, P.; Narayan, S.; Matschinsky, F.M.; Bennett, M.J.; Stanley, C.A.; Smith, T.J. Green tea polyphenols control dysregulated glutamate dehydrogenase in transgenic mice by hijacking the ADP activation site. J. Biol. Chem. 2011, 286, 34164–34174. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, G.M.; Yielding, K.L.; Curran, J.F. The influence of diethylstilbestrol and adenosine diphosphate on pyridine nucleotide coenzyme binding by glutamic dehydrogenase. J. Biol. Chem. 1962, 237, 1704–1708. [Google Scholar] [CrossRef]
- Li, M.; Li, C.; Allen, A.; Stanley, C.A.; Smith, T.J. The structure and allosteric regulation of mammalian glutamate dehydrogenase. Arch. Biochem. Biophys. 2012, 519, 69–80. [Google Scholar] [CrossRef]
- Borgnia, M.J.; Banerjee, S.; Merk, A.; Matthies, D.; Bartesaghi, A.; Rao, P.; Pierson, J.; Earl, L.A.; Falconieri, V.; Subramaniam, S.; et al. Using Cryo-EM to Map Small Ligands on Dynamic Metabolic Enzymes: Studies with Glutamate Dehydrogenase. Mol. Pharmacol. 2016, 89, 645–651. [Google Scholar] [CrossRef]
- Smith, T.J.; Peterson, P.E.; Schmidt, T.; Fang, J.; Stanley, C.A. Structures of bovine glutamate dehydrogenase complexes elucidate the mechanism of purine regulation. J. Mol. Biol. 2001, 307, 707–720. [Google Scholar] [CrossRef]
- Peterson, P.E.; Smith, T.J. The structure of bovine glutamate dehydrogenase provides insights into the mechanism of allostery. Structure 1999, 7, 769–782. [Google Scholar] [CrossRef]
- Nassar, O.M.; Li, C.; Stanley, C.A.; Pettitt, B.M.; Smith, T.J. Glutamate dehydrogenase: Structure of a hyperinsulinism mutant, corrections to the atomic model, and insights into a regulatory site. Proteins 2019, 87, 41–50. [Google Scholar] [CrossRef]
- Frieden, C. Glutamic dehydrogenase. II. The effect of various nucleotides on the association-dissociation and kinetic properties. J. Biol. Chem. 1959, 234, 815–820. [Google Scholar] [CrossRef]
- Smith, E.L.; Pisziewicz, D. Bovine glutamate dehydrogenase. The pH dependence of native and nitrated enzyme in the presence of allosteric modifiers. J. Biol. Chem. 1973, 248, 3089–3092. [Google Scholar] [CrossRef]
- Oroguchi, T.; Nakasako, M. Changes in hydration structure are necessary for collective motions of a multi-domain protein. Sci. Rep. 2016, 6, 26302. [Google Scholar] [CrossRef]
- Stanley, C.A.; Lieu, Y.K.; Hsu, B.Y.; Burlina, A.B.; Greenberg, C.R.; Hopwood, N.J.; Perlman, K.; Rich, B.H.; Zammarchi, E.; Poncz, M. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N. Engl. J. Med. 1998, 338, 1352–1357. [Google Scholar] [CrossRef]
- Stanley, C.A.; Fang, J.; Kutyna, K.; Hsu, B.Y.; Ming, J.E.; Glaser, B.; Poncz, M. Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome: Predominance of mutations in exons 11 and 12 of the glutamate dehydrogenase gene. HI/HA Contributing Investigators. Diabetes 2000, 49, 667–673. [Google Scholar] [CrossRef]
- Stanley, C.A. Hyperinsulinism/hyperammonemia syndrome: Insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol. Genet. Metab. 2004, 81 (Suppl. 1), S45–S51. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.3.0a0; Schrodinger, LLC: New York, NY, USA, 2015.
- Sittel, F.; Jain, A.; Stock, G. Principal component analysis of molecular dynamics: On the use of Cartesian vs. internal coordinates. J. Chem. Phys. 2014, 141, 014111. [Google Scholar] [CrossRef]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. A Molecular Dynamics Approach to Explore the Intramolecular Signal Transduction of PPAR-alpha. Int. J. Mol. Sci. 2019, 20, 1666. [Google Scholar] [CrossRef] [PubMed]
- Wolfram Research, Inc. Mathematica; Version 12.2; Wolfram Research, Inc.: Champaign, IL, USA, 2020. [Google Scholar]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 1991, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Lange, O.F.; Grubmuller, H. Generalized correlation for biomolecular dynamics. Proteins 2006, 62, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Skjaerven, L.; Yao, X.Q.; Scarabelli, G.; Grant, B.J. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinform. 2014, 15, 399. [Google Scholar] [CrossRef]
- Yao, X.Q.; Malik, R.U.; Griggs, N.W.; Skjaerven, L.; Traynor, J.R.; Sivaramakrishnan, S.; Grant, B.J. Dynamic Coupling and Allosteric Networks in the alpha Subunit of Heterotrimeric G Proteins. J. Biol. Chem. 2016, 291, 4742–4753. [Google Scholar] [CrossRef]
- Scarabelli, G.; Grant, B.J. Kinesin-5 allosteric inhibitors uncouple the dynamics of nucleotide, microtubule, and neck-linker binding sites. Biophys. J. 2014, 107, 2204–2213. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Ringner, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, S.; Hyeon, C. Mapping the intramolecular signal transduction of G-protein coupled receptors. Proteins 2014, 82, 727–743. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef]
- Scarabelli, G.; Grant, B.J. Mapping the structural and dynamical features of kinesin motor domains. PLoS Comput. Biol. 2013, 9, e1003329. [Google Scholar] [CrossRef]
- Gasper, P.M.; Fuglestad, B.; Komives, E.A.; Markwick, P.R.; McCammon, J.A. Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc. Natl. Acad. Sci. USA 2012, 109, 21216–21222. [Google Scholar] [CrossRef]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA: Protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
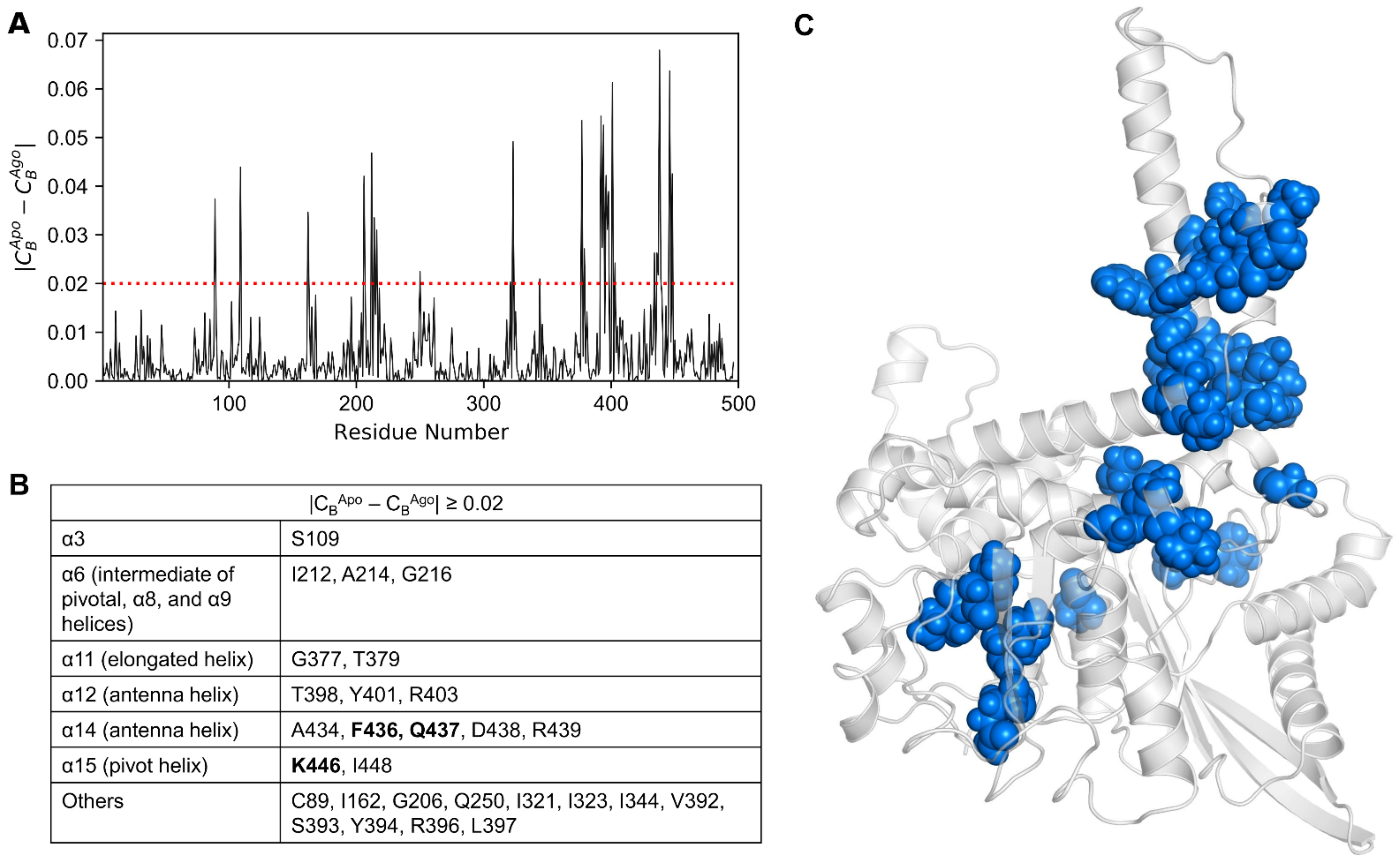
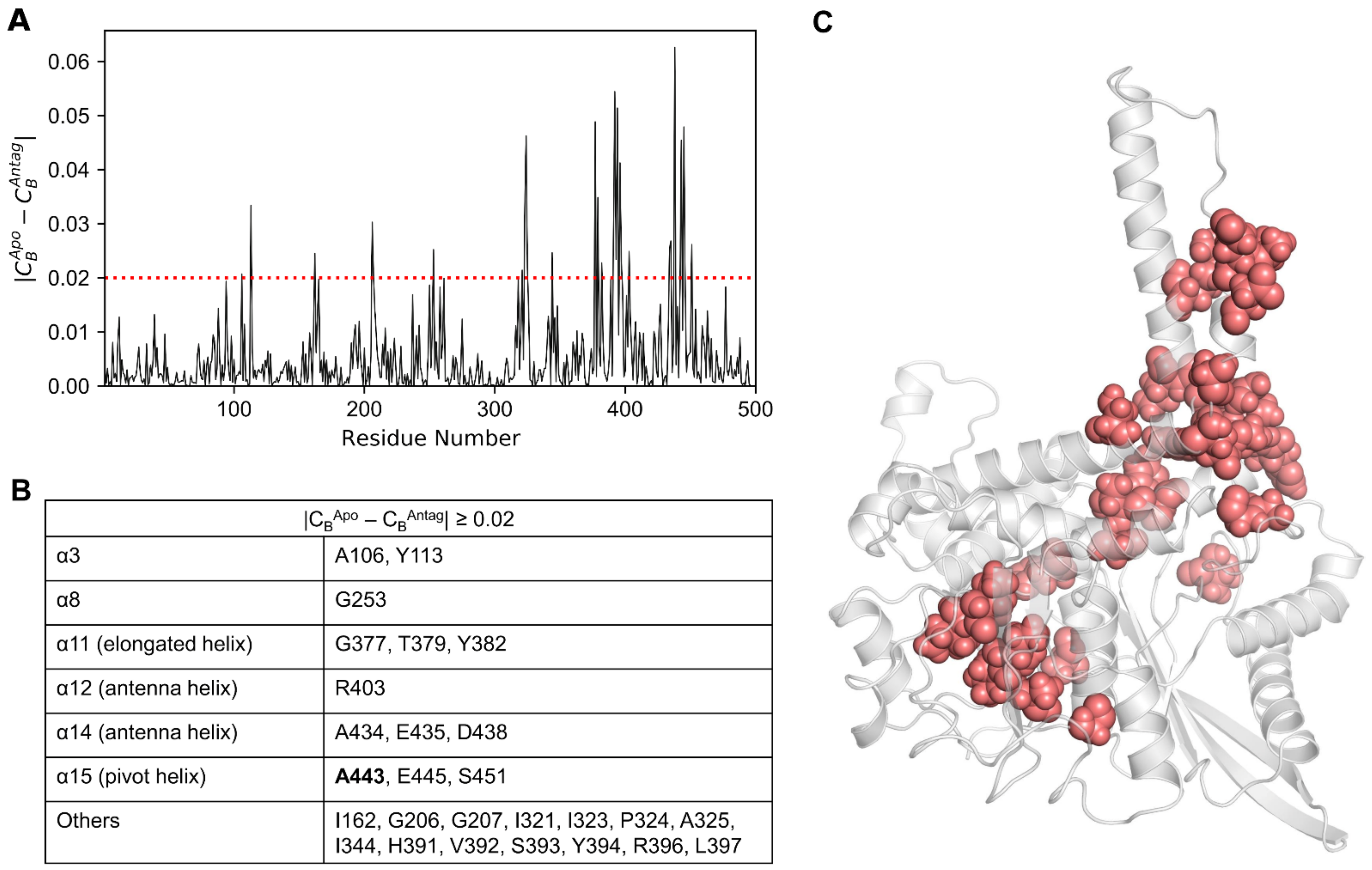
| CB ≥ 0.05 | |
| CBApo ≥ 0.05 | P167, *Y262, #E346, Y382, L397, F399 |
| CBAgo ≥ 0.05 | R94, K114, R211, R217, #E346, N406, &A443, S451 |
| CBAntag ≥ 0.05 | *Y262, K329, Y394, &A443, I448, S450 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. Mapping the Intramolecular Communications among Different Glutamate Dehydrogenase States Using Molecular Dynamics. Biomolecules 2021, 11, 798. https://doi.org/10.3390/biom11060798
Basith S, Manavalan B, Shin TH, Lee G. Mapping the Intramolecular Communications among Different Glutamate Dehydrogenase States Using Molecular Dynamics. Biomolecules. 2021; 11(6):798. https://doi.org/10.3390/biom11060798
Chicago/Turabian StyleBasith, Shaherin, Balachandran Manavalan, Tae Hwan Shin, and Gwang Lee. 2021. "Mapping the Intramolecular Communications among Different Glutamate Dehydrogenase States Using Molecular Dynamics" Biomolecules 11, no. 6: 798. https://doi.org/10.3390/biom11060798
APA StyleBasith, S., Manavalan, B., Shin, T. H., & Lee, G. (2021). Mapping the Intramolecular Communications among Different Glutamate Dehydrogenase States Using Molecular Dynamics. Biomolecules, 11(6), 798. https://doi.org/10.3390/biom11060798