



Animals 2026, 16(3), 422; https://doi.org/10.3390/ani16030422 - 29 Jan 2026
Viewed by 807
Abstract
►
Show Figures
Splenic nodules in dogs that were historically classified under the broad term “fibrohistiocytic nodules” are now recognised as distinct entities within likely a biological continuum. These include lymphoid hyperplasia extending to indolent lymphoma and complex hyperplasia to stromal sarcoma. However, the molecular mechanisms
[...] Read more.
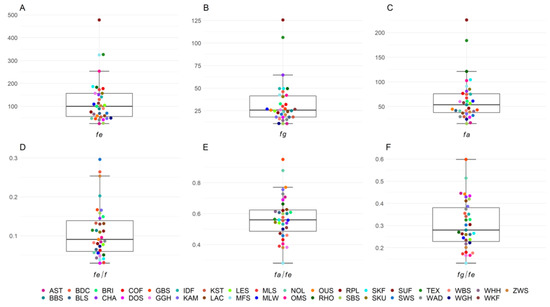
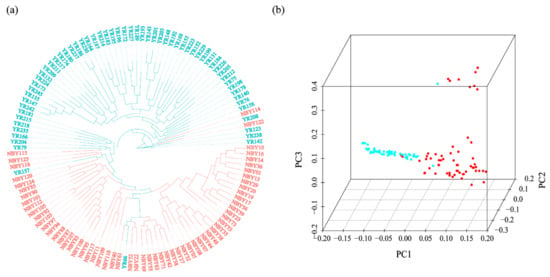
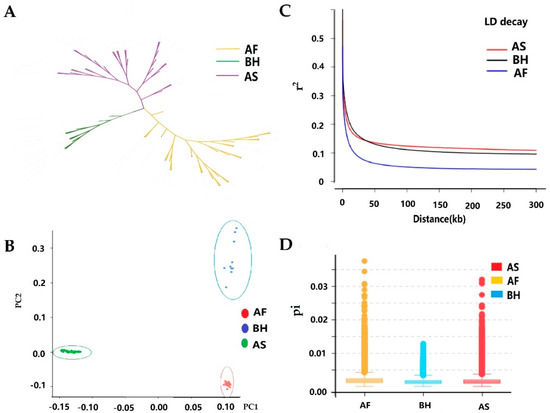
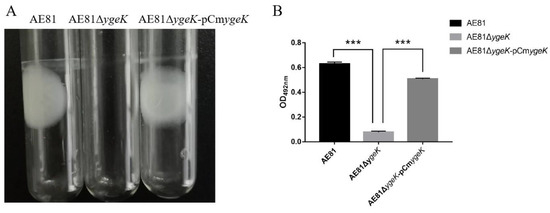
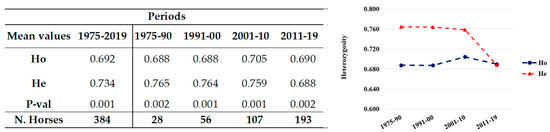
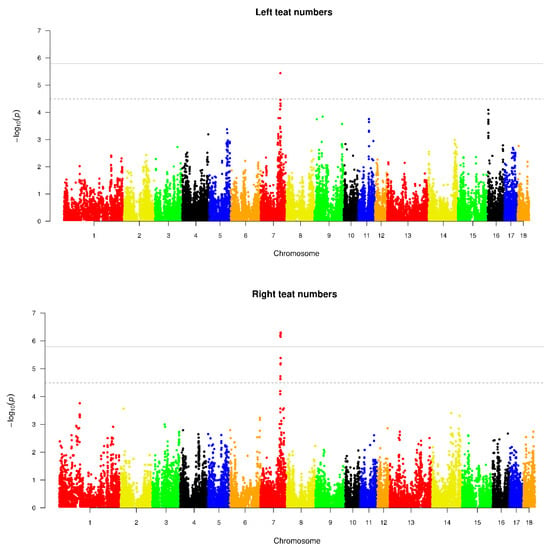
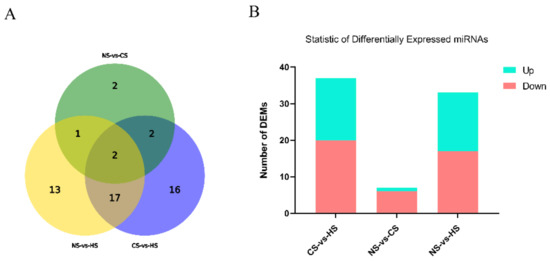
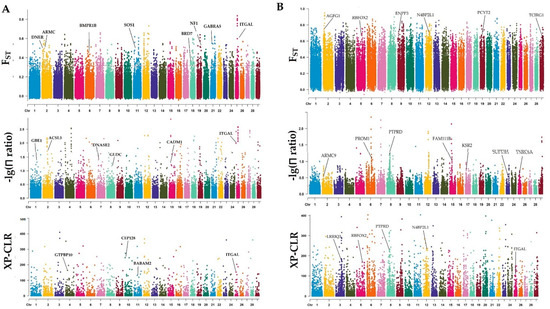
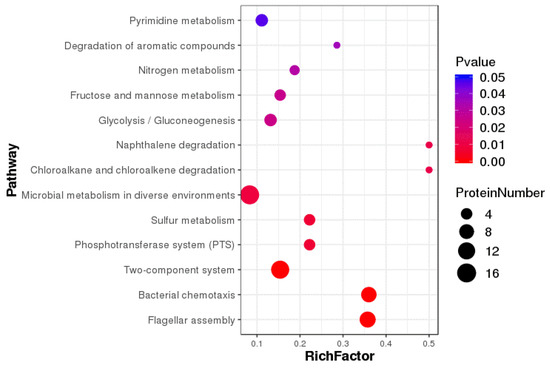
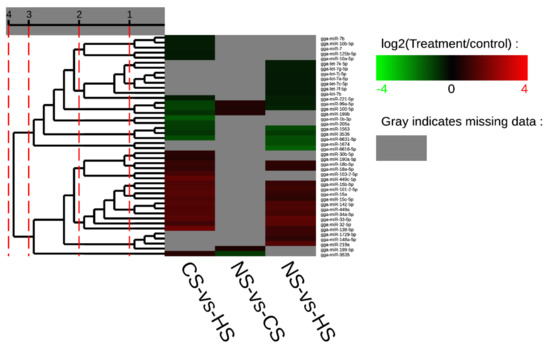
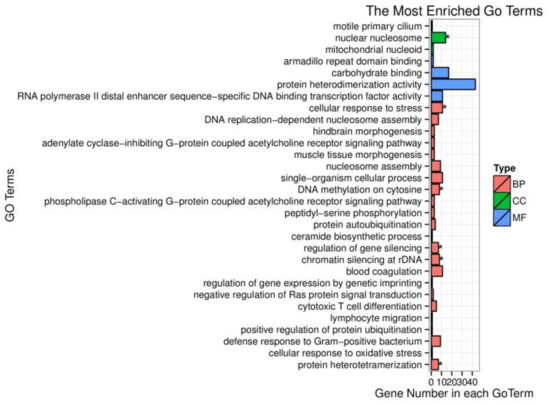
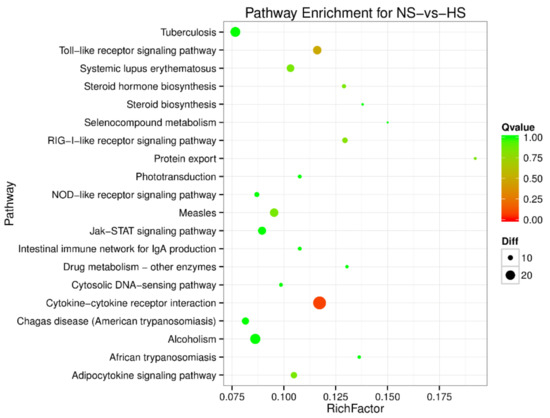
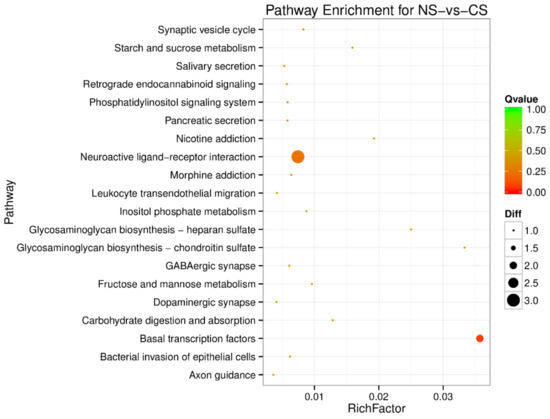
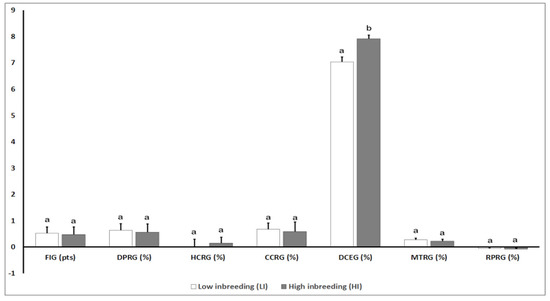
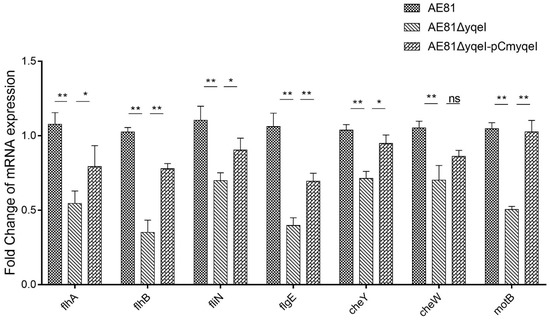
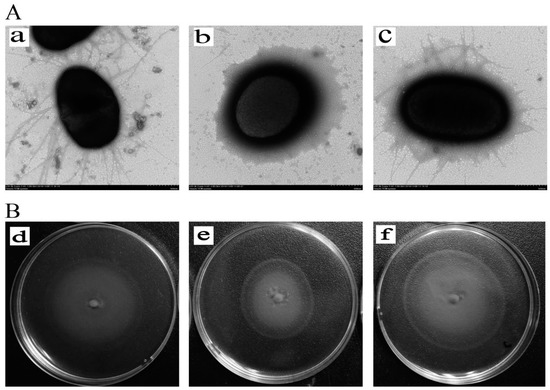
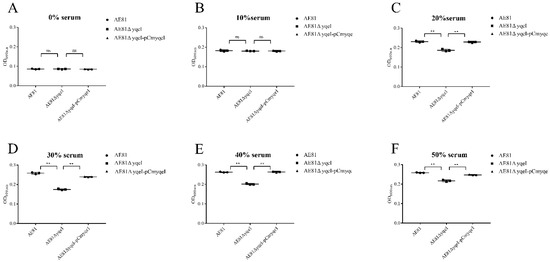
Splenic nodules in dogs that were historically classified under the broad term “fibrohistiocytic nodules” are now recognised as distinct entities within likely a biological continuum. These include lymphoid hyperplasia extending to indolent lymphoma and complex hyperplasia to stromal sarcoma. However, the molecular mechanisms underpinning these proposed progressions remain largely unexplored, particularly at the genomic and transcriptomic levels. This study aimed to delineate and compare the transcriptomic landscapes of four distinct canine splenic nodules through differential gene expression profiling. RNA sequencing was performed on twelve formalin-fixed, paraffin-embedded (FFPE) splenic tissue samples obtained from dogs diagnosed with lymphoid hyperplasia, complex hyperplasia, histiocytic sarcoma, and stromal sarcoma, with normal canine spleen serving as a control tissue. Comparative transcriptomic analysis identified 47 differentially expressed genes (DEGs) between splenic nodules and normal spleen, including CSRP1, SLC40A1, C1QA, C1QC, DLA-12, FTL, FXYD6, MPEG1, OAS3, CSF1, and JMJD6. Furthermore, 39 DEGs were significantly altered among the four splenic lesion types, such as MLC1, ERAS, MOV10L1, LOC102152143, COL4A1, COL4A2, COL12A1, NOTCH3, PLOD2, CPXM2, MRC1, GALNT5, TIMP1, and TFPI2. Many of these genes have previously been implicated in tumorigenesis and metastasis in other malignancies. These findings suggest that dysregulated gene expression may contribute to the activation of stromal cells and macrophages within the spleen, facilitating malignant transformation. Overall, these findings deliver novel transcriptomic insights into canine splenic tumorigenesis that may improve diagnostic precision, inform prognostic assessment, and support the development of targeted therapeutic strategies in veterinary oncology.
Full article

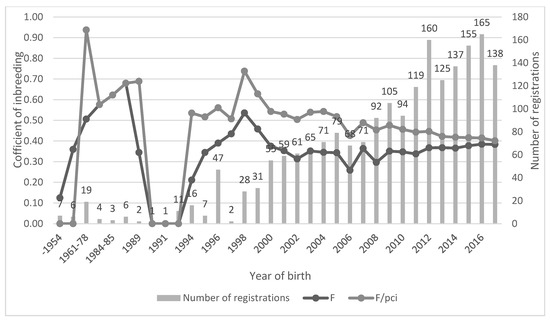
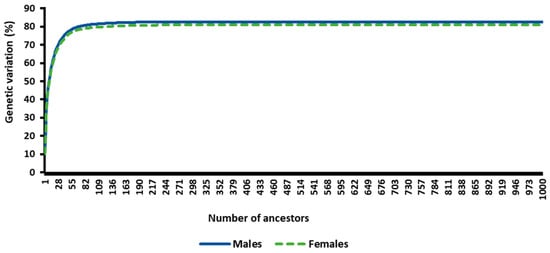
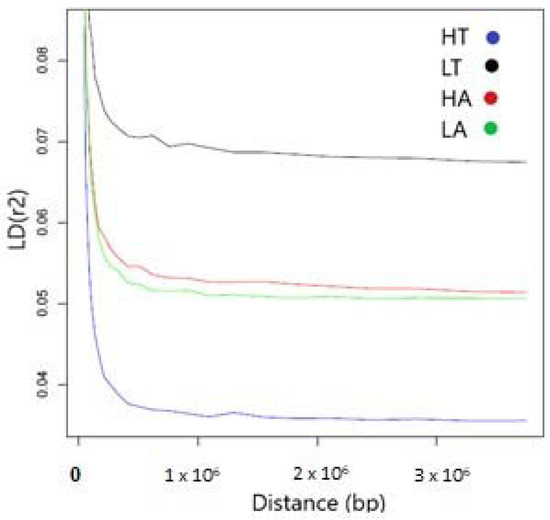
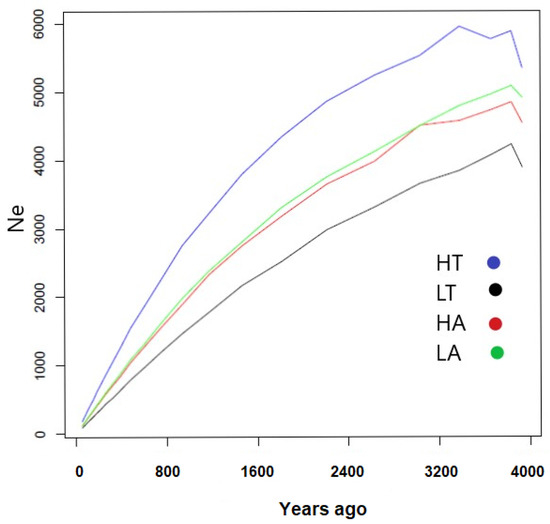
Figure 1








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}