
Genome-Wide Population Structure and Selection Signatures of Yunling Goat Based on RAD-seq

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval Statement
2.2. Sample Collection and Genomic DNA Extraction
2.3. RAD-seq Data Analysis and Genome Alignment
2.4. Phylogenetic Tree Construction and Population Structure Analysis
2.5. Selection Signature Analysis and Enrichment Analysis
3. Results
3.1. Identify SNPs and InDels
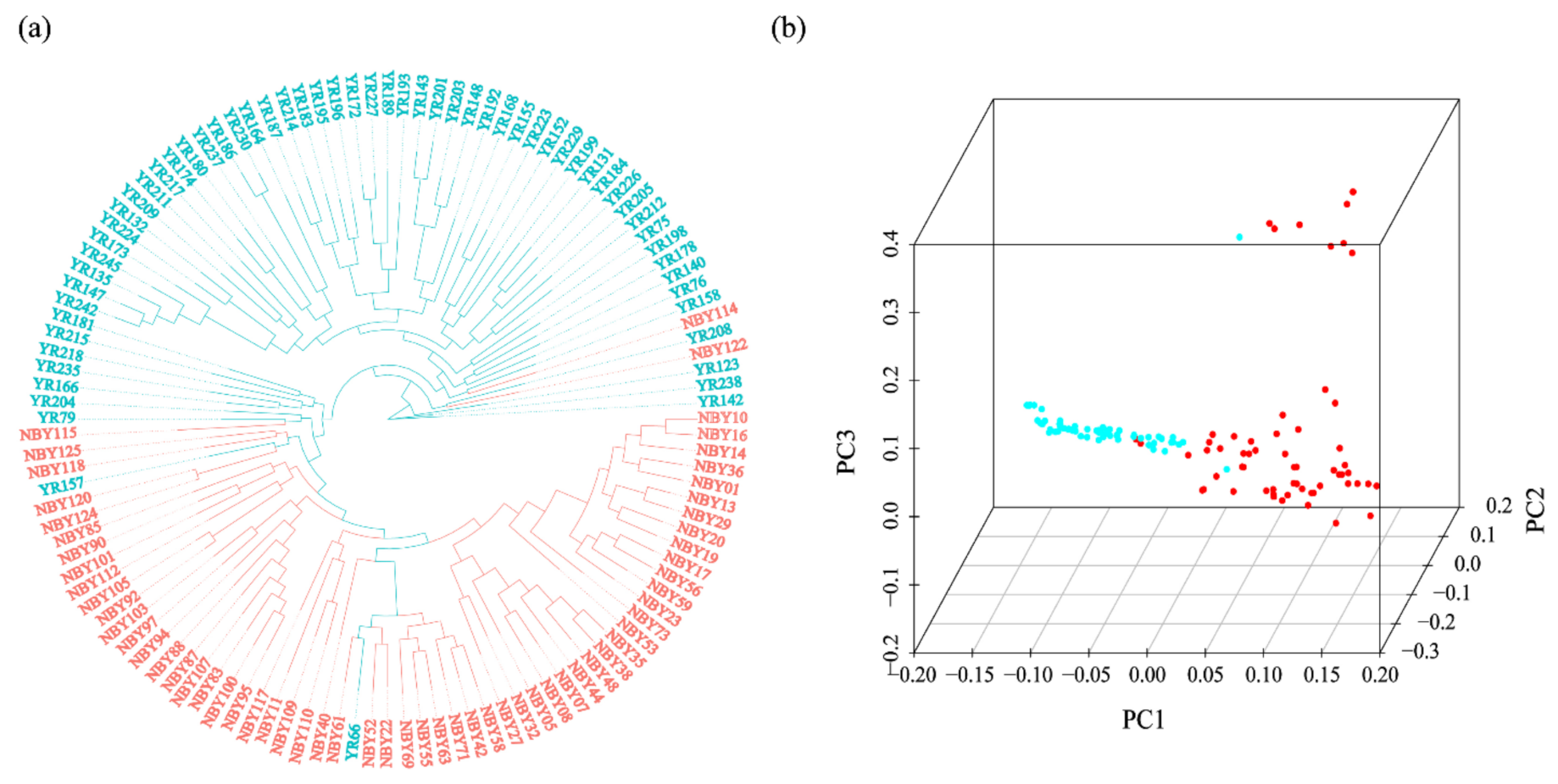
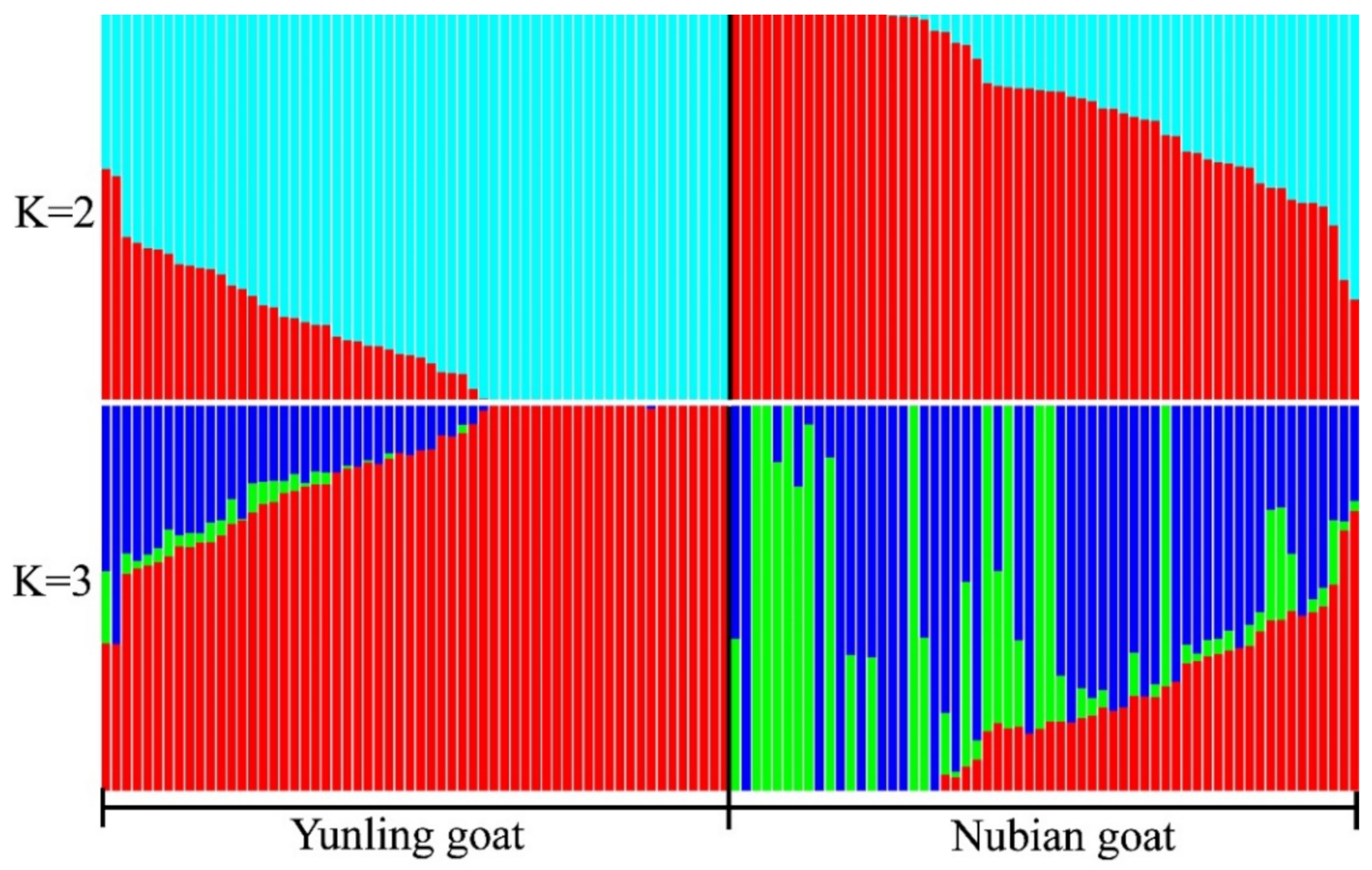
3.2. Phylogenetic Tree and Population Structure between Two Goat Breeds
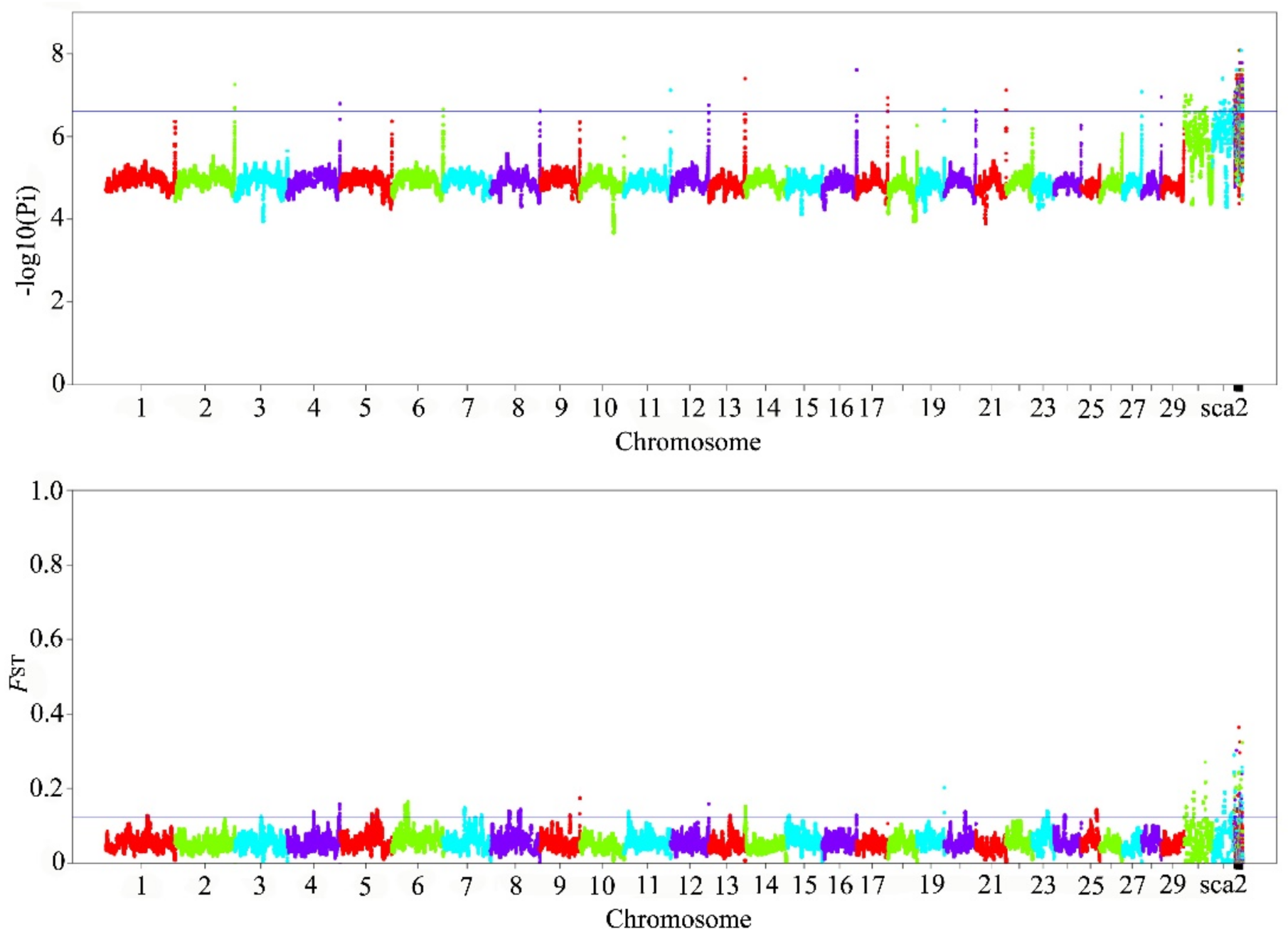
3.3. Identify Candidate Genes and Pathways Associated with Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, Y.; Fu, W.; Cai, D.; Heller, R.; Zheng, Z.; Wen, J.; Li, H.; Wang, X.; Alshawi, A.; Sun, Z.; et al. Ancient Genomes Reveal the Evolutionary History and Origin of Cashmere-Producing Goats in China. Mol. Biol. Evol. 2020, 37, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Jie, W.; Yulin, C. Study on Chinese goat breed resources and genetic diversity. J. Domest. Anim. Ecol. 2005, 26, 4–6. [Google Scholar]
- Jensen, K.; Paxton, E.; Waddington, D.; Talbot, R.; Darghouth, M.A.; Glass, E.J. Differences in the transcriptional responses induced by Theileria annulata infection in bovine monocytes derived from resistant and susceptible cattle breeds. Int. J. Parasitol. 2008, 38, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Glass, E.J.; Jensen, K. Resistance and susceptibility to a protozoan parasite of cattle—Gene expression differences in macrophages from different breeds of cattle. Vet. Immunol. Immunopathol. 2007, 120, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Glass, E.J.; Crutchley, S.; Jensen, K. Living with the enemy or uninvited guests: Functional genomics approaches to investigating host resistance or tolerance traits to a protozoan parasite, Theileria annulata, in cattle. Vet. Immunol. Immunopathol. 2012, 148, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Arsenopoulos, K.V.; Fthenakis, G.C.; Katsarou, E.I.; Papadopoulos, E. Haemonchosis: A Challenging Parasitic Infection of Sheep and Goats. Animals 2021, 11, 363. [Google Scholar] [CrossRef] [PubMed]
- Rahmatalla, S.A.; Arends, D.; Reissmann, M.; Ahmed, A.S.; Wimmers, K.; Reyer, H.; Brockmann, G.A. Whole genome population genetics analysis of Sudanese goats identifies regions harboring genes associated with major traits. BMC Genet. 2017, 18, 92. [Google Scholar] [CrossRef]
- Yunnan Commission of Animal Genetic Resources. Animal Genetic Resources in Yunnan; Yunnan Science and Technology Press: Kunming, China, 2015; pp. 181–228. [Google Scholar]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Tsuchihashi, Z.; Dracopoli, N.C. Progress in high throughput SNP genotyping methods. Pharm. J. 2002, 2, 103–110. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Morley, M.; Molony, C.M.; Weber, T.M.; Devlin, J.L.; Ewens, K.G.; Spielman, R.S.; Cheung, V.G. Genetic analysis of genome-wide variation in human gene expression. Nature 2004, 430, 743–747. [Google Scholar] [CrossRef]
- Orita, R.; Nagano, Y.; Kawamura, Y.; Kimura, K.; Kobayashi, G. Genetic diversity and population structure of razor clam Sinonovacula constricta in Ariake Bay, Japan, revealed using RAD-Seq SNP markers. Sci. Rep. 2021, 11, 7761. [Google Scholar] [CrossRef]
- Cariou, M.; Henri, H.; Martinez, S.; Duret, L.; Charlat, S. How consistent is RAD-seq divergence with DNA-barcode based clustering in insects? Mol. Ecol. Resour. 2020, 20, 1294–1298. [Google Scholar] [CrossRef]
- Miller, J.M.; Moore, S.S.; Stothard, P.; Liao, X.; Coltman, D.W. Harnessing cross-species alignment to discover SNPs and generate a draft genome sequence of a bighorn sheep (Ovis canadensis). BMC Genom. 2015, 16, 397. [Google Scholar] [CrossRef]
- Baxter, S.W.; Davey, J.W.; Johnston, J.S.; Shelton, A.M.; Heckel, D.G.; Jiggins, C.D.; Blaxter, M.L. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS ONE 2011, 6, e19315. [Google Scholar] [CrossRef]
- Pfender, W.F.; Saha, M.C.; Johnson, E.A.; Slabaugh, M.B. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor. Appl. Genet. 2011, 122, 1467–1480. [Google Scholar] [CrossRef]
- Palaiokostas, C.; Cariou, S.; Bestin, A.; Bruant, J.-S.; Haffray, P.; Morin, T.; Cabon, J.; Allal, F.; Vandeputte, M.; Houston, R.D. Genome-wide association and genomic prediction of resistance to viral nervous necrosis in European sea bass (Dicentrarchus labrax) using RAD sequencing. Genet. Sel. Evol. 2018, 50, 30. [Google Scholar] [CrossRef]
- Zhai, Z.; Zhao, W.; He, C.; Yang, K.; Tang, L.; Liu, S.; Zhang, Y.; Huang, Q.; Meng, H. SNP discovery and genotyping using restriction-site-associated DNA sequencing in chickens. Anim. Genet. 2015, 46, 216–219. [Google Scholar] [CrossRef]
- Wang, H.Z.; Li, G.H.; Zhang, Y.P.; Zhang, H.Y.; Yan, J.M.; Su, Y.J.; Wang, K.H.; Han, W.; Zou, J.M. Conservation Status Evaluation of Two Luyuan Chicken Populations Based on RAD-seq Simplified Genome Sequencing Technology Acta Vet. Zootech. Sin. 2017, 48, 818–825. [Google Scholar]
- Yong, L.; Ze-Pu, M.; Xiao-Yun, M.; Man-Man, Y.; Qiang, W.; Xiao-Feng, G.; Hua, Z.; Ming, F. The Application of RAD-seq Technology on Genomic Selection of Fertility Traits for Large White Pigs (Sus scrofa). J. Agric. Biotechnol. 2017, 25, 1508–1515. [Google Scholar]
- Ying, Z.; Shaoyi, H.; Yongxian, D.; Zhimin, Y.; Chengyu, G. Study on the characteristics of Yunling goats. Chin. Livest. Poult. Breed. 2016, 12, 54–55. [Google Scholar]
- Yiduan, L.; Limin, S.; Jun, D.; Wenkun, X. Introduction of genetic resources of local goat and sheep breeds in Yunnan Province. Yunnan Agric. 2021, 4, 82–85. [Google Scholar]
- Etter, P.D.; Bassham, S.; Hohenlohe, P.A.; Johnson, E.A.; Cresko, W.A. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol. Biol. 2011, 772, 157–178. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Collins, D.W.; Jukes, T.H. Rates of transition and transversion in coding sequences since the human-rodent divergence. Genomics 1994, 20, 386–396. [Google Scholar] [CrossRef]
- Wakeley, J. The excess of transitions among nucleotide substitutions: New methods of estimating transition bias underscore its significance. Trends Ecol. Evol. 1996, 11, 158–163. [Google Scholar] [CrossRef]
- Li, W.H.; Wu, C.I.; Luo, C.C. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Mol. Evol. 1984, 21, 58–71. [Google Scholar] [CrossRef]
- Halim, D.; Wilson, M.P.; Oliver, D.; Brosens, E.; Verheij, J.B.G.M.; Han, Y.; Nanda, V.; Lyu, Q.; Doukas, M.; Stoop, H.; et al. Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc. Natl. Acad. Sci. USA 2017, 114, E2739–E2747. [Google Scholar] [CrossRef]
- Zamorano Cuervo, N.; Osseman, Q.; Grandvaux, N. Virus Infection Triggers MAVS Polymers of Distinct Molecular Weight. Viruses 2018, 10, 56. [Google Scholar] [CrossRef]
- Randall, K.L.; Lambe, T.; Goodnow, C.C.; Cornall, R.J. The essential role of DOCK8 in humoral immunity. Dis. Markers 2010, 29, 141–150. [Google Scholar] [CrossRef]
- Chan, S.W.; Fowler, K.J.; Choo, K.H.; Kalitsis, P. Spef1, a conserved novel testis protein found in mouse sperm flagella. Gene 2005, 353, 189–199. [Google Scholar] [CrossRef]
- Kang, H.; Hwang, S.C.; Park, Y.S.; Oh, J.S. Cdc25B phosphatase participates in maintaining metaphase II arrest in mouse oocytes. Mol. Cells 2013, 35, 514–518. [Google Scholar] [CrossRef]
- Lin, X.; Luo, J.; Zhang, L.; Wang, W.; Gou, D. MiR-103 controls milk fat accumulation in goat (Capra hircus) mammary gland during lactation. PLoS ONE 2013, 8, e79258. [Google Scholar] [CrossRef]
- Secco, B.; Camiré, E.; Brière, M.-A.; Caron, A.; Billong, A.; Gélinas, Y.; Lemay, A.-M.; Tharp, K.M.; Lee, P.L.; Gobeil, S.; et al. Amplification of Adipogenic Commitment by VSTM2A. Cell Rep. 2017, 18, 93–106. [Google Scholar] [CrossRef]
- Berger, A.H.; Chen, M.; Morotti, A.; Janas, J.A.; Niki, M.; Bronson, R.T.; Taylor, B.S.; Ladanyi, M.; Van Aelst, L.; Politi, K.; et al. DOK2 inhibits EGFR-mutated lung adenocarcinoma. PLoS ONE 2013, 8, e79526. [Google Scholar] [CrossRef]
- Miyagaki, H.; Yamasaki, M.; Takahashi, T.; Kurokawa, Y.; Miyata, H.; Nakajima, K.; Takiguchi, S.; Fujiwara, Y.; Mori, M.; Doki, Y. DOK2 as a marker of poor prognosis of patients with gastric adenocarcinoma after curative resection. Ann. Surg. Oncol. 2012, 19, 1560–1567. [Google Scholar] [CrossRef]
- Wen, X.; Zhou, M.; Guo, Y.; Zhu, Y.; Li, H.; Zhang, L.; Yu, L.; Wang, X.; Peng, X. Expression and significance of DOK2 in colorectal cancer. Oncol. Lett. 2015, 9, 241–244. [Google Scholar] [CrossRef][Green Version]
- Huang, J.; Peng, X.; Zhang, K.; Li, C.; Su, B.; Zhang, Y.; Yu, W. Co-expression and significance of Dok2 and Ras p21 protein activator 1 in breast cancer. Oncol. Lett. 2017, 14, 5386–5392. [Google Scholar] [CrossRef][Green Version]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. Identification and characterization of HAOX1, HAOX2, and HAOX3, three human peroxisomal 2-hydroxy acid oxidases. J. Biol. Chem. 2000, 275, 12590–12597. [Google Scholar] [CrossRef] [PubMed]
- Barton, P.J.; Townsend, P.J.; Brand, N.J.; Yacoub, M.H. Localization of the fast skeletal muscle troponin I gene (TNNI2) to 11p15.5: Genes for troponin I and T are organized in pairs. Ann. Hum. Genet. 1997, 61, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Gunther, K. The CRP/MLP/TLP family of LIM domain proteins: Acting by connecting. Bioessays 2003, 25, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, K.Y.; Kida, Y.S.; Sato, T.; Minami, M.; Ogura, T. Csrp1 regulates dynamic cell movements of the mesendoderm and cardiac mesoderm through interactions with Dishevelled and Diversin. Proc. Natl. Acad. Sci. USA 2007, 104, 11274–11279. [Google Scholar] [CrossRef]
- Salhab, M.; Patani, N.; Jiang, W.; Mokbel, K. High TIMM17A expression is associated with adverse pathological and clinical outcomes in human breast cancer. Breast Cancer 2012, 19, 153–160. [Google Scholar] [CrossRef]
- Miao, Q.; Huang, S.Y.; Qin, S.Y.; Yu, X.; Yang, Y.; Yang, J.F.; Zhu, X.Q.; Zou, F.C. Genetic characterization of Toxoplasma gondii in Yunnan black goats (Capra hircus) in southwest China by PCR-RFLP. Parasites Vectors 2015, 8, 57. [Google Scholar] [CrossRef]
- Park, S.D.E.; Magee, D.A.; McGettigan, P.A.; Teasdale, M.D.; Edwards, C.J.; Lohan, A.J.; Murphy, A.; Braud, M.; Donoghue, M.T.; Liu, Y.; et al. Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol. 2015, 16, 234. [Google Scholar] [CrossRef]
- Jia, X.J.; Chen, Y.L.; Wang, S.J.; Lu, Z.T.; Zhang, C.L.; Zhang, Z.J.; Ren, C.H. Genetic diversity analysis of microsatellite DNA of nine goat breeds. J. Anhui Agric. Univ. 2019, 46, 779–784. [Google Scholar]
- Burren, A.; Neuditschko, M.; Signer-Hasler, H.; Frischknecht, M.; Reber, I.; Menzi, F.; Drogemuller, C.; Flury, C. Genetic diversity analyses reveal first insights into breed-specific selection signatures within Swiss goat breeds. Anim. Genet. 2016, 47, 727–739. [Google Scholar] [CrossRef]
- Kim, E.S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.M.; Rothschild, M.F. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity 2016, 116, 255–264. [Google Scholar] [CrossRef]
- Brito, L.F.; Jafarikia, M.; Grossi, D.A.; Kijas, J.W.; Porto-Neto, L.R.; Ventura, R.V.; Salgorzaei, M.; Schenkel, F.S. Characterization of linkage disequilibrium, consistency of gametic phase and admixture in Australian and Canadian goats. BMC Genet. 2015, 16, 67. [Google Scholar] [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef]
- Taye, M.; Lee, W.; Caetano-Anolles, K.; Dessie, T.; Hanotte, O.; Mwai, O.A.; Kemp, S.; Cho, S.; Oh, S.J.; Lee, H.-K.; et al. Whole genome detection of signature of positive selection in African cattle reveals selection for thermotolerance. Anim. Sci. J. 2017, 88, 1889–1901. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; Van Binsbergen, R.; Brøndum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C.; et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014, 46, 858–865. [Google Scholar] [CrossRef]
- Xu, L.; Bickhart, D.M.; Cole, J.B.; Schroeder, S.G.; Song, J.; Tassell, C.P.; Sonstegard, T.S.; Liu, G.E. Genomic signatures reveal new evidences for selection of important traits in domestic cattle. Mol. Biol. Evol. 2015, 32, 711–725. [Google Scholar] [CrossRef]
- Li, M.; Tian, S.; Jin, L.; Zhou, G.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.L.; Chen, L.; Ma, J.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013, 45, 1431–1438. [Google Scholar] [CrossRef]
- Mashima, R.; Arimura, S.; Kajikawa, S.; Oda, H.; Nakae, S.; Yamanashi, Y. Dok adaptors play anti-inflammatory roles in pulmonary homeostasis. Genes Cells 2013, 18, 56–65. [Google Scholar] [CrossRef]
- Sun, P.; Li, R.; Meng, Y.; Xi, S.; Wang, Q.; Yang, X.; Peng, X.; Cai, J. Introduction to DOK2 and its potential role in cancer. Physiol. Res. 2021, 70, 671–685. [Google Scholar] [CrossRef]
- Randall, K.L.; Lambe, T.; Johnson, A.L.; Treanor, B.; Kucharska, E.; Domaschenz, H.; Whittle, B.; Tze, L.E.; Enders, A.; Crockford, T.L.; et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat. Immunol. 2009, 10, 1283–1291. [Google Scholar] [CrossRef]
- Chen, L.P.; Zhang, J.; Wei, X.L.; Chen, N.; Huang, C.X.; Xu, M.X.; Wang, W.M.; Wang, H.L. Megalobrama amblycephala cardiac troponin T variants: Molecular cloning, expression and response to nitrite. Gene 2013, 527, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Kamisago, M.; Sharma, S.D.; DePalma, S.R.; Solomon, S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, E.D.; et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000, 343, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.D.; Tsai, W.Y.; Horng, L.S.; Tsai, H.J. Molecular structure and developmental expression of three muscle-type troponin T genes in zebrafish. Dev. Dyn. 2003, 227, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, R.; Xu, X. Myofibrillogenesis in the developing zebrafish heart: A functional study of tnnt2. Dev. Biol. 2009, 331, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Xu, H.; Li, R.; Gao, S.; Chen, N.; Luo, J.; Jiang, Y. Genetic Basis of Phenotypic Differences between Chinese Yunling Black Goats and Nubian Goats Revealed by Allele-Specific Expression in Their F1 Hybrids. Front. Genet. 2019, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Goyache, F.; Perez-Pardal, L.; Fernandez, I.; Traore, A.; Menendez-Arias, N.A.; Alvarez, I. Ancient autozygous segments subject to positive selection suggest adaptive immune responses in West African cattle. Gene 2021, 803, 145899. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Li, Z.; Wang, P.; Fan, P.; Zhang, Y.; Zhang, Q.; Wang, Y.; Xu, X.; Liu, B. Differences of immune responses between Tongcheng (Chinese local breed) and Large White pigs after artificial infection with highly pathogenic porcine reproductive and respiratory syndrome virus. Virus Res. 2016, 215, 84–93. [Google Scholar] [CrossRef]
- Xing, J.; Xing, F.; Zhang, C.; Zhang, Y.; Wang, N.; Li, Y.; Yang, L.; Jiang, C.; Zhang, C.; Wen, C.; et al. Genome-wide gene expression profiles in lung tissues of pig breeds differing in resistance to porcine reproductive and respiratory syndrome virus. PLoS ONE 2014, 9, e86101. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Zhao, Y.L.; He, J.; Wang, W.; Davies, K.E.; Nose, V.; Xiao, S. UTRN on chromosome 6q24 is mutated in multiple tumors. Oncogene 2007, 26, 6220–6228. [Google Scholar] [CrossRef][Green Version]
- Vizkeleti, L.; Kiss, T.; Koroknai, V.; Ecsedi, S.; Papp, O.; Szasz, I.; Adany, R.; Balazs, M. Altered integrin expression patterns shown by microarray in human cutaneous melanoma. Melanoma Res. 2017, 27, 180–188. [Google Scholar] [CrossRef]
- Rákosy, Z.; Ecsedi, S.; Toth, R.; Vízkeleti, L.; Herandez-Vargas, H.; Lazar, V.; Emri, G.; Szatmári, I.; Herceg, Z.; Ádány, R.; et al. Integrative genomics identifies gene signature associated with melanoma ulceration. PLoS ONE 2013, 8, e54958. [Google Scholar] [CrossRef]
- Zhou, S.; Ouyang, W.; Zhang, X.; Liao, L.; Pi, X.; Yang, R.; Mei, B.; Xu, H.; Xiang, S.; Li, J. UTRN inhibits melanoma growth by suppressing p38 and JNK/c-Jun signaling pathways. Cancer Cell Int. 2021, 21, 88. [Google Scholar] [CrossRef]
- Garibyan, L.; Fisher, D.E. How Sunlight Causes Melanoma. Curr. Oncol. Rep. 2010, 12, 319–326. [Google Scholar] [CrossRef]
- Zaidi, M.R.; Davis, S.; Noonan, F.P.; Graff-Cherry, C.; Hawley, T.S.; Walker, R.L.; Feigenbaum, L.; Fuchs, E.; Lyakh, L.; Young, H.A.; et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature 2011, 469, 548–553. [Google Scholar] [CrossRef]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; Van den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef]
- Yanai, H.; Savitsky, D.; Tamura, T.; Taniguchi, T. Regulation of the cytosolic DNA-sensing system in innate immunity: A current view. Curr. Opin. Immunol. 2009, 21, 17–22. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Trebak, M.; Kinet, J.P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef]
- Blitzer, R.D.; Iyengar, R.; Landau, E.M. Postsynaptic signaling networks: Cellular cogwheels underlying long-term plasticity. Biol. Psychiatry 2005, 57, 113–119. [Google Scholar] [CrossRef]
- Chen, Z.; Gibson, T.B.; Robinson, F.; Silvestro, L.; Pearson, G.; Xu, B.; Wright, A.; Vanderbilt, C.; Cobb, M.H. MAP kinases. Chem. Rev. 2001, 101, 2449–2476. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Chromosome | Start | End | Gene Dosage | Gene Name |
|---|---|---|---|---|
| chr4 | 118360001 | 121760000 | 1 | VSTM2A |
| chr7 | 85940001 | 88020000 | 9 | ADAMTS19, KIAA1024L, CHSY3, HINT1, LYRM7, DC42SE2, RAPGEF6, FNIP1, MEIKIN |
| chr8 | 42100001 | 44360000 | 7 | SMARCA2, DMRT2, DMRT3, DMRT1, KANK1, DOCK8, PGM5 |
| chr8 | 67820001 | 70010000 | 27 | GFRA2, DOK2, XPO7, NPM2, FGF17, DMTN, FAM160B2, NUDT18, HR, REEP4, LGI3, SFTPC, BMP1, PHYHIP, MIR320, POLR3D, PIWIL2, SLC39A14, PPP3CC, SORBS3, PDLIM2, C8H8orf58, CCAR2, BIN3, EGR3, PEBP4, RHOBTB2 |
| chr9 | 68190001 | 70570000 | 11 | PHACTR2, LTV1, ZC2HC1B, PLAGL1, SF3B5, STX11, UTRN, EPM2A, FBXO30, SHPRH, GRM1 |
| chr12 | 87130001 | 89210000 | 0 | / |
| chr13 | 48860001 | 51230000 | 19 | HAO1, ADRA1D, SMOX, RNF24, TRNAE-UUC, PANK2, MIR103, MAVS, AP5S1, CDC25B, CENPB, SPEF1, C13H20orf27, HSPA12B, SIGLEC1, ADAM33, GFRA4, ATRN, C13H20orf194 |
| chr16 | 78640001 | 80720000 | 17 | PPP1R12B, UBE2T, LGR6, PTPN7, ARL8A, GPR37L1, NAV1, IPO9, LMOD1,TIMM17A, RNPEP, ELF3, CSRP1, PHLDA3, TNNI1, LAD1, TNNT2 |
| Chromosome | Gene | Gene Description or Function | References |
|---|---|---|---|
| chr4 | VSTM2A | Associated with fat production | [47] |
| chr8 | DOK2 | Acts as a suppressor in lung, gastric, colorectal, and ovarian cancer | [48,49,50,51] |
| chr8 | DOCK8 | Associated with humoral immunity | [43] |
| chr13 | MIR103 | Affects milk fat accumulation in the mammary gland of goats during lactation | [46] |
| chr13 | MAVS | An essential component of virus-activated signaling pathways and is involved in immunity | [42] |
| chr13 | HAO1 | Associated with calcium binding, alters the deposition of bone and cartilage | [52] |
| chr13 | SPEF1 | Associated with reproduction | [44] |
| chr13 | CDC25B | Important for oocyte meiosis, associated with reproduction | [45] |
| chr16 | TNNT2 | Troponin T2, which mediates muscle contraction | [53] |
| chr16 | CSRP1 | Associated with skeletal muscle growth | [54,55] |
| chr16 | TIMM17A | Associated with breast cancer | [56] |
| chr16 | LMOD1 | Associated with the contractility of smooth muscle cells, and its loss can lead to intestinal hypoperistalsis syndrome | [41] |
| Category | GO ID | Term | Genes | p-Value |
|---|---|---|---|---|
| BP | GO:0060903 | Positive regulation of meiosis I | PIWIL2, DMRT1 | 0.0093 |
| BP | GO:0010591 | Regulation of lamellipodium assembly | DMTN, BIN3 | 0.0231 |
| BP | GO:0042274 | Ribosomal small subunit biogenesis | LTV1, ZC2HC1B | 0.0231 |
| BP | GO:1900025 | Negative regulation of substrate adhesion-dependent cell spreading | KANK1, DMTN | 0.0411 |
| CC | GO:0016010 | Dystrophin-associated glycoprotein complex | PGM5, UTRN | 0.0194 |
| CC | GO:0005861 | Troponin complex | TNNT2, TNNI1 | 0.0271 |
| CC | GO:0005737 | Cytoplasm | KANK1, DMRT1, DOCK8, XPO7, PHYHIP, SORBS3, CCAR2, PPP3CC, IPO9, BIN3, PPP1R12B, UTRN, PHLDA3 | 0.0466 |
| MF | GO:0003779 | Actin binding | DMTN, SPEF1, LMOD1, PHACTR2, UTRN | 0.0153 |
| MF | GO:0038023 | Signaling receptor activity | ATRN, GFRA2, GFRA4 | 0.0265 |
| MF | GO:0017166 | Vinculin binding | UTRN, SORBS3 | 0.0386 |
| KEGG | chx04623 | Cytosolic DNA-sensing pathway | POLR3D, MAVS | 0.0260 |
| KEGG | chx04010 | MAPK signaling pathway | CDC25B, PPP3CC, PTPN7, FGF17 | 0.0295 |
| KEGG | chx04720 | Long-term potentiation | PPP3CC, GRM1 | 0.0298 |
| KEGG | chx04020 | Calcium signaling pathway | PPP3CC, GRM1, ADRA1D | 0.0446 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Li, R.; Sun, J.; Li, C.; Xiao, H.; Chen, S. Genome-Wide Population Structure and Selection Signatures of Yunling Goat Based on RAD-seq. Animals 2022, 12, 2401. https://doi.org/10.3390/ani12182401
Chen Y, Li R, Sun J, Li C, Xiao H, Chen S. Genome-Wide Population Structure and Selection Signatures of Yunling Goat Based on RAD-seq. Animals. 2022; 12(18):2401. https://doi.org/10.3390/ani12182401
Chicago/Turabian StyleChen, Yuming, Rong Li, Jianshu Sun, Chunqing Li, Heng Xiao, and Shanyuan Chen. 2022. "Genome-Wide Population Structure and Selection Signatures of Yunling Goat Based on RAD-seq" Animals 12, no. 18: 2401. https://doi.org/10.3390/ani12182401
APA StyleChen, Y., Li, R., Sun, J., Li, C., Xiao, H., & Chen, S. (2022). Genome-Wide Population Structure and Selection Signatures of Yunling Goat Based on RAD-seq. Animals, 12(18), 2401. https://doi.org/10.3390/ani12182401