
Astaxanthin Inhibits Acetaldehyde-Induced Cytotoxicity in SH-SY5Y Cells by Modulating Akt/CREB and p38MAPK/ERK Signaling Pathways

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
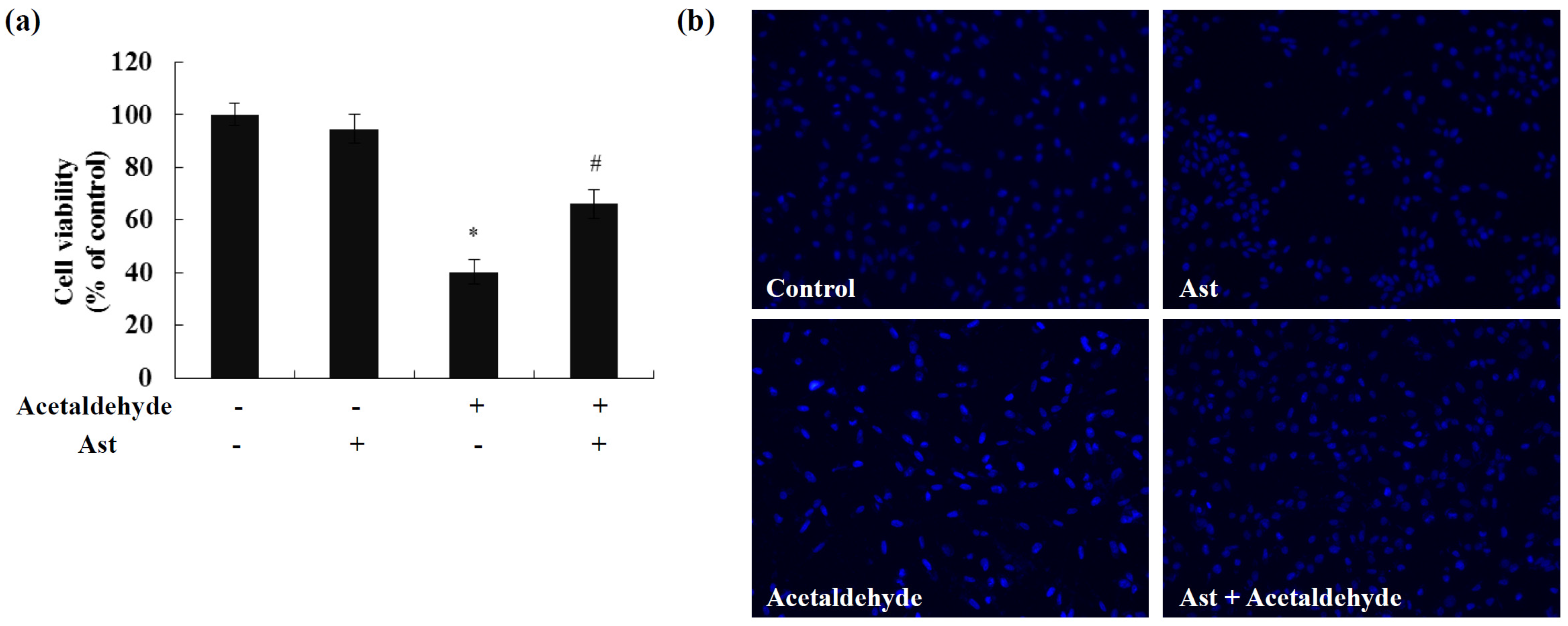
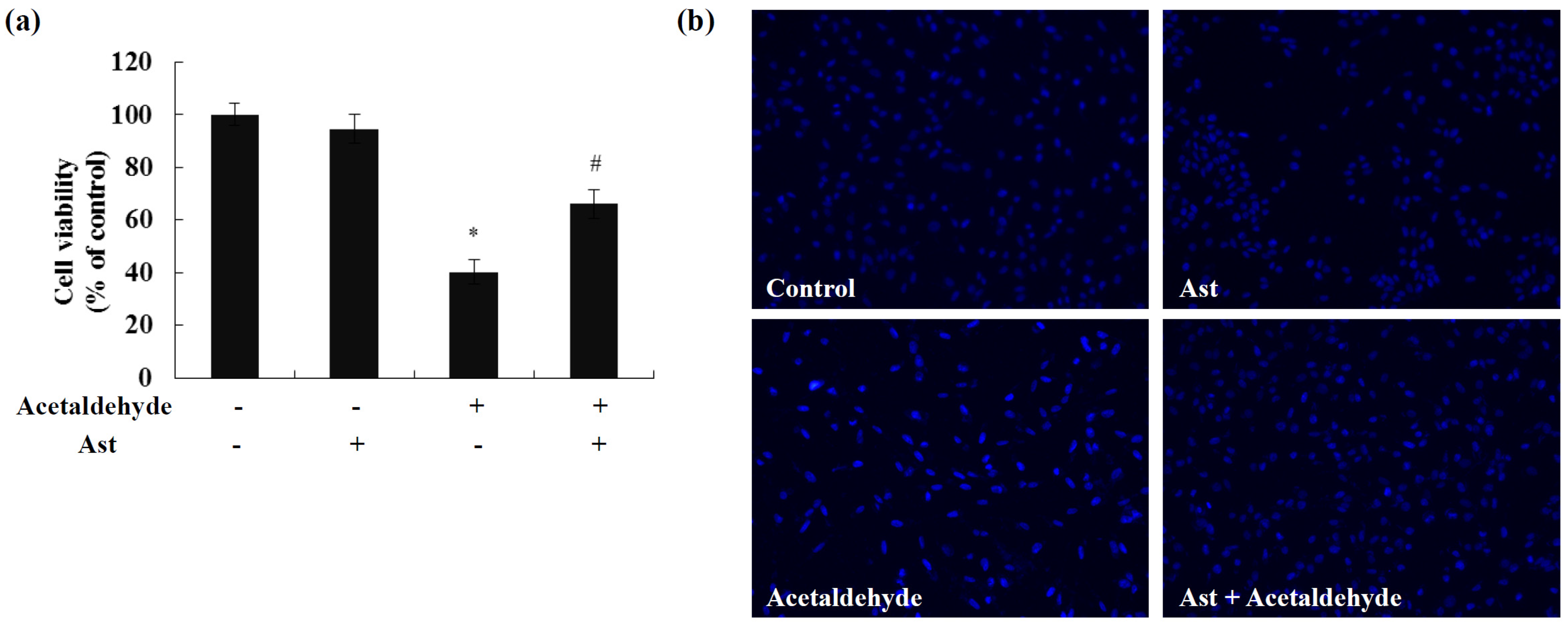
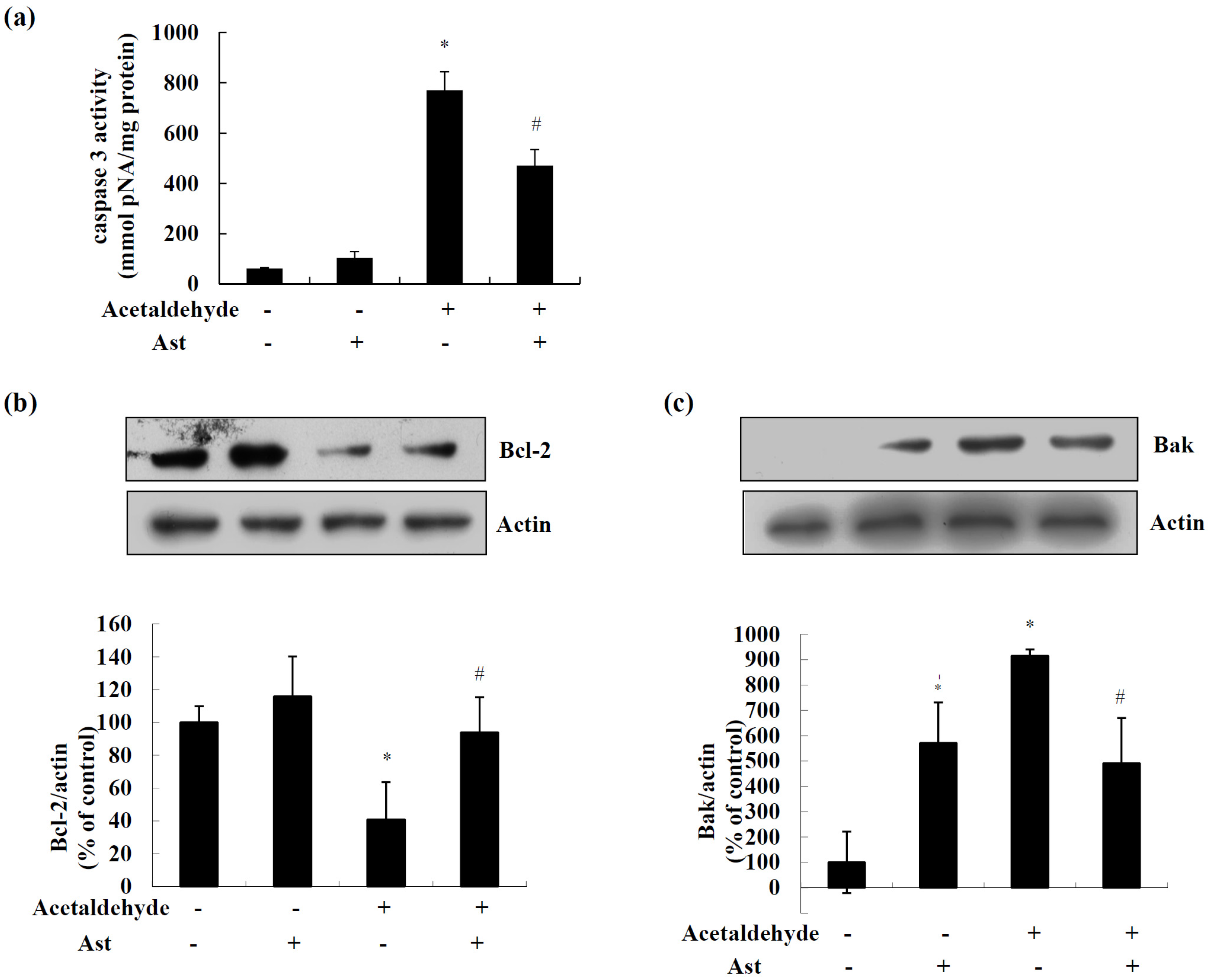
2.1. Ast Reduces Acetaldehyde-Induced Cytotoxicity and Apoptosis
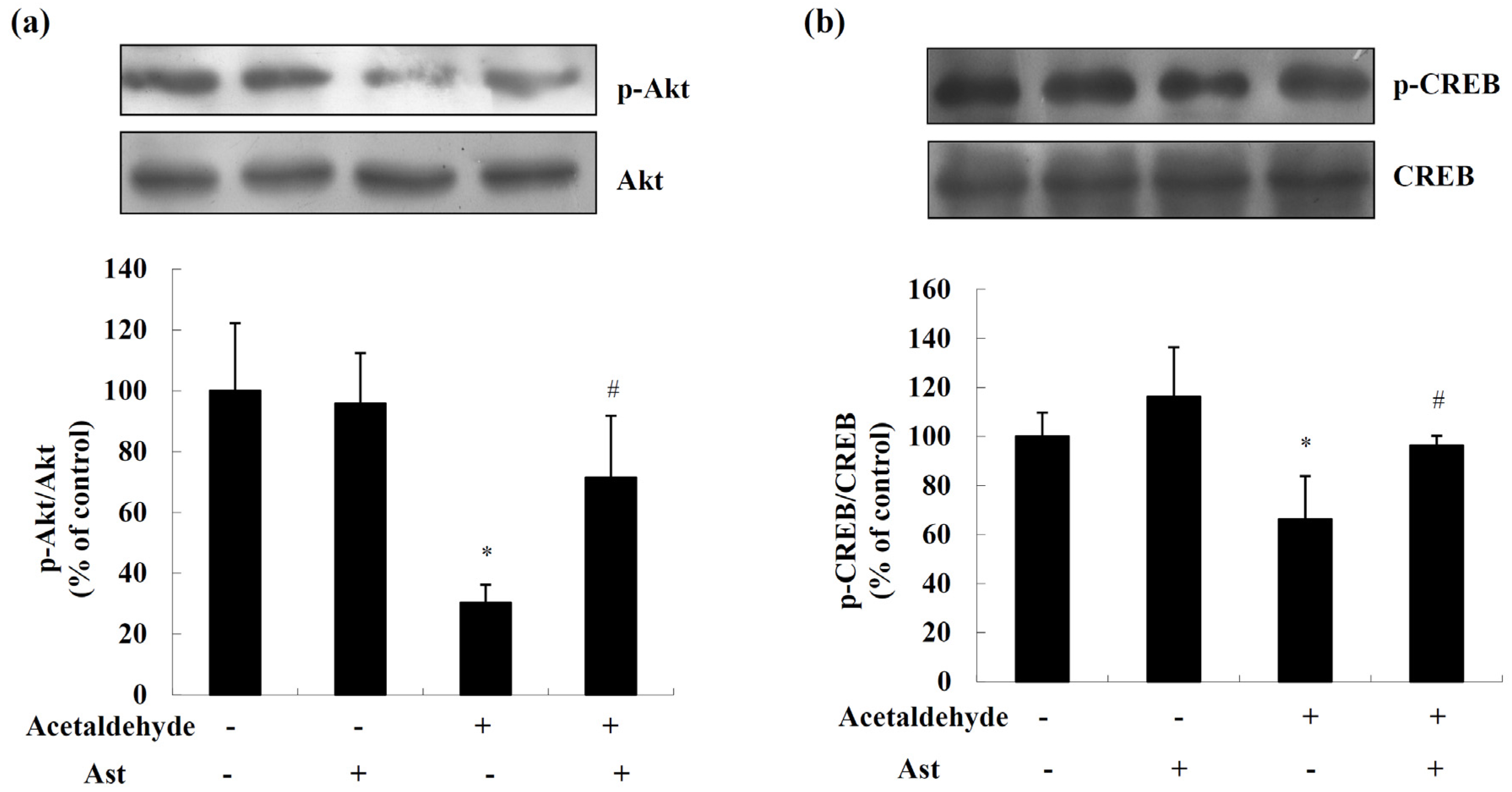
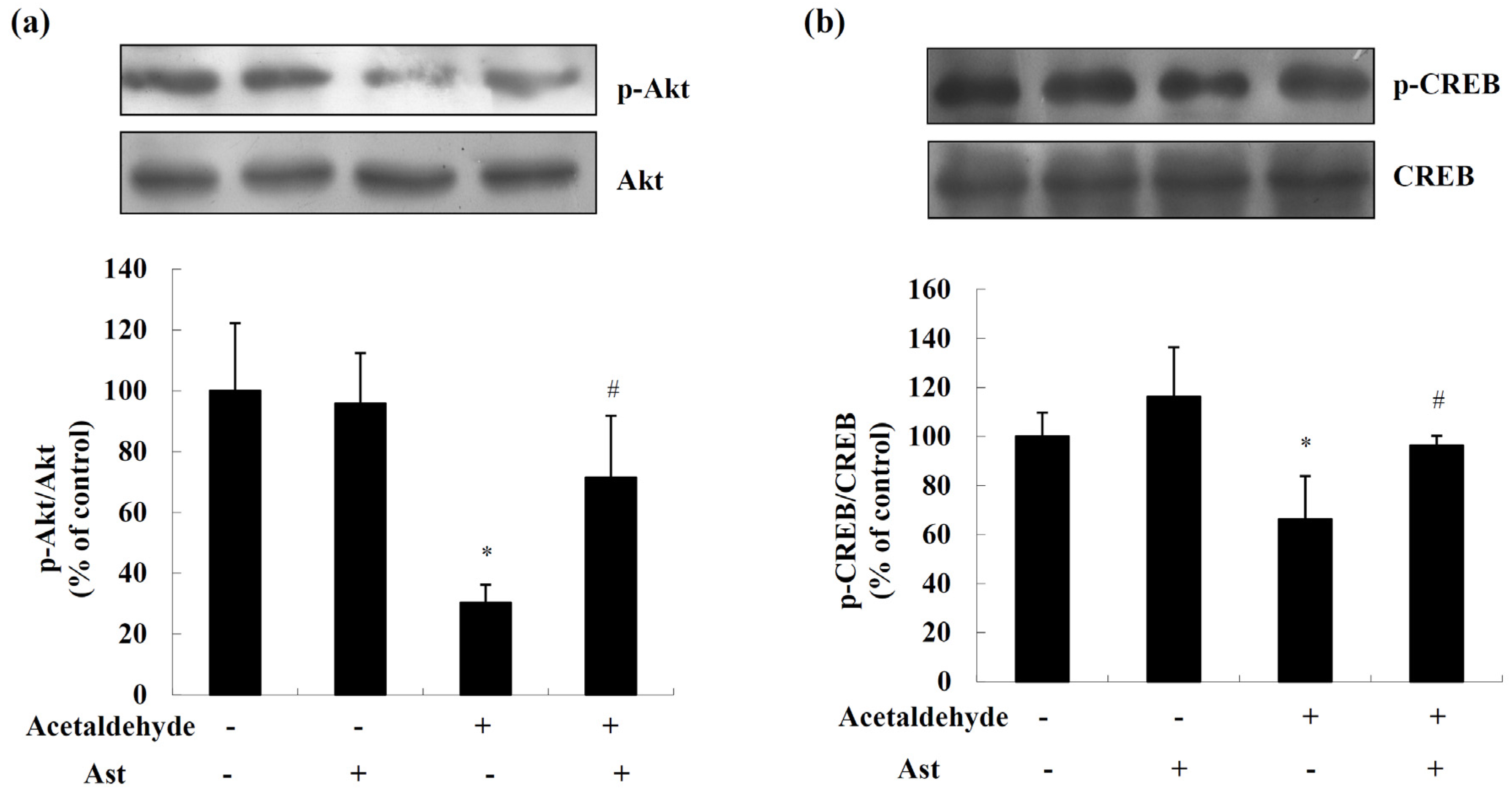
2.2. AST Ameliorates Acetaldehyde-Induced Inhibition of Akt and CREB
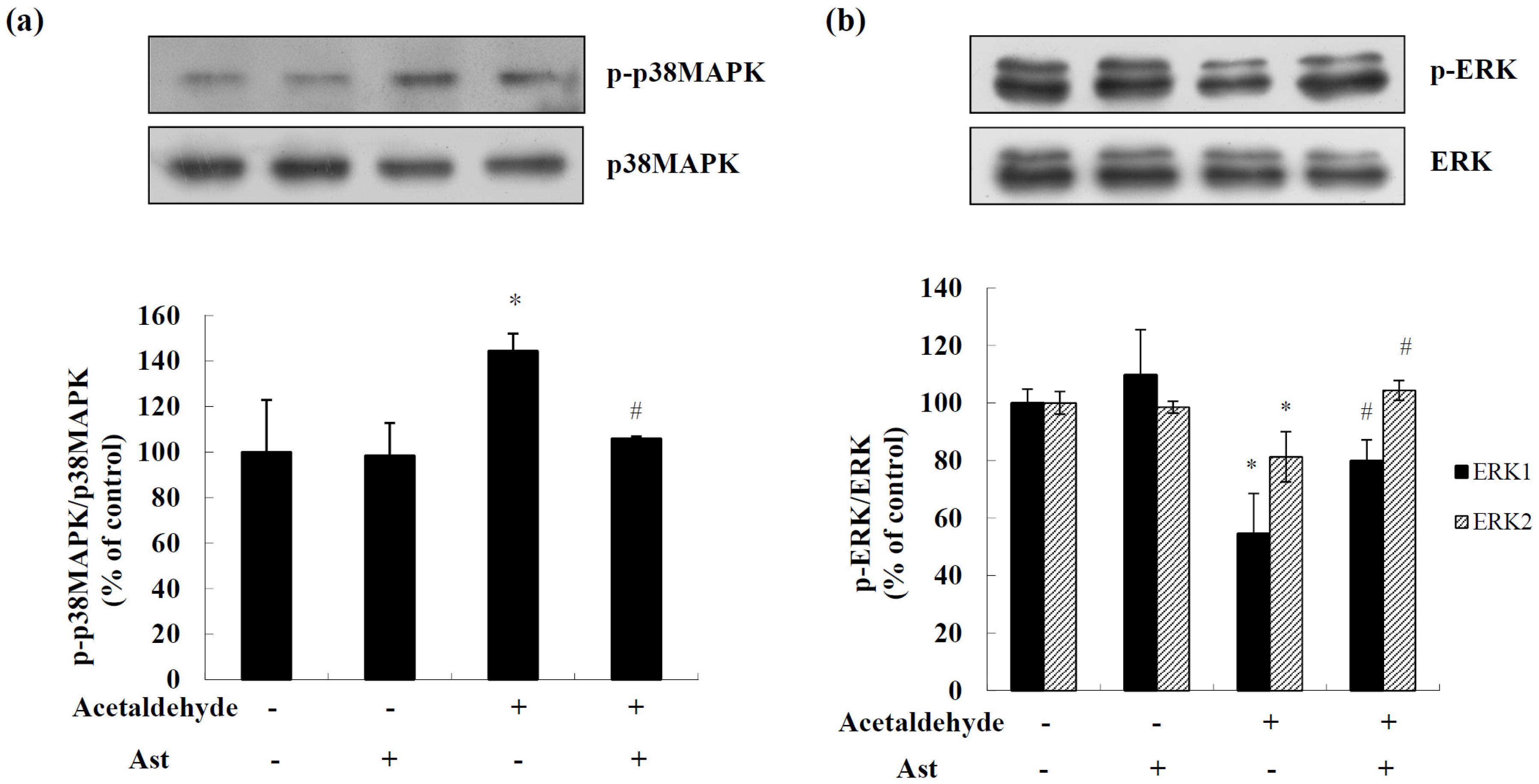
2.3. Effects of AST on the Activation of MAPKs in Acetaldehyde-Treated Cells
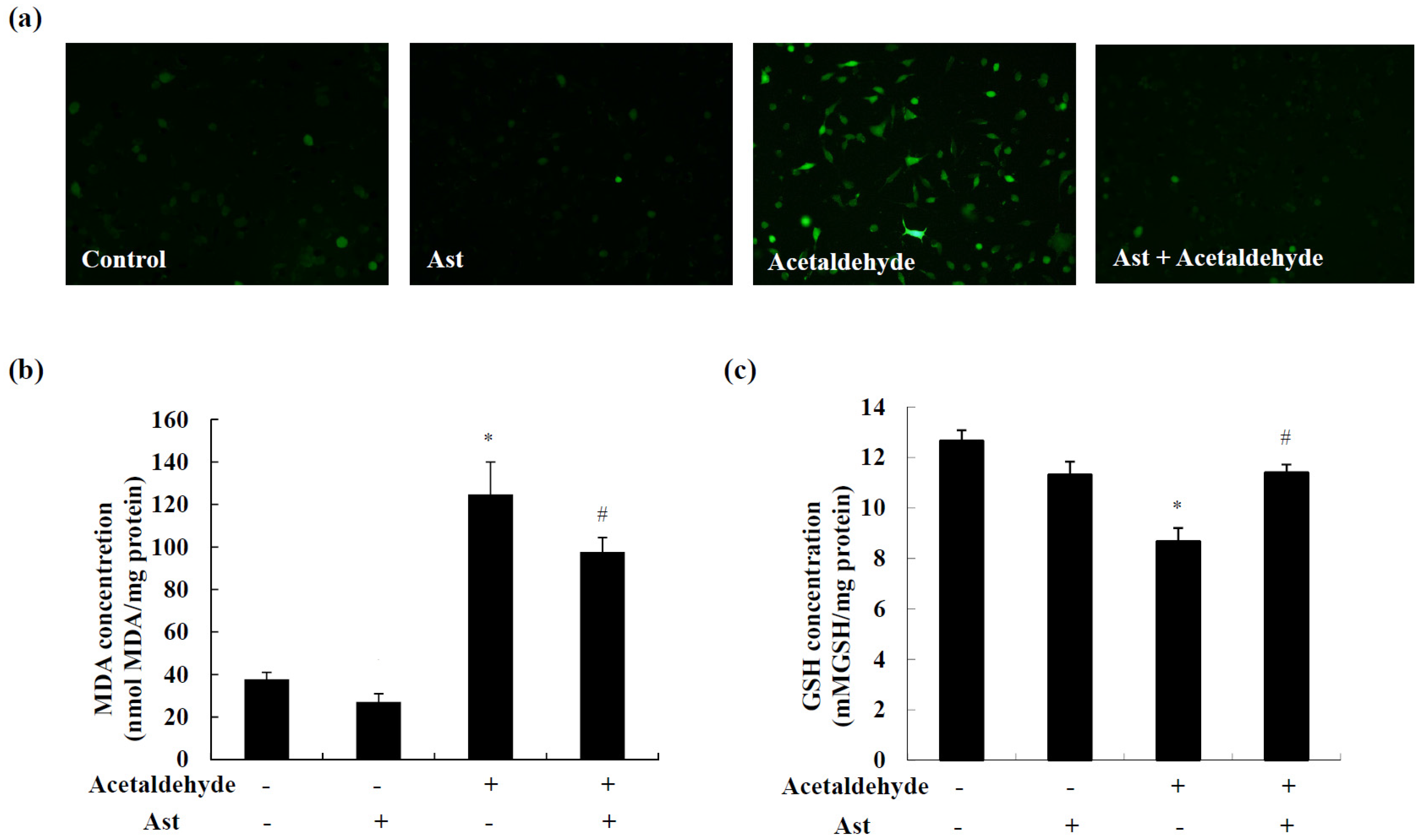
2.4. Ast Reduces Acetaldehyde-Induced Oxidative Stress
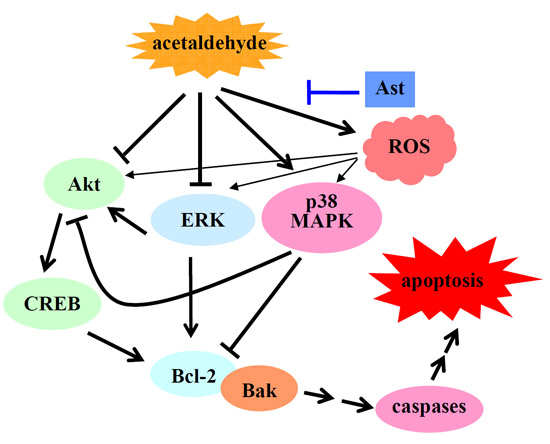
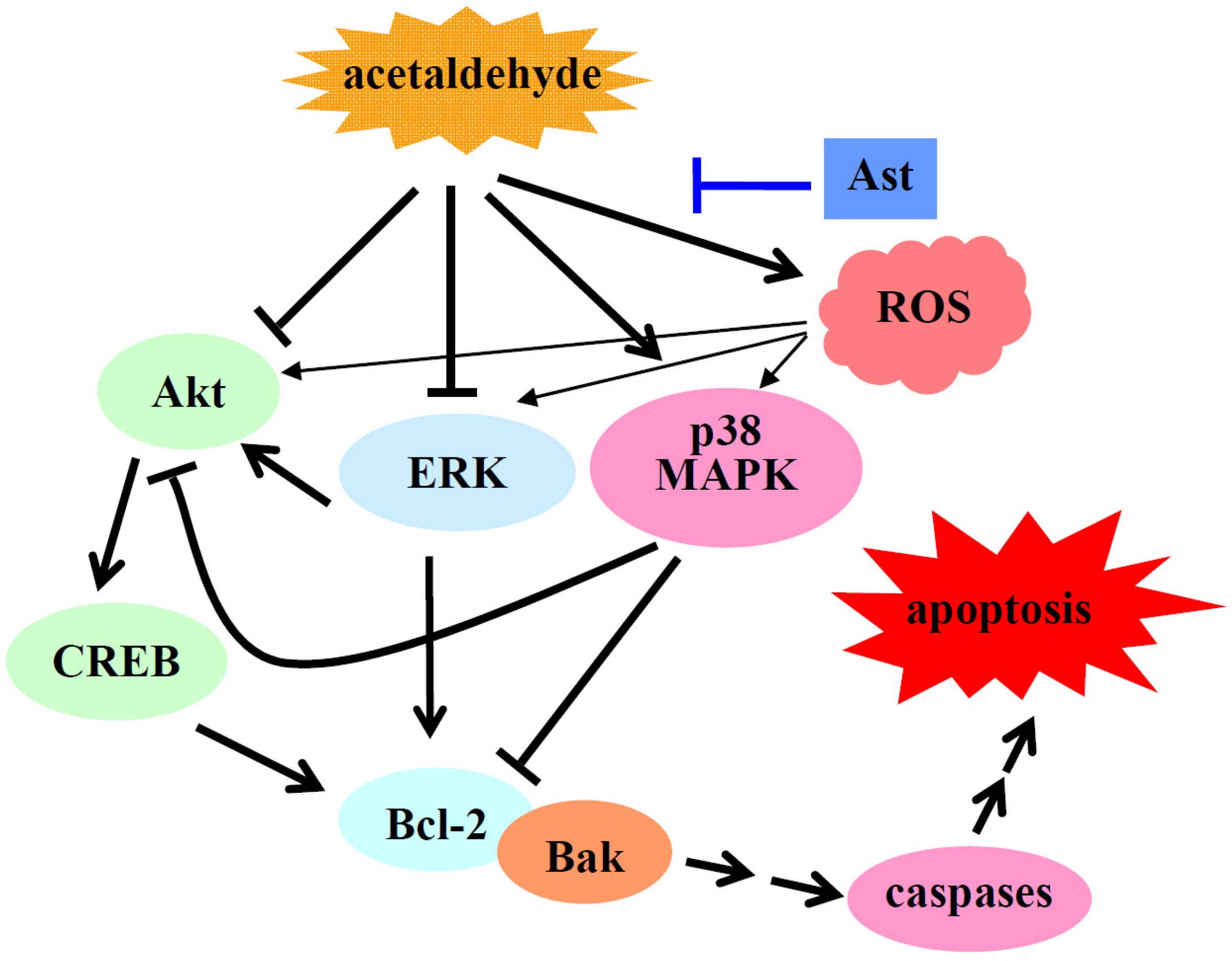
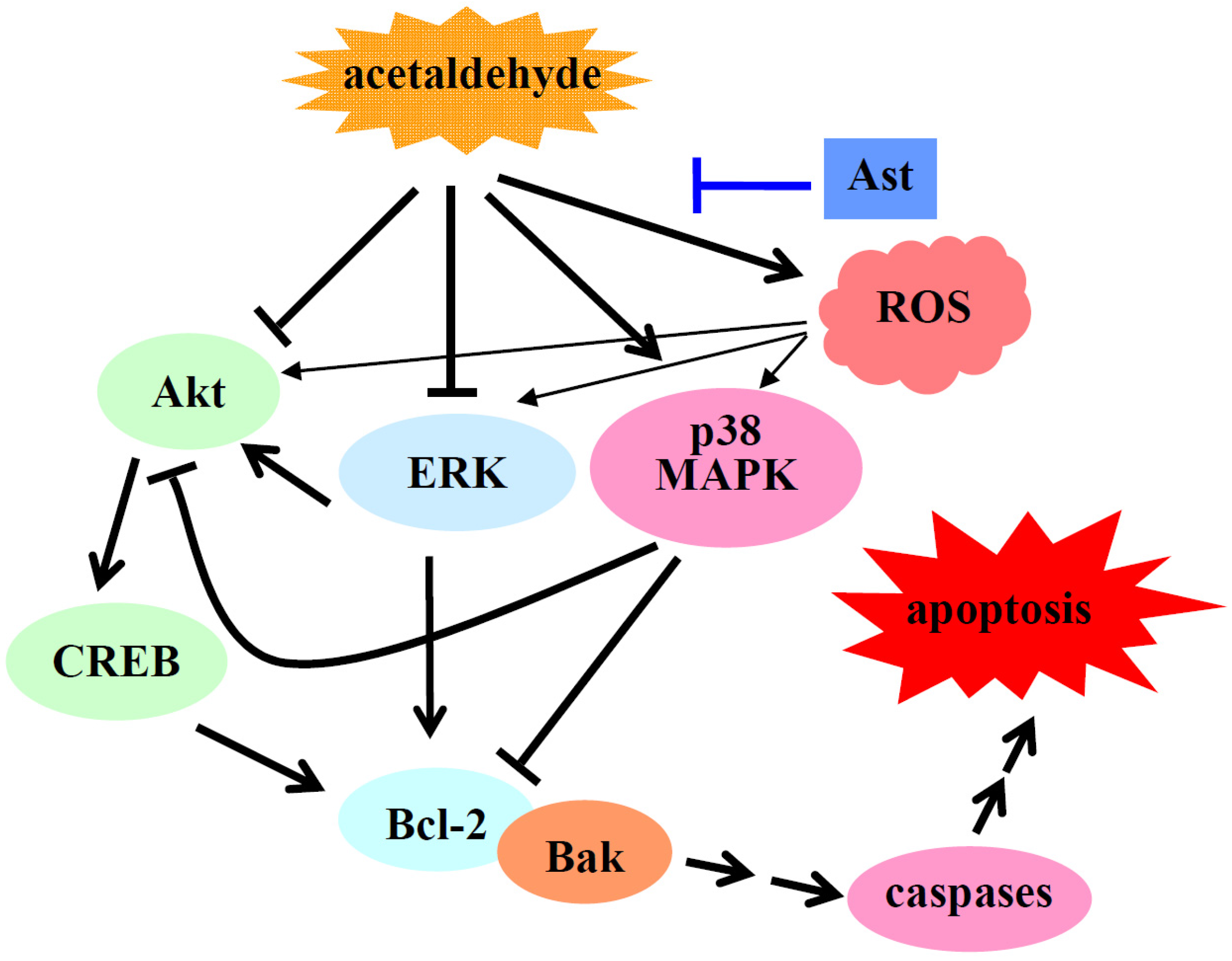
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Viability Assay
4.3. Hoechst 33258 Nuclear Staining Assay
4.4. Caspase 3 Activity
4.5. Measurement of Intracellular Oxidation Stress
4.6. Western Blotting Analysis
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CREB | cyclic AMP-responsive element binding protein |
| MAPK | mitogen-activated protein kinase |
| ERK | extracellular signal-regulated kinases |
| ADH | alcohol dehydrogenase |
| ALDH | aldehyde dehydrogenase |
| Ast | astaxanthin |
| ROS | reactive oxygen species |
| MDA | malondialdehyde |
| GSH | glutathione |
| FBS | fetal bovine serum |
| DMEM | Dulbecco’s modified eagle’s medium |
| DCFH-DA | 2′,7′-dichlorodihydrofluorescin diacetate |
| PVDF | polyvinylidene difluoride |
References
- Harper, C. The neuropathology of alcohol-related brain damage. Alcohol Alcohol. 2009, 44, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Langballe, E.M.; Ask, H.; Holmen, J.; Stordal, E.; Saltvedt, I.; Selbæk, G.; Fikseaunet, A.; Bergh, S.; Nafstad, P.; Tambs, K. Alcohol consumption and risk of dementia up to 27 years later in a large, population-based sample: The HUNT study, Norway. Eur. J. Epidemiol. 2015, 30, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwood, D.G.; Kalechstein, A.; Barker, W.W.; Strauman, S.; George-Hyslop, P.S.; Iglesias, C.; Loewenstein, D.; Duara, R. The effect of alcohol and tobacco consumption, and apolipoprotein E genotype, on the age of onset in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2010, 25, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Crabb, D.W.; Liangpunsakul, S. Acetaldehyde generating enzyme systems: Roles of alcohol dehydrogenase, CYP2E1 and catalase, and speculations on the role of other enzymes and processes. Novartis Found. Symp. 2007, 285, 4–16, discussion 16–22, 198–199. [Google Scholar] [PubMed]
- Peng, G.S.; Yin, S.J. Effect of the allelic variants of aldehyde dehydrogenase ALDH2*2 and alcohol dehydrogenase ADH1B*2 on blood acetaldehyde concentrations. Hum. Genom. 2009, 3, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Holownia, A.; Ledig, M.; Brszko, J.J.; Ménez, J.F. Acetaldehyde cytotoxicity in cultured rat astrocytes. Brain Res. 1999, 833, 202–208. [Google Scholar] [CrossRef]
- Aberle, N.S., II; Burd, L.; Zhao, B.H.; Ren, J. Acetaldehyde-induced cardiac contractile dysfunction may bealleviated by vitamin B1 but not by vitamins B6 or B12. Alcohol Alcohol. 2004, 39, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, K.K.; Samak, G.; Shukla, P.K.; Mir, H.; Gangwar, R.; Manda, B.; Isse, T.; Kawamoto, T.; Salaspuro, M.; Kaihovaara, P.; et al. ALDH2 deficiency promotes ethanol-induced gut barrier dysfunction and fatty liver in mice. Alcohol. Clin. Exp. Res. 2015, 39, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Holownia, A.; Ledig, M.; Mapoles, J.; Ménez, J.-F. Acetaldehyde-induced growth inhibition in cultured rat astroglial cells. Alcohol 1996, 13, 93–97. [Google Scholar] [CrossRef]
- Tokuda, K.; Izumi, Y.; Zorumski, C.F. Locally-generated acetaldehyde is involved in ethanol-mediated LTP inhibition in the hippocampus. Neurosci. Lett. 2013, 537, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Menegola, E.; Broccia, M.L.; Renzo, F.D.; Giavini, E. Acetaldehyde in vitro exposure and apoptosis: A possible mechanism of terato genesis. Alcohol 2001, 23, 35–39. [Google Scholar] [CrossRef]
- Tong, M.; Longato, L.; Nguyen, Q.G.L.; Chen, W.C.; Spaisman, A.; Monte, S.M. Acetaldehyde-mediated neurotoxicity: Relevance to fetal alcohol spectrum disorders. Oxid. Med. Cell. Longev. 2011, 2011, 213–286. [Google Scholar] [CrossRef]
- Jung, T.W.; Lee, J.Y.; Shim, W.S.; Kang, E.S.; Kim, J.S.; Ahn, C.W.; Lee, H.C.; Cha, B.S. Adiponectin protects human neuroblastoma SH-SY5Y cells against acetaldehyde-induced cytotoxicity. Biochem. Pharmacol. 2006, 72, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Clavijo-Cornejo, D.; Gutiérrez-Carrera, M.; Palestino-Domínguez, M.; Dominguez-Pereza, M.; Nuño, N.; Souza, V.; Miranda, R.; Kershenobich, D.; Gutiérrez-Ruiz, MC.; Bucio, L.; et al. Acetaldehyde targets superoxide dismutase 2 in liver cancer cells inducing transient enzyme impairment and a rapid transcriptional recovery. Food Chem. Toxicol. 2014, 69, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Li, Y.; Fanning, K.; Netzel, M.; Schenk, P.M. Effect of drying, storage temperature and air exposure on astaxanthin stability from Haematococcus pluvialis. Food Res. Int. 2015, 74, 231–236. [Google Scholar] [CrossRef]
- Nakajima, Y.; Inokuchi, Y.; Shimazawa, M.; Otsubo, K.; Ishibashi, T.; Hara, H. Astaxanthin, a dietary carotenoid, protects retinal cells against oxidative stress in-vitro and in mice in-vivo. J. Pharm. Pharmacol. 2008, 60, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Mcnulty, H.P.; Byun, J.; Lockwood, S.F.; Jacob, R.F.; Mason, R.P. Differential effects of carotenoids on lipid peroxidation due to membrane interactions: X-ray diffraction analysis. Biochim. Biophys. Acta 2007, 1768, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Marin, D.P.; Bolin, A.P.; Macedo Rde, C.; Sampaio, S.C.; Otton, R. ROS production in neutrophils from alloxan-induced diabetic rats treated in vivo with astaxanthin. Int. Immunopharmacol. 2011, 11, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Shibata, T.; Hisaka, S.; Osawa, T. Astaxanthin inhibits reactive oxygen species-mediated cellular toxicity in dopaminergic SH-SY5Y cells via mitochondria-targeted protective mechanism. Brain Res. 2009, 1254, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Chew, B.P.; Park, J.S.; Wong, M.W.; Wong, T.S. A comparison of the anticancer activities of dietary beta-carotene, canthaxanthin and astaxanthin in mice in vivo. Anticancer Res. 1999, 19, 1849–1853. [Google Scholar] [PubMed]
- Pashkow, F.J.; Watumull, D.G.; Campbell, C.L. Astaxanthin: A novel potential treatment for oxidative stress and inflammation in cardiovascular disease. Am. J. Cardiol. 2008, 101, 58D–68D. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Zhang, L.; Gross, M.; Tomita, Y. Immunomodulating actions of carotenoids: Enhancement of in vivo and in vitro antibody production to T-dependent antigens. Nutr. Cancer 1994, 21, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.P.; Liu, S.Y.; Sun, H.; Wu, X.M.; Li, J.J.; Zhu, L. Neuroprotective effect of astaxanthin on H2O2-induced neurotoxicity in vitro and on focal cerebral ischemia in vivo. Brain Res. 2010, 1360, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Kuo, C.C.; Chou, J.; Delvolve, A.; Jackson, S.N.; Post, J.; Woods, A.S.; Hoffer, B.J.; Wang, Y.; Harvey, B.K. Astaxanthin reduces ischemic brain injury in adult rats. FASEB J. 2009, 23, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Tsuji, S.; Satoh, A.; Ishikura, M.; Shirasawa, T.; Shimizu, T. Protective effects of astaxanthin on 6-hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J. Neurochem. 2008, 107, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Huang, A.; Hu, J.; Zhong, Z.; Liu, Y.; Li, Z.; Pan, X.; Liu, Z. Neuroprotective effect of astaxanthin against glutamate-induced cytotoxicity in HT22 cells: Involvement of the Akt/GSK-3β pathway. Neuroscience 2015, 303, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.T.; Zhao, Y.; Zhang, X. Acetaldehyde Induces Cytotoxicity of SH-SY5Y cells via inhibition of Akt activation and induction of oxidative stress. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.Y.; Peng, T.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2, release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.Y.; Liu, C.M.; Tsai, H.H.; Jong, Y.J.; Chen, I.J.; Lo, Y.C. KMUP-1 attenuates serum deprivation-induced neurotoxicity in SH-SY5Y cells: Roles of PKG, PI3K/AKT and Bcl-2/Bax pathways. Toxicology 2010, 268, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, H.; Zhang, X.; Chen, L.; Zhao, X.; Bai, X.; Zhang, J. Nobiletin protects against cerebral ischemia via activating the p-Akt, p-CREB, BDNF and Bcl-2 pathway and ameliorating BBB permeability in rat. Brain Res. Bull. 2013, 96, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Lee, R.D.; An, S.M.; Kim, S.S.; Rhee, G.S.; Kwack, S.J.; Seok, J.H.; Chae, S.Y.; Park, C.H.; Choi, Y.W.; Kim, H.S.; et al. Neurotoxic effects of alcohol and acetaldehyde during embryonic development. J. Toxicol. Environ. Health A 2015, 68, 2147–2162. [Google Scholar] [CrossRef] [PubMed]
- Dewson, G.; Kluck, R.M. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J. Cell Sci. 2009, 22, 2801–2808. [Google Scholar] [CrossRef]
- Selzner, M.; Rudiger, H.A.; Selzner, N.; Thomas, D.W.; Sindram, D.; Clavien, P.A. Transgenic mice overexpressing human Bcl-2 are resistant to hepatic ischemia and reperfusion. J. Hepatol. 2002, 36, 218–225. [Google Scholar] [CrossRef]
- Song, X.D.; Zhang, J.J.; Wang, M.R.; Liu, W.B.; Gu, X.B.; Lv, C.J. Astaxanthin induces mitochondria-mediated apoptosis in rat hepatocellular carcinoma CBRH-7919 cells. Biol. Pharm. Bull. 2011, 34, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dai, W.; Xia, Y.; Chen, K.; Li, S.; Liu, T.; Zhang, R.; Wang, J.; Lu, W.; Zhou, Y.; et al. Astaxanthin Inhibits Proliferation and Induces Apoptosis of Human Hepatocellular Carcinoma Cells via Inhibition of NF-κB P65 and Wnt/Β-Catenin in Vitro. Mar. Drug 2015, 13, 6064–6081. [Google Scholar] [CrossRef] [PubMed]
- Graupner, V.; Alexander, E.; Overkamp, T.; Rothfuss, O.; de Laurenzi, V.; Gillissen, B.F.; Daniel, P.T.; Schulze-Osthoff, K.; Essmann, F. Differential regulation of the proapoptotic multidomain protein Bak by p53 and p73 at the promoter level. Cell Death Differ. 2011, 18, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, B.; Lin, S.; Jing, L.; Mao, C.; Xu, P.; Lv, C.; Liu, W.; Zuo, J. Astaxanthin inhibits apoptosis in alveolar epithelial cells type II in vivo and in vitro through the ROS-dependent mitochondrial signalling pathway. J. Cell. Mol. Med. 2014, 18, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Q.; Chong, Z.Z.; Maiese, K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol. Pharmacol. 2003, 64, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Socodato, R.; Brito, R.; Portugal, C.C.; de Oliveira, N.A.; Calaza, K.C.; Paes-de-Carvalho, R. The nitric oxide-cGKII system relays death and survival signals during embryonic retinal development via AKT-induced CREB1 activation. Cell Death Differ. 2014, 21, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ping, F.F.; Lv, W.T.; Feng, J.Y.; Shang, J. Interleukin-18 directly protects cortical neurons by activating PI3K/AKT/NF-κB/CREB pathways. Cytokine 2014, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S. CREB couples neurotrophin signals to survival messages. Neuron 2000, 25, 11–14. [Google Scholar] [CrossRef]
- Gomez-Lazaro, M.; Galindo1, M.F.; Concannon, C.G.; Segura, M.F.; Fernandez-Gomez, F.J.; Llecha, N.; Comella, J.X.; Prehn, J.H.; Jordan, J. 6-Hydroxydopamine activates the mitochondrial apoptosis pathway through p38 MAPK-mediated, p53-independent activation of Bax and PUMA. J. Neurochem. 2008, 104, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Sun, X.B.; Xu, Y.X.; Zhao, H.; Zhu, Q.Y.; Zhu, C.Q. Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against beta-amyloid-induced cytotoxicity in SH-SY5Y cells. Brain Res. 2010, 1360, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.; Leng, X.; Lu, H.; Yang, H.; Song, N.; Tan, L.; Jiang, Y.; Guo, C. Depletion of intracellular zinc induces apoptosis of cultured hippocampal neurons through suppression of ERK signaling pathway and activation of caspase-3. Neurosci. Lett. 2013, 552, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Liu, Q.; Dou, Y.; Hsieh, Y.; Liu, Y.; Tao, R.; Zhu, D.; Lou, Y. Activating glucocorticoid receptor-ERK signaling pathway contributes to ginsenoside Rg1 protection against β-amyloid peptide-induced human endothelial cells apoptosis. J. Ethnopharmacol. 2013, 147, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.Y.; Yang, Y.; Shi, K.J.; Luo, H.; Duan, J.; An, J.J.; Wu, P.; Ci, Y.; Shi, L.; Xu, C. The p38MAPK-regulated PKD1/CREB/Bcl-2 pathway contributes to selenite-induced colorectal cancer cell apoptosis in vitro and in vivo. Cancer Lett. 2014, 354, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Oh, J.E.; Kim, S.W.; Chun, Y.J.; Kim, M.Y. Ceramide induces p38MAPK-dependent apoptosis and Bax translocation via inhibition of AKT in HL-60 cells. Cancer Lett. 2008, 260, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.Z.; Hu, J.X.; Wu, H.; Li, Y.L.; Chen, H.L.; Bai, H.; Hai, C.X. ROS-activated p38MAPK/ERK-AKT cascade plays a central role in palmitic acid-stimulated hepatocyte proliferation. Free Radic. Biol. Med. 2011, 51, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.H.; Fu, Q.C.; Shi, D.; Bu, H.L.; Song, Z.P.; Xiong, B.R.; Shu, B.; Xiang, H.B.; Xu, B.; Manyande, A.; et al. Activation of spinal chemokine receptor CXCR3 mediates bone cancer pain through an AKT-ERK crosstalk pathway in rats. Exp. Neurol. 2015, 263, 39–49. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J. Signal Transduct. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, T.; Zhao, Y.; Zhang, X.; Lin, X. Astaxanthin Inhibits Acetaldehyde-Induced Cytotoxicity in SH-SY5Y Cells by Modulating Akt/CREB and p38MAPK/ERK Signaling Pathways. Mar. Drugs 2016, 14, 56. https://doi.org/10.3390/md14030056
Yan T, Zhao Y, Zhang X, Lin X. Astaxanthin Inhibits Acetaldehyde-Induced Cytotoxicity in SH-SY5Y Cells by Modulating Akt/CREB and p38MAPK/ERK Signaling Pathways. Marine Drugs. 2016; 14(3):56. https://doi.org/10.3390/md14030056
Chicago/Turabian StyleYan, Tingting, Yan Zhao, Xia Zhang, and Xiaotong Lin. 2016. "Astaxanthin Inhibits Acetaldehyde-Induced Cytotoxicity in SH-SY5Y Cells by Modulating Akt/CREB and p38MAPK/ERK Signaling Pathways" Marine Drugs 14, no. 3: 56. https://doi.org/10.3390/md14030056