Metabolome Mining
Share This Topical Collection
Editor
Topical Collection Information
Dear Colleagues,
In current state-of-the-art metabolomics, the majority of metabolite features in metabolomics datasets are never fully assigned (Level 1: a validated assignment often against a reference standard) and mapped to known metabolic networks. Assignments can range from a Level 1 validated assignment to a putative assignment (Level 2), tentative assignment (Level 3), molecular formula assignment (Level 4), and unique metabolite feature (Level 5). Useful interpretable information can still be gleamed from these general levels of assignment, which represent chemical annotations on metabolite features. Metabolome mining is defined as the use of metabolite features, with chemical and other annotations, to derive metabolic information that is interpretable in a biological or biomedical context.
This Topical Collection covers the method developments, software, applications, and evaluation of metabolome mining.
Prof. Hunter Moseley
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 250 words) can be sent to the Editorial Office for assessment.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Metabolites is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- metabolome mining
- partial metabolite identification
- chemical classification
- chemical annotation
- metabolite annotation
- metabolic mapping
- chemical substructure
Published Papers (4 papers)
Open AccessArticle
Advanced Machine Learning for Comparative Synovial Fluid Analysis in Osteoarthritis and Rheumatoid Arthritis
by
Karolina Krystyna Kopeć, Gabrieleanselmo Uccheddu, Paweł Chodnicki, Antonio Noto, Cristina Piras, Martina Spada, Luigi Atzori and Vassilios Fanos
Cited by 3 | Viewed by 2633
Abstract
Osteoarthritis (OA) and rheumatoid arthritis (RA) are joint diseases that share similar clinical features but have different etiologies, making a differential diagnosis particularly challenging.
Background/Objectives: Utilizing advanced machine learning (ML) techniques on metabolomic data, this study aimed to identify key metabolites in
[...] Read more.
Osteoarthritis (OA) and rheumatoid arthritis (RA) are joint diseases that share similar clinical features but have different etiologies, making a differential diagnosis particularly challenging.
Background/Objectives: Utilizing advanced machine learning (ML) techniques on metabolomic data, this study aimed to identify key metabolites in synovial fluid (SF) that could aid in distinguishing between OA and RA.
Methods: Metabolite data from the MetaboLights database (MTBLS564), analyzed using nuclear magnetic resonance (NMR), were processed using normalization, a principal component analysis (PCA), and a partial least squares discriminant analysis (PLS-DA) to reveal prominent clustering.
Results: Decision forests and random forest classifiers, optimized using genetic algorithms (GAs), highlighted a selection of a few metabolites—primarily glutamine, pyruvate, and proline—with significant discriminative power. A Shapley additive explanations (SHAP) analysis confirmed these metabolites to be pivotal predictors, offering a streamlined approach for clinical diagnostics.
Conclusions: Our findings suggest that a minimal set of key metabolites can effectively be relied upon to distinguish between OA and RA, supported by an optimized ML model achieving high accuracy. This workflow could streamline diagnostic efficiency and enhance clinical decision-making in rheumatology.
Full article
►▼
Show Figures
Open AccessArticle
Mining Small Molecules from Teredinibacter turnerae Strains Isolated from Philippine Teredinidae
by
Jamaine B. Villacorta, Camille V. Rodriguez, Jacquelyn E. Peran, Jeremiah D. Batucan, Gisela P. Concepcion, Lilibeth A. Salvador-Reyes and Hiyas A. Junio
Cited by 4 | Viewed by 4938
Abstract
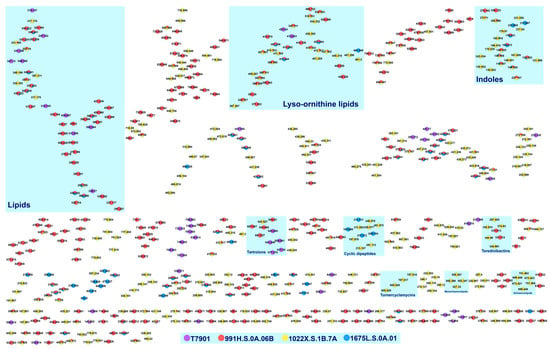
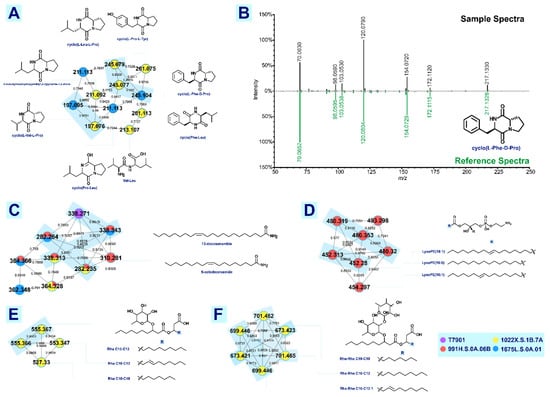
Endosymbiotic relationship has played a significant role in the evolution of marine species, allowing for the development of biochemical machinery for the synthesis of diverse metabolites. In this work, we explore the chemical space of exogenous compounds from shipworm endosymbionts using LC-MS-based metabolomics.
[...] Read more.
Endosymbiotic relationship has played a significant role in the evolution of marine species, allowing for the development of biochemical machinery for the synthesis of diverse metabolites. In this work, we explore the chemical space of exogenous compounds from shipworm endosymbionts using LC-MS-based metabolomics. Priority
T. turnerae strains (1022X.S.1B.7A, 991H.S.0A.06B, 1675L.S.0A.01) that displayed antimicrobial activity, isolated from shipworms collected from several sites in the Philippines were cultured, and fractionated extracts were subjected for profiling using ultrahigh-performance liquid chromatography with high-resolution mass spectrometry quadrupole time-of-flight mass analyzer (UHPLC-HRMS QTOF).
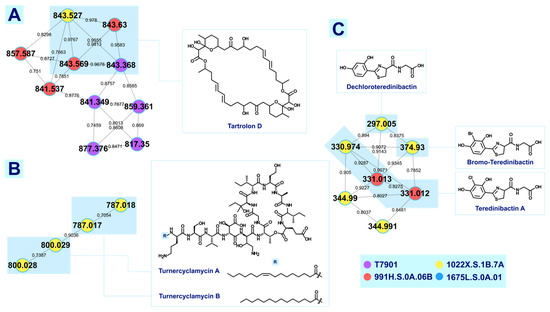
T. turnerae T7901 was used as a reference microorganism for dereplication analysis. Tandem MS data were analyzed through the Global Natural Products Social (GNPS) molecular networking, which resulted to 93 clusters with more than two nodes, leading to four putatively annotated clusters: lipids, lysophosphatidylethanolamines, cyclic dipeptides, and rhamnolipids. Additional clusters were also annotated through molecular networking with cross-reference to previous publications. Tartrolon D cluster with analogues, turnercyclamycins A and B; teredinibactin A, dechloroteredinibactin, and two other possible teredinibactin analogues; and oxylipin (E)-11-oxooctadec-12-enoic acid were putatively identified as described. Molecular networking also revealed two additional metabolite clusters, annotated as lyso-ornithine lipids and polyethers. Manual fragmentation analysis corroborated the putative identification generated from GNPS. However, some of the clusters remained unclassified due to the limited structural information on marine natural products in the public database. The result of this study, nonetheless, showed the diversity in the chemical space occupied by shipworm endosymbionts. This study also affirms the use of bioinformatics, molecular networking, and fragmentation mechanisms analysis as tools for the dereplication of high-throughput data to aid the prioritization of strains for further analysis.
Full article
►▼
Show Figures
Open AccessArticle
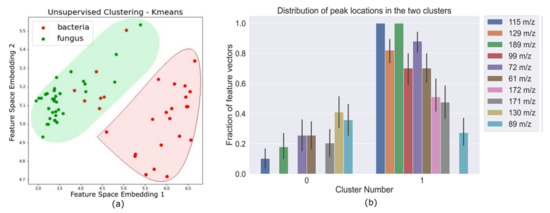
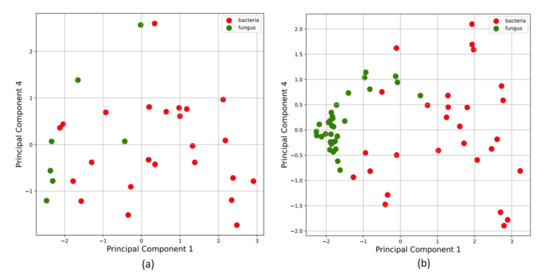
Machine Learning Approaches to Identify Discriminative Signatures of Volatile Organic Compounds (VOCs) from Bacteria and Fungi Using SPME-DART-MS
by
Mehak Arora, Stephen C. Zambrzycki, Joshua M. Levy, Annette Esper, Jennifer K. Frediani, Cassandra L. Quave, Facundo M. Fernández and Rishikesan Kamaleswaran
Cited by 25 | Viewed by 5437
Abstract
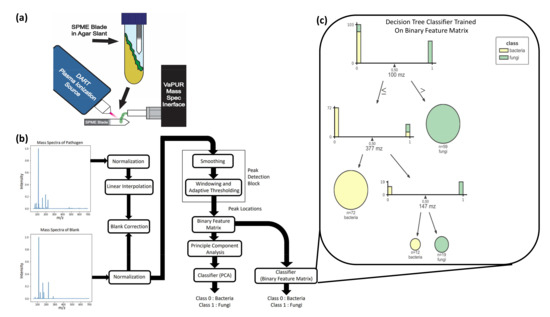
Point-of-care screening tools are essential to expedite patient care and decrease reliance on slow diagnostic tools (e.g., microbial cultures) to identify pathogens and their associated antibiotic resistance. Analysis of volatile organic compounds (VOC) emitted from biological media has seen increased attention in recent
[...] Read more.
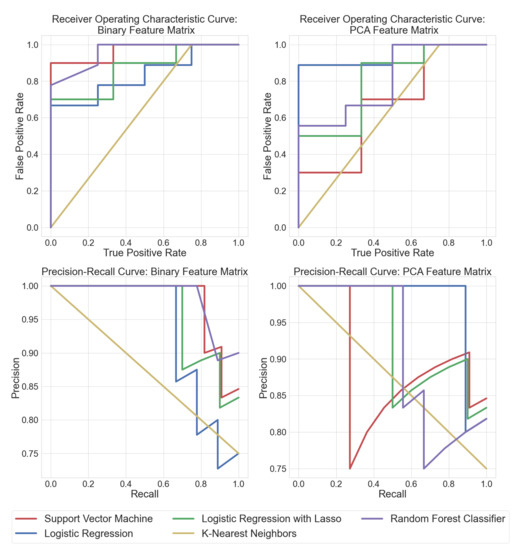
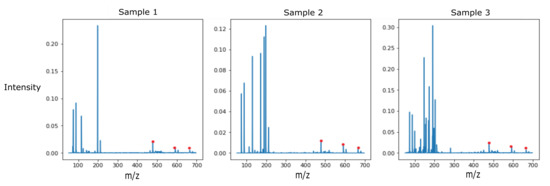
Point-of-care screening tools are essential to expedite patient care and decrease reliance on slow diagnostic tools (e.g., microbial cultures) to identify pathogens and their associated antibiotic resistance. Analysis of volatile organic compounds (VOC) emitted from biological media has seen increased attention in recent years as a potential non-invasive diagnostic procedure. This work explores the use of solid phase micro-extraction (SPME) and ambient plasma ionization mass spectrometry (MS) to rapidly acquire VOC signatures of bacteria and fungi. The MS spectrum of each pathogen goes through a preprocessing and feature extraction pipeline. Various supervised and unsupervised machine learning (ML) classification algorithms are trained and evaluated on the extracted feature set. These are able to classify the type of pathogen as bacteria or fungi with high accuracy, while marked progress is also made in identifying specific strains of bacteria. This study presents a new approach for the identification of pathogens from VOC signatures collected using SPME and ambient ionization MS by training classifiers on just a few samples of data. This ambient plasma ionization and ML approach is robust, rapid, precise, and can potentially be used as a non-invasive clinical diagnostic tool for point-of-care applications.
Full article
►▼
Show Figures
Open AccessArticle
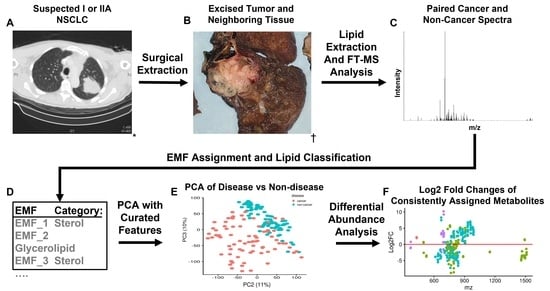
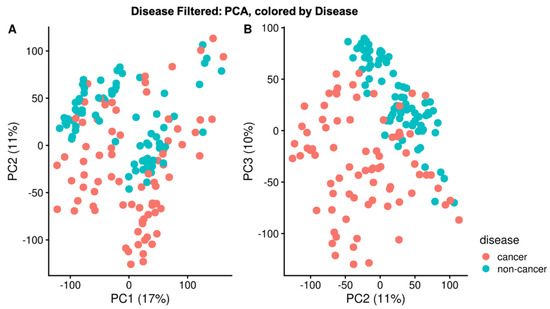
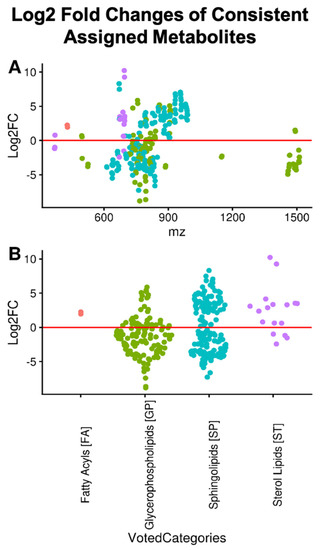
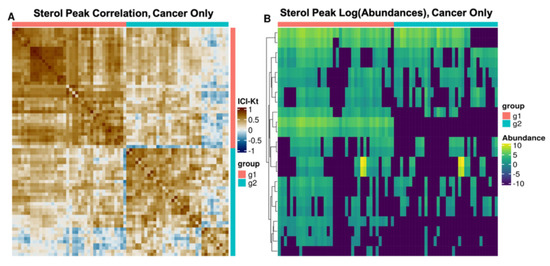
Untargeted Lipidomics of Non-Small Cell Lung Carcinoma Demonstrates Differentially Abundant Lipid Classes in Cancer vs. Non-Cancer Tissue
by
Joshua M. Mitchell, Robert M. Flight and Hunter N. B. Moseley
Cited by 14 | Viewed by 4701
Abstract
Lung cancer remains the leading cause of cancer death worldwide and non-small cell lung carcinoma (NSCLC) represents 85% of newly diagnosed lung cancers. In this study, we utilized our untargeted assignment tool Small Molecule Isotope Resolved Formula Enumerator (SMIRFE) and ultra-high-resolution Fourier transform
[...] Read more.
Lung cancer remains the leading cause of cancer death worldwide and non-small cell lung carcinoma (NSCLC) represents 85% of newly diagnosed lung cancers. In this study, we utilized our untargeted assignment tool Small Molecule Isotope Resolved Formula Enumerator (SMIRFE) and ultra-high-resolution Fourier transform mass spectrometry to examine lipid profile differences between paired cancerous and non-cancerous lung tissue samples from 86 patients with suspected stage I or IIA primary NSCLC. Correlation and co-occurrence analysis revealed significant lipid profile differences between cancer and non-cancer samples. Further analysis of machine-learned lipid categories for the differentially abundant molecular formulas identified a high abundance sterol, high abundance and high m/z sphingolipid, and low abundance glycerophospholipid metabolic phenotype across the NSCLC samples. At the class level, higher abundances of sterol esters and lower abundances of cardiolipins were observed suggesting altered stearoyl-CoA desaturase 1 (SCD1) or acetyl-CoA acetyltransferase (ACAT1) activity and altered human cardiolipin synthase 1 or lysocardiolipin acyltransferase activity respectively, the latter of which is known to confer apoptotic resistance. The presence of a shared metabolic phenotype across a variety of genetically distinct NSCLC subtypes suggests that this phenotype is necessary for NSCLC development and may result from multiple distinct genetic lesions. Thus, targeting the shared affected pathways may be beneficial for a variety of genetically distinct NSCLC subtypes.
Full article
►▼
Show Figures






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}