Dear Colleagues,
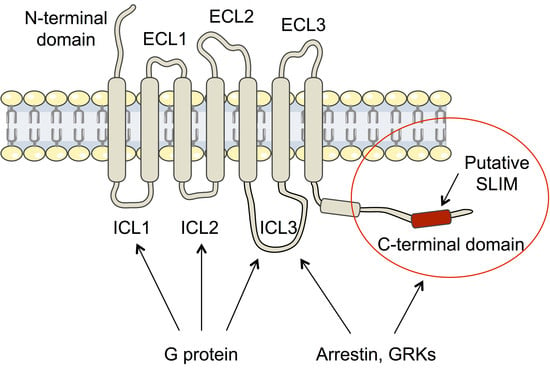
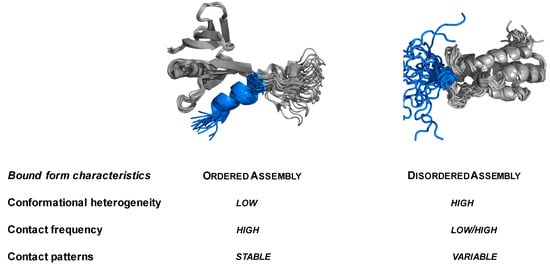
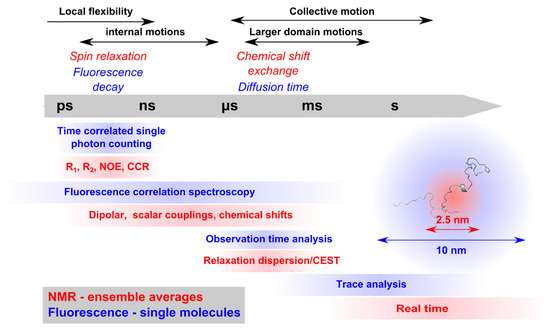
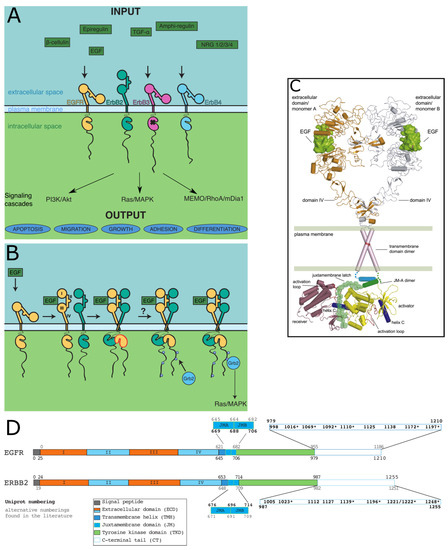
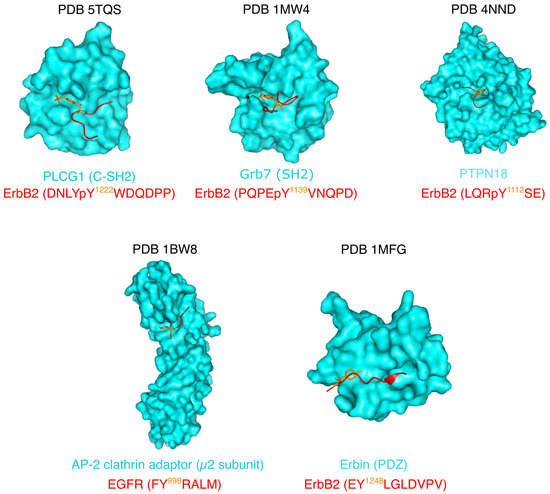
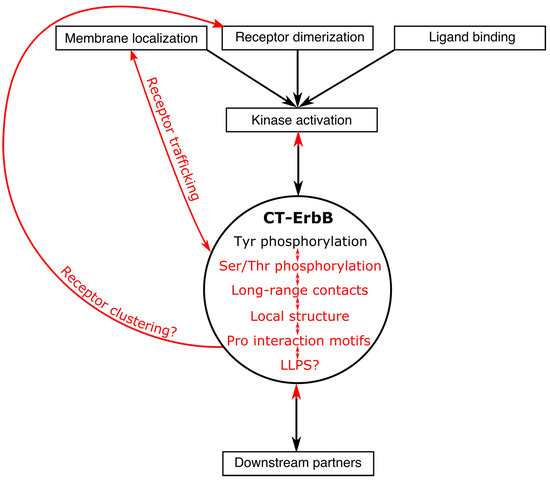
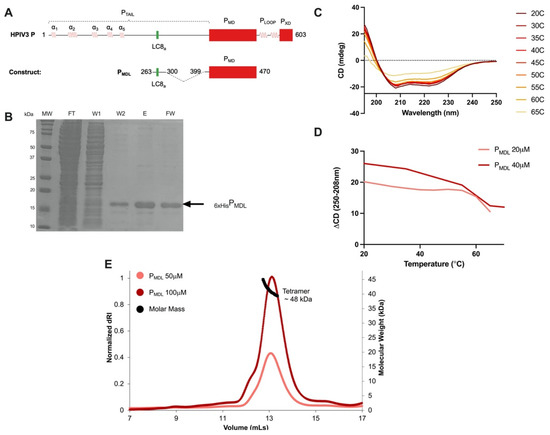
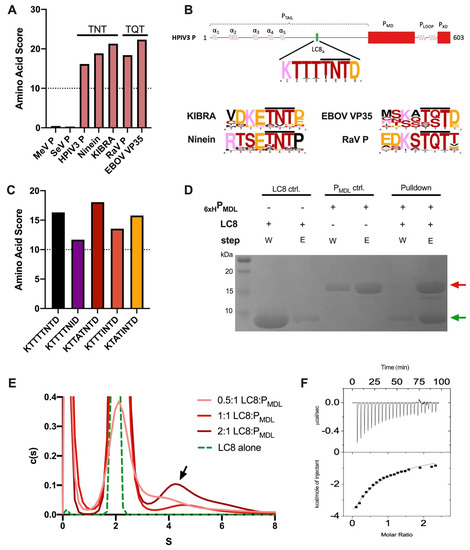
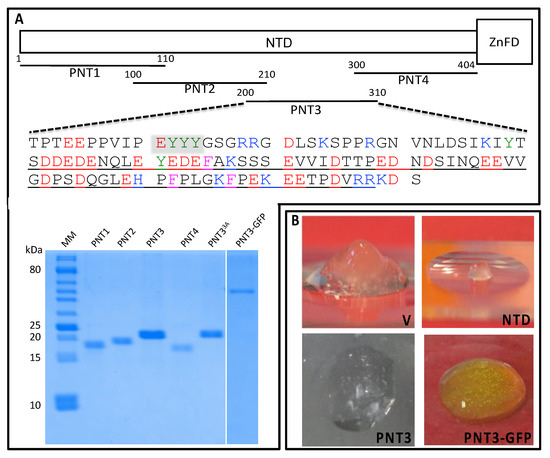
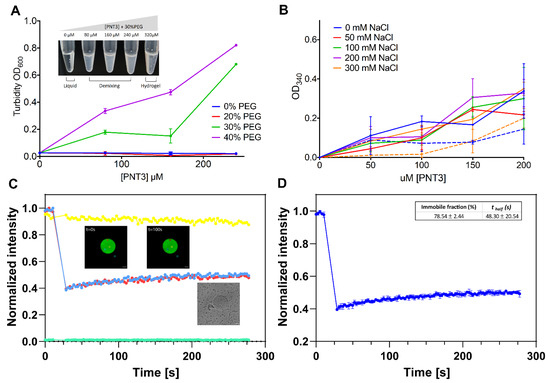
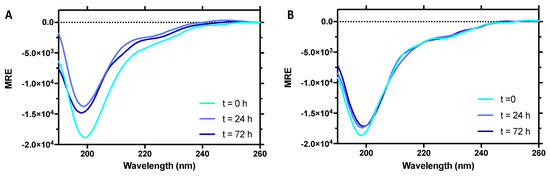
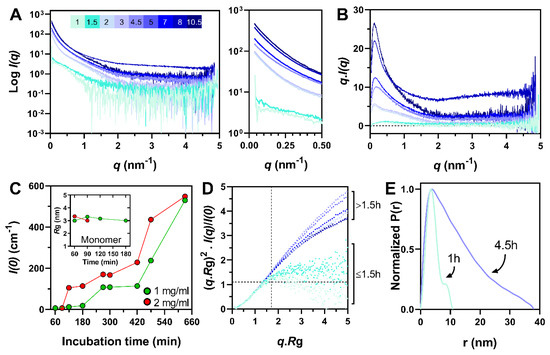
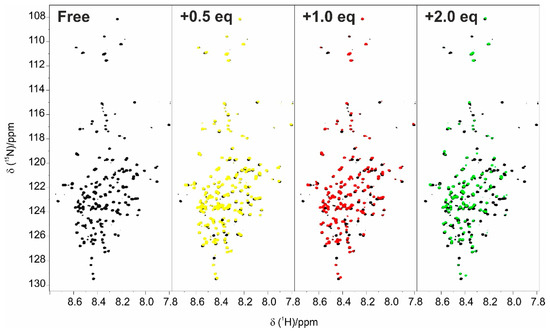
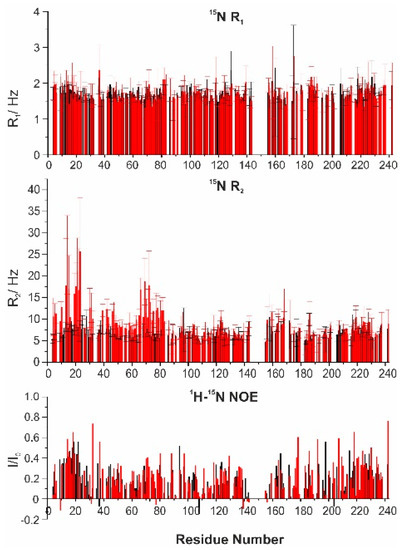
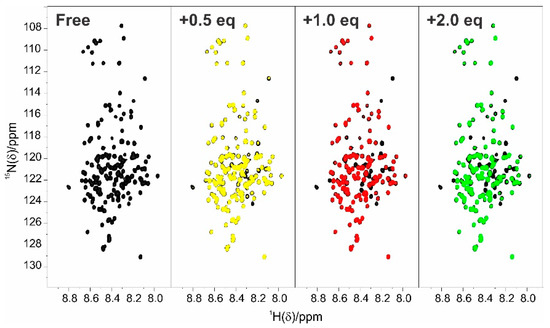
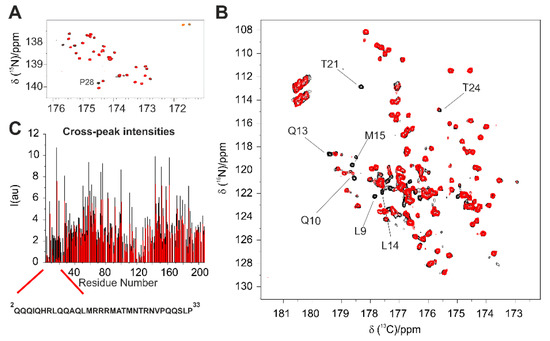
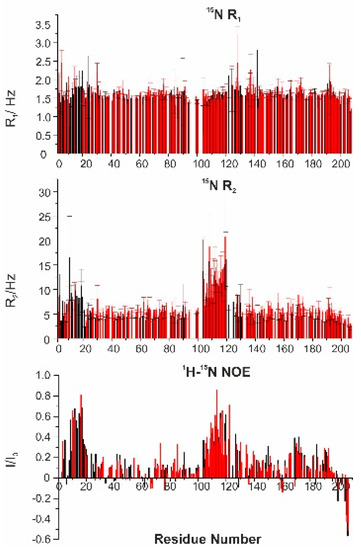
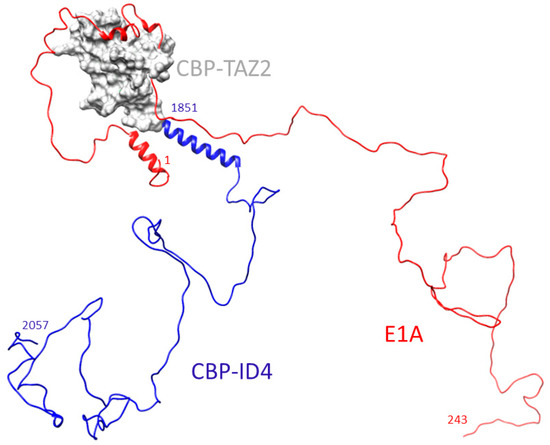
Intrinsically disordered proteins (IDPs) are fascinating multifaceted proteins. Their discovery dates back to the mid-1990s, and it is now well established that IDPs are very common in the protein realm. They are particularly abundant in the proteome of eukaryotes, with this prevalence being tied to the higher complexity of the latter compared to prokaryotes. The lack of stable secondary and tertiary structure that typifies these proteins and that is encoded by their amino acid sequence makes these proteins very elusive; thus, IDPs and their complexes are very difficult to characterize at the structural level due to their inherent flexibility. Although IDPs lack a fixed 3D structure, they can feature transiently populated secondary structure elements, which are often conserved and linked to function. In particular, these elements often serve as binding sites for partners and attenuate the entropic cost of the disorder-to-order transition triggered by binding to the partner. Many IDPs conserve a considerable extent of residual disorder upon binding to a partner, leading to so-called “fuzzy complexes”. By tuning the extent of preconfiguration and/or of fuzziness, IDPs can finely tune their affinity towards multiple partners. They can establish interactions ranging from low to high affinity, while conserving specificity in all cases. All these features are also modified by frequent post-translational modifications due to their high flexibility and accessibility. As such, they are the target of more than 300 PTMs that ultimately modulate their structural features and their ability to interact with partners. Thanks to these peculiar biophysical and biochemical features, IDPs are widely involved in signaling and regulation. Their action is also highly dependent on their expression level and local concentration in the cell. In the last five years, a growing number of studies has thus addressed the ability of IDPs to undergo liquid–liquid phase separation (LLPS), a phenomenon that underlies the formation of membrane-less organelles (MLOs). As a consequence, the deregulation of processes implying IDPs is at the origin of a multitude of diseases such as cancer, neurodegeneration, cardiovascular diseases, and diabetes. In order to better understand the mechanisms behind these diseases, it is necessary to decipher their structural bases. This Topical Collection on “Protein Intrinsic Disorder: Role in Signaling, Regulation and Membrane-Less Organelle Formation” aims at further deepening our understanding of how the physicochemical properties of IDPs enable them to play a crucial role in the regulation of many critical biological processes.
Dr. Nathalie Sibille
Dr. Sonia Longhi
Dr. Carine Van Heijenoort
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 250 words) can be sent to the Editorial Office for assessment.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Biomolecules is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}