
Molecular Determinants of Selectivity in Disordered Complexes May Shed Light on Specificity in Protein Condensates


Abstract
:1. Introduction
2. Results
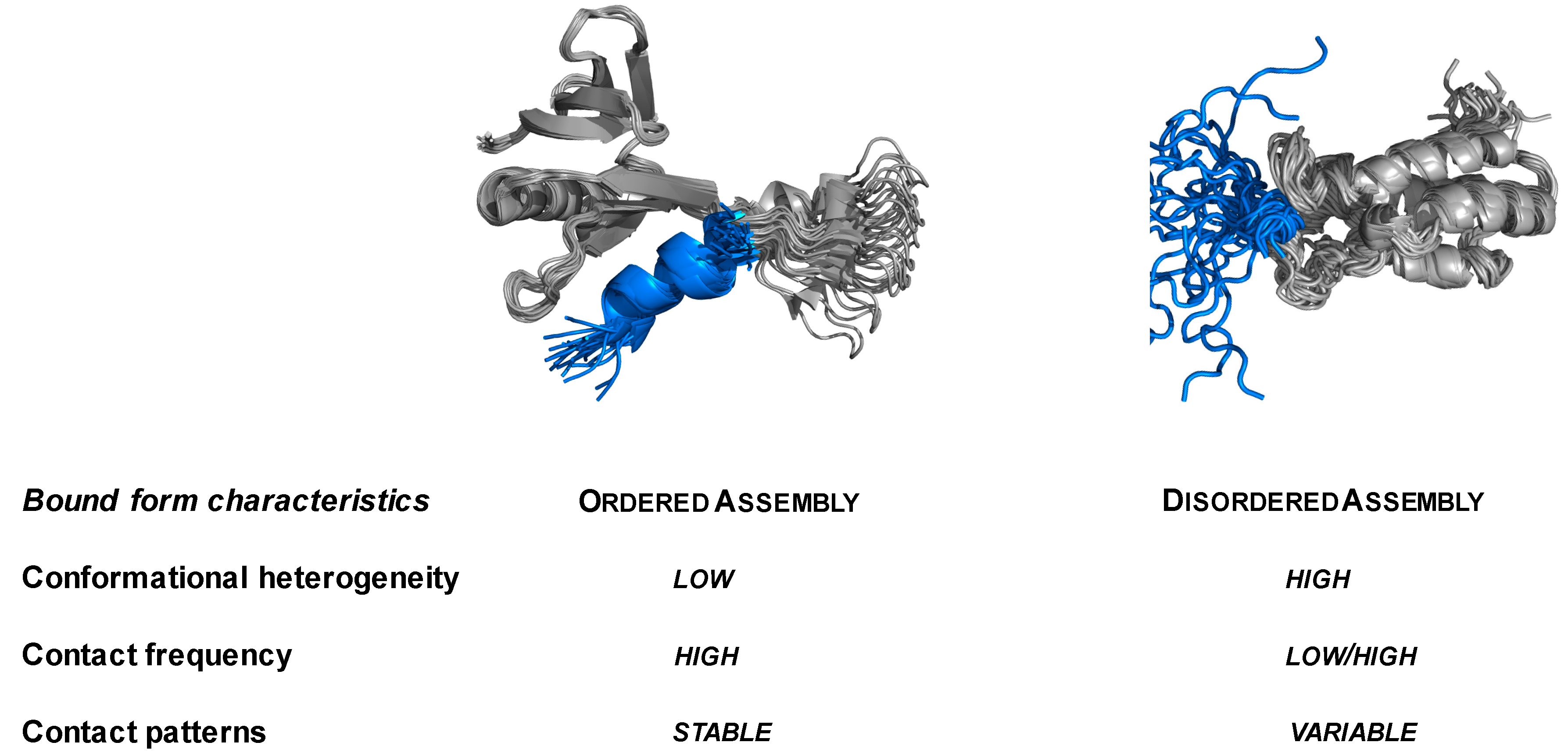
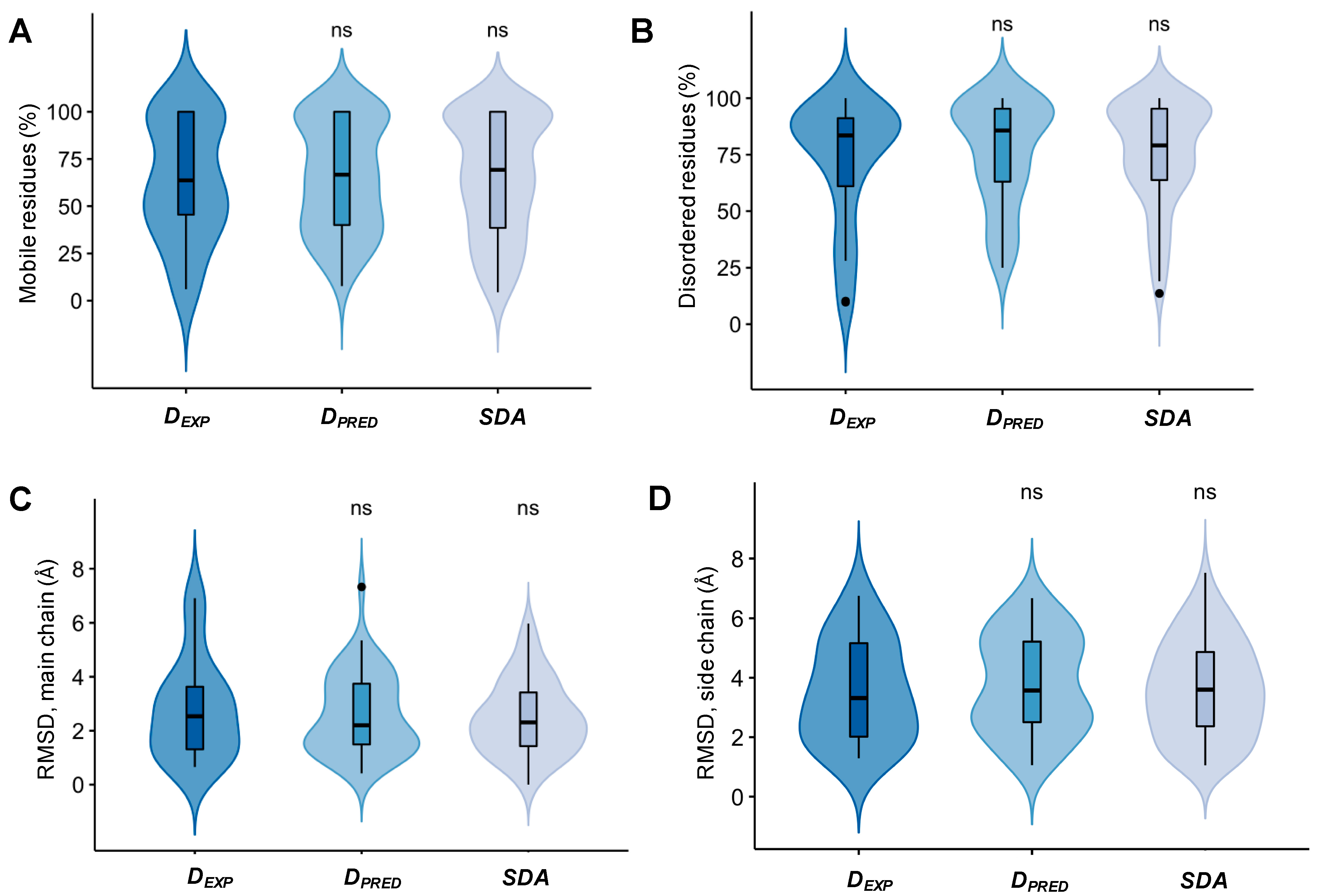
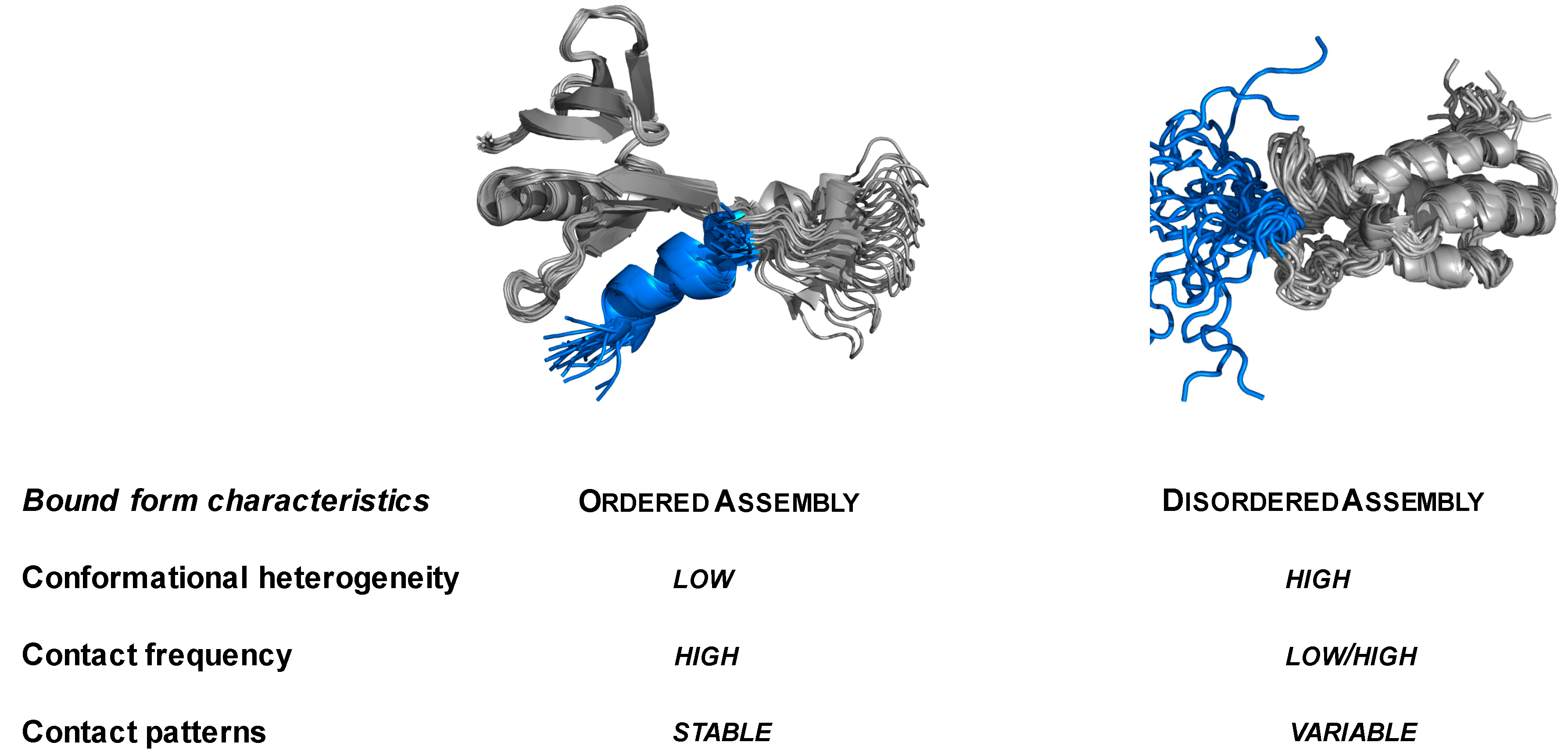
2.1. Specific Complexes of Droplet-Driver Proteins Are Conformationally Heterogeneous
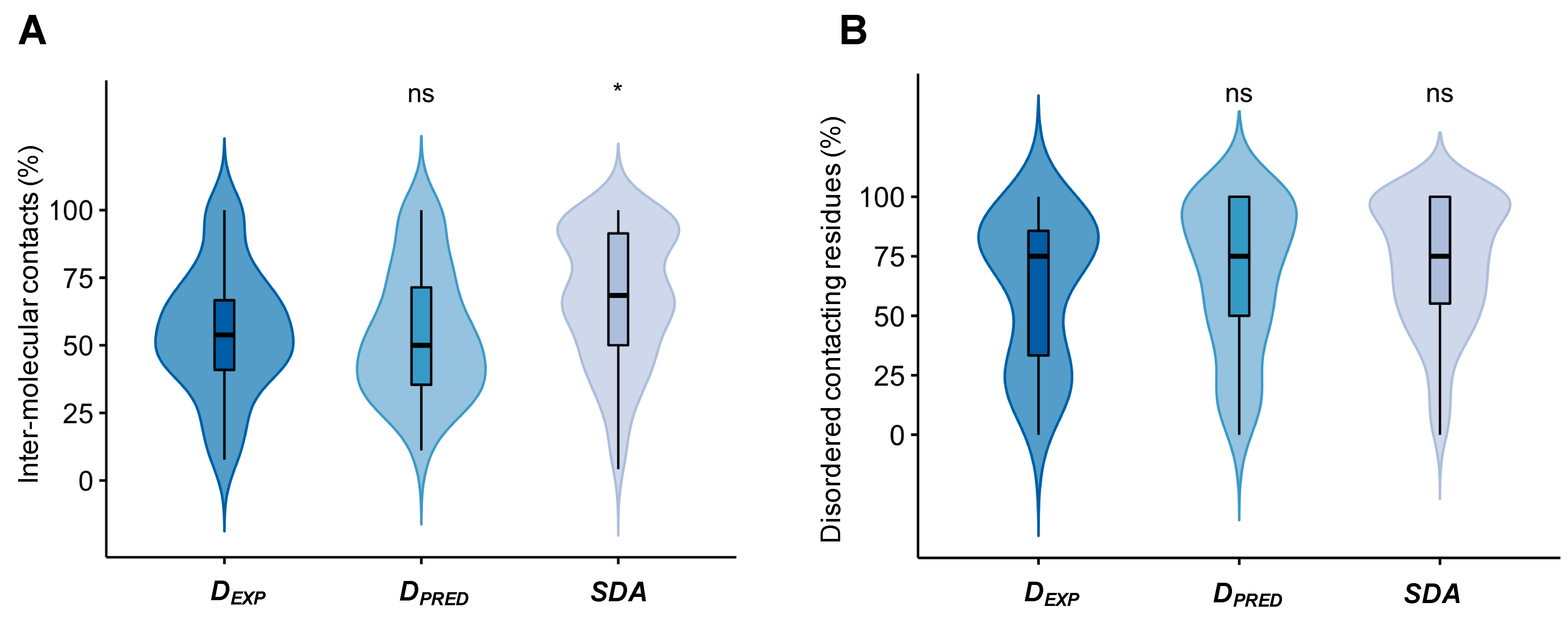
2.2. Droplet-Forming Proteins Sample Multiple Binding Configurations in Specific Assemblies
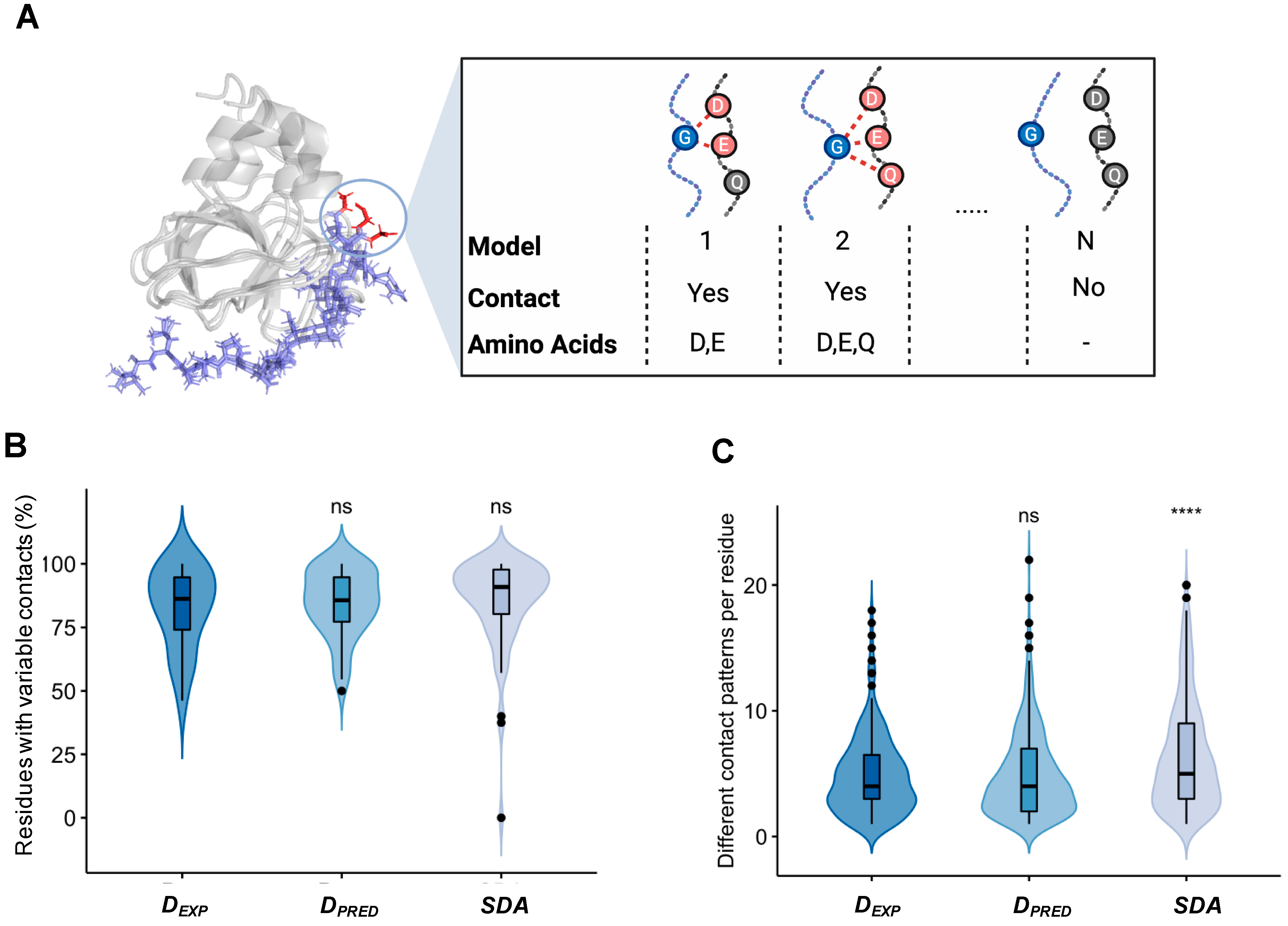
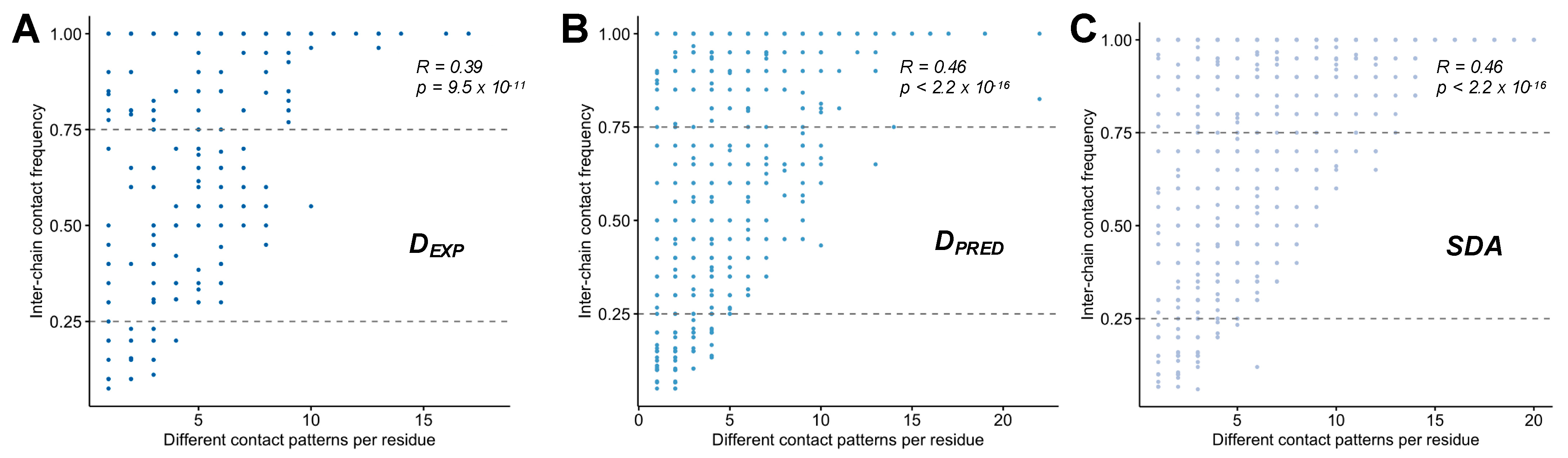
2.3. Different Contact Patterns Mediate Specific Interactions in Disordered Assemblies
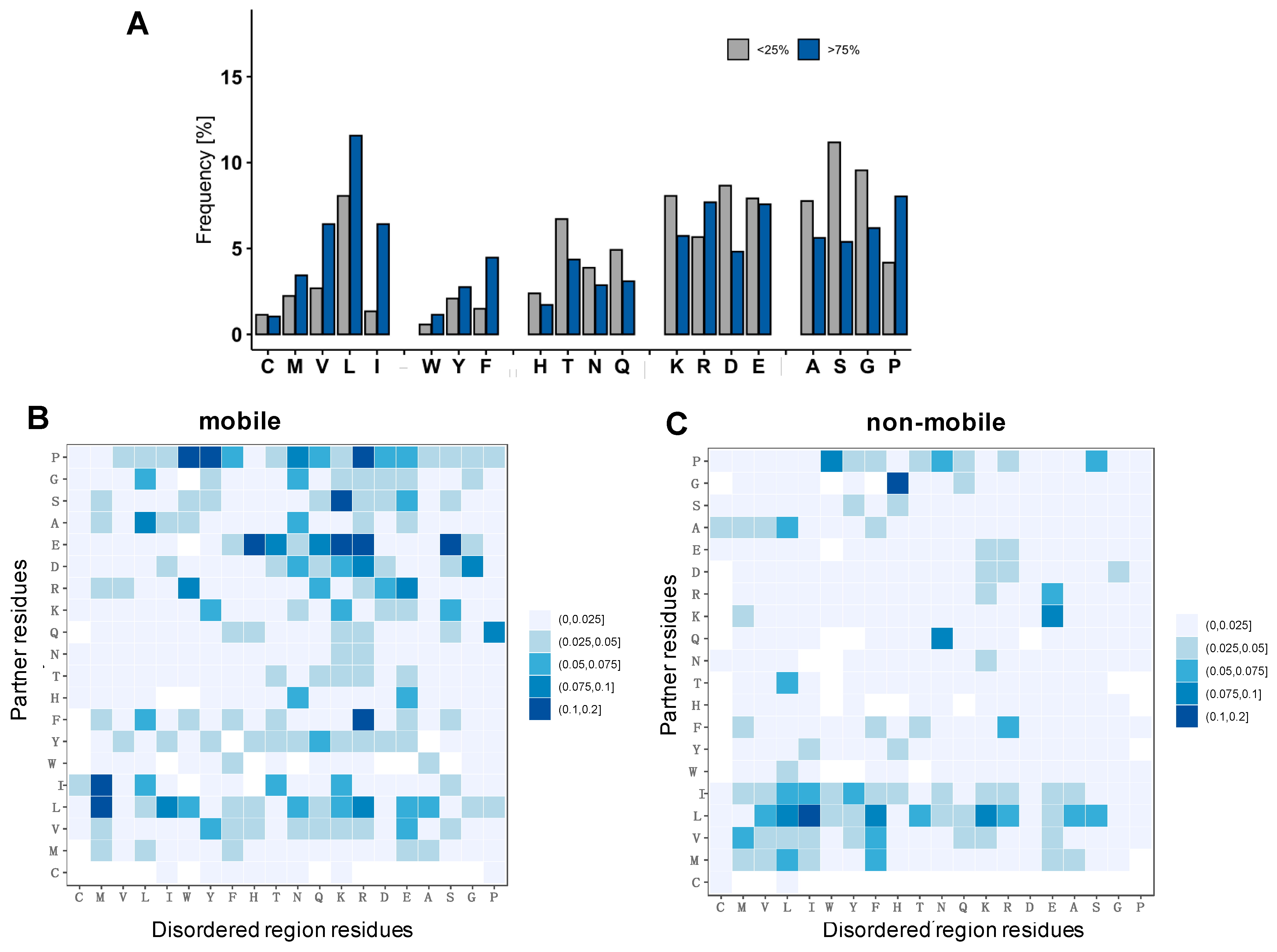
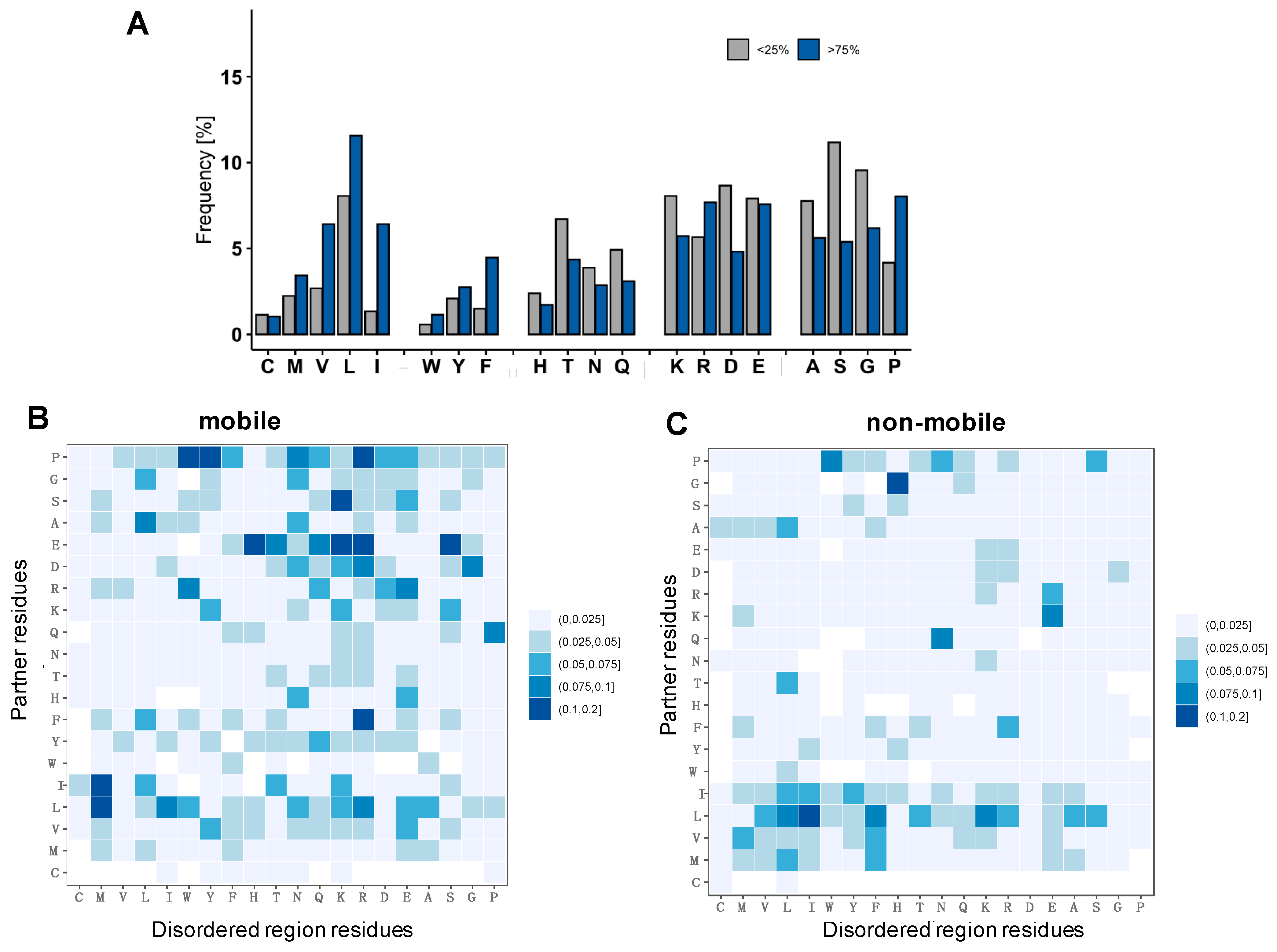
2.4. Specific Interactions Can Be Realized via a Wide Range of Sequence Motifs
3. Discussion and Conclusions
4. Methods
4.1. Dataset of Disordered Protein Complexes
4.2. Assessing Structural Heterogeneity in Conformational Ensembles
4.3. Characterization the Variability of Inter-Chain Contacts
4.4. Composition of the Interface
4.5. Frequency of Contacting Residues
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2020, 22, 215–235. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Vendruscolo, M. Generic nature of the condensed states of proteins. Nat. Cell Biol. 2021, 23, 587–594. [Google Scholar] [CrossRef]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2021, 22, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Dall’Agnese, A.; Young, R.A. Biomolecular Condensates in the Nucleus. Trends Biochem. Sci. 2020, 45, 961–977. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Dyakov, B.J.A.; Zhang, J.; Knight, J.D.R.; Vernon, R.M.; Forman-Kay, J.D.; Gingras, A.C. Properties of Stress Granule and P-Body Proteomes. Mol. Cell 2019, 76, 286–294. [Google Scholar] [CrossRef]
- Lin, Y.; Protter, D.S.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Tsang, B.; Vernon, R.M.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 2019, 365, 825–829. [Google Scholar] [CrossRef]
- Fuxreiter, M. Classifying the Binding Modes of Disordered Proteins. Int. J. Mol. Sci. 2020, 21, 8615. [Google Scholar] [CrossRef]
- Gianni, S.; Freiberger, M.I.; Jemth, P.; Ferreiro, D.U.; Wolynes, P.G.; Fuxreiter, M. Fuzziness and Frustration in the Energy Landscape of Protein Folding, Function, and Assembly. Acc. Chem. Res. 2021, 54, 1251–1259. [Google Scholar] [CrossRef]
- Hatos, A.; Monzon, A.M.; Tosatto, S.C.E.; Piovesan, D.; Fuxreiter, M. FuzDB: A new phase in understanding fuzzy interactions. Nucleic Acids Res. 2021, 50, D509–D517. [Google Scholar] [CrossRef]
- Miskei, M.; Horvath, A.; Vendruscolo, M.; Fuxreiter, M. Sequence-Based Prediction of Fuzzy Protein Interactions. J. Mol. Biol. 2020, 432, 2289–2303. [Google Scholar] [CrossRef]
- Horvath, A.; Miskei, M.; Ambrus, V.; Vendruscolo, M.; Fuxreiter, M. Sequence-based prediction of protein binding mode landscapes. PLoS Comp. Biol. 2020, 16, e1007864. [Google Scholar] [CrossRef] [PubMed]
- Di Lello, P.; Jenkins, L.M.M.; Jones, T.N.; Nguyen, B.D.; Hara, T.; Yamaguchi, H.; Dikeakos, J.D.; Appella, E.; Legault, P.; Omichinski, J.G. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell. 2006, 22, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell. 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Heikaus, C.C.; Kisselev, L.; Vernon, R.; Herbig, E.; Pacheco, D.; Warfield, L.; Littlefield, P.; Baker, D.; Klevit, R.E.; et al. The acidic transcription activator Gcn4 binds the mediator subunit Gal11/Med15 using a simple protein interface forming a fuzzy complex. Mol. Cell 2011, 44, 942–953. [Google Scholar] [CrossRef] [Green Version]
- Warfield, L.; Tuttle, L.M.; Pacheco, D.; Klevit, R.E.; Hahn, S. A sequence-specific transcription activator motif and powerful synthetic variants that bind Mediator using a fuzzy protein interface. Proc. Natl. Acad. Sci. USA 2014, 111, E3506–E3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, K.; Huang, Q.; Yu, C.; Shen, B.; Sevilla, C.; Shi, M.; Hermjakob, H.; Chen, Y.; Li, T. PhaSepDB: A database of liquid-liquid phase separation related proteins. Nucleic Acids Res. 2020, 48, D354–D359. [Google Scholar] [CrossRef] [PubMed]
- Delaforge, E.; Kragelj, J.; Tengo, L.; Palencia, A.; Milles, S.; Bouvignies, G.; Salvi, N.; Blackledge, M.; Jensen, M.R. Deciphering the Dynamic Interaction Profile of an Intrinsically Disordered Protein by NMR Exchange Spectroscopy. J. Am. Chem. Soc. 2018, 140, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Tosatto, S.C.E. Mobi 2.0: An improved method to define intrinsic disorder, mobility and linear binding regions in protein structures. Bioinformatics 2018, 34, 122–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Vernon, R.M.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife 2018, 7, e31486. [Google Scholar] [CrossRef] [PubMed]
- Hatos, A.; Hajdu-Soltesz, B.; Monzon, A.M.; Palopoli, N.; Alvarez, L.; Aykac-Fas, B.; Bassot, C.; Benitez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef] [Green Version]
- Jalihal, A.P.; Pitchiaya, S.; Xiao, L.; Bawa, P.; Jiang, X.; Bedi, K.; Parolia, A.; Cieslik, M.; Ljungman, M.; Chinnaiyan, A.M.; et al. Multivalent Proteins Rapidly and Reversibly Phase-Separate upon Osmotic Cell Volume Change. Mol. Cell 2020, 79, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.C.; Brangwynne, C.P. Inverse size scaling of the nucleolus by a concentration-dependent phase transition. Curr. Biol. 2015, 25, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.; Yuan, H.; Liang, F.X.; Yang, Y.M.; Westley, J.; Petzold, C.; Dancel-Manning, K.; Deng, Y.; Sall, J.; Sehgal, P.B. Human Antiviral Protein MxA Forms Novel Metastable Membraneless Cytoplasmic Condensates Exhibiting Rapid Reversible Tonicity-Driven Phase Transitions. J. Virol. 2019, 93, e01014-19. [Google Scholar] [CrossRef]
- Wu, H.; Fuxreiter, M. The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 2016, 165, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Jain, A. Osmotic Stress Triggers Phase Separation. Mol. Cell 2020, 79, 876–877. [Google Scholar] [CrossRef]
- Freiberger, M.I.; Wolynes, P.G.; Ferreiro, D.U.; Fuxreiter, M. Frustration in Fuzzy Protein Complexes Leads to Interaction Versatility. J. Phys. Chem. B 2021, 125, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, D.; Minervini, G.; Tosatto, S.C. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016, 44, W367–W374. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datase | Main-Chain RMSD (Å) | Side-Chain RMSD (Å) | ||
|---|---|---|---|---|
| fC ≤ 0.25 | fC ≥ 0.75 | fC ≤ 0.25 | fC ≥ 0.75 | |
| DEXP | 2.71 | 1.93 | 3.52 | 2.91 |
| DPRED | 2.88 | 1.77 | 4.77 | 2.65 |
| SDA | 3.10 | 1.81 | 4.54 | 2.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monzon, A.M.; Piovesan, D.; Fuxreiter, M. Molecular Determinants of Selectivity in Disordered Complexes May Shed Light on Specificity in Protein Condensates. Biomolecules 2022, 12, 92. https://doi.org/10.3390/biom12010092
Monzon AM, Piovesan D, Fuxreiter M. Molecular Determinants of Selectivity in Disordered Complexes May Shed Light on Specificity in Protein Condensates. Biomolecules. 2022; 12(1):92. https://doi.org/10.3390/biom12010092
Chicago/Turabian StyleMonzon, Alexander Miguel, Damiano Piovesan, and Monika Fuxreiter. 2022. "Molecular Determinants of Selectivity in Disordered Complexes May Shed Light on Specificity in Protein Condensates" Biomolecules 12, no. 1: 92. https://doi.org/10.3390/biom12010092
APA StyleMonzon, A. M., Piovesan, D., & Fuxreiter, M. (2022). Molecular Determinants of Selectivity in Disordered Complexes May Shed Light on Specificity in Protein Condensates. Biomolecules, 12(1), 92. https://doi.org/10.3390/biom12010092