Feature Papers in 'Biomacromolecules: Proteins'
Share This Topical Collection
Editor
 Dr. Adrián Velázquez Campoy
Dr. Adrián Velázquez Campoy
Dr. Adrián Velázquez Campoy
E-Mail
Website1
Website2
Collection Editor
1. Institute of Biocomputation and Physics of Complex Systems (BIFI), Universidad de Zaragoza, 50018 Zaragoza, Spain
2. Departamento de Bioquímica y Biología Molecular y Celular, Universidad de Zaragoza, 50009 Zaragoza, Spain
Interests: protein biophysics; protein interactions; protein stability and folding; cooperativity and allostery; biological calorimetry; drug discovery
Special Issues, Collections and Topics in MDPI journals
Topical Collection Information
Dear Colleagues,
This Topical Collection, “Feature Papers in Biomacromolecules: Proteins”, aims to collect high-quality research articles and comprehensive reviews on all aspects of proteins. It is dedicated to recent advances in the research area of proteins, and comprises a selection of exclusive papers from the Editorial Board Members (EBMs) of the Proteins Section, as well as invited papers from relevant experts. We also welcome senior experts in the field to make contributions to this Topical Collection. We aim to represent our Section as an attractive open-access publishing platform for biochemistry research.
Dr. Adrián Velázquez Campoy
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 250 words) can be sent to the Editorial Office for assessment.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Biomolecules is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Published Papers (13 papers)
Open AccessArticle
Non-Vesicular Extracellular Particle (NVEP) Proteomes from Diverse Biological Sources Reveal Specific Marker Composition with Varying Enrichment Levels
by
Wasifa Naushad, Bryson C. Okeoma, Carlos Gartner, Yulica Santos-Ortega, Calvin P. H. Vary, Lakmini S. Premadasa, Alessio Noghero, Jack T. Stapleton, Ionita C. Ghiran, Mahesh Mohan and Chioma M. Okeoma
Viewed by 1404
Abstract
Extracellular particles (EPs), an umbrella term encompassing membrane-enclosed extracellular vesicles (EVs) and non-vesicular extracellular particles ([NVEPs], previously described as extracellular condensates [ECs]) contain a complex cargo of biomolecules, including DNA, RNA, proteins, and lipids, reflecting the physiological state of their cell of origin.
[...] Read more.
Extracellular particles (EPs), an umbrella term encompassing membrane-enclosed extracellular vesicles (EVs) and non-vesicular extracellular particles ([NVEPs], previously described as extracellular condensates [ECs]) contain a complex cargo of biomolecules, including DNA, RNA, proteins, and lipids, reflecting the physiological state of their cell of origin. Identifying proteins associated with EPs that regulate host responses to physiological and pathophysiological processes is of critical importance. Here, we report the findings of our study to gain insight into the proteins associated with NVEPs. We used samples from human semen, the rat brain, and the rhesus macaque (RM) brain and blood to assess the physical properties and proteome profiles of NVEPs from these specimens. The results show significant differences in the zeta potential, concentration, and size of NVEPs across different species. We identified 938, 51, and 509 total proteins from NVEPs isolated from rat brain tissues, RM blood, and human seminal plasma, respectively. The species-specific protein networks show distinct biological themes, while the species-conserved protein interactome was identified with six proteins (ALB, CST3, FIBA/FGA, GSTP1, PLMN/PLG, PPIA) associated with NVEPs in all samples. The six NVEP-associated proteins are prone to aggregation and formation of wide, insoluble, unbranched filaments with a cross-beta sheet quaternary structure, such as amyloid fibrils. Protein-to-function analysis indicates that the six identified proteins are linked to the release of dopamine, immune-mediated inflammatory disease, replication of RNA viruses, HIV-HCV co-infection, and inflammation. These interesting findings have created an opportunity to evaluate NVEPs for their potential use as biomarkers of health and disease. Additional in-depth studies are needed to clarify when and how these proteins sustain their physiological role or transition to pathogenic roles.
Full article
►▼
Show Figures
Open AccessArticle
Anionic Lipid Catalyzes the Generation of Cytotoxic Insulin Oligomers
by
Jhinuk Saha, Audrey Wolszczak, Navneet Kaur, Malitha C. Dickwella Widanage, Samuel D. McCalpin, Riqiang Fu, Jamel Ali and Ayyalusamy Ramamoorthy
Cited by 1 | Viewed by 1207
Abstract
The misfolding and aggregation of proteins into amyloidogenic assemblies are key features of several metabolic and neurodegenerative diseases. Human insulin has long been known to form amyloid fibrils under various conditions, which affects its bioavailability and function. Clinically, insulin aggregation at recurrent injection
[...] Read more.
The misfolding and aggregation of proteins into amyloidogenic assemblies are key features of several metabolic and neurodegenerative diseases. Human insulin has long been known to form amyloid fibrils under various conditions, which affects its bioavailability and function. Clinically, insulin aggregation at recurrent injection sites poses a challenge for diabetic patients who rely on insulin therapy. Furthermore, decreased responsiveness to insulin in type 2 diabetic (T2D) patients may lead to its overproduction and accumulation as aggregates. Earlier reports have reported that various factors such as pH, temperature, agitation, and the presence of lipids or other proteins influence insulin aggregation. Our present study aims to elucidate the effects of non–micellar anionic DMPG (1,2–dimyristoyl–sn–glycero–3–phosphoglycerol) lipids on insulin aggregation. Distinct pathways of insulin aggregation and intermediate formation were observed in the presence of DMPG using a ThT fluorescence assay. The formation of soluble intermediates alongside large insulin fibrils was observed in insulin incubated with DMPG via TEM, DLS, and NMR as opposed to insulin aggregates generated without lipids.
13C magic angle spinning solid–state NMR and FTIR experiments indicated that lipids do not alter the conformation of insulin fibrils but do alter the time scale of motion of aromatic and aliphatic side chains. Furthermore, the soluble intermediates were found to be more cytotoxic than fibrils generated with or without lipids. Overall, our study elucidates the importance of anionic lipids in dictating the pathways and intermediates associated with insulin aggregation. These findings could be useful in determining various approaches to avoid toxicity and enhance the effectiveness of insulin in therapeutic applications.
Full article
►▼
Show Figures
Open AccessArticle
Proteomic Profiling of Inflammatory Protein Dysregulation in HLA-B27-Positive Ankylosing Spondylitis: Molecular Signatures and Potential Biomarkers
by
Yuzhu Yan, Jihan Wang, Yangyang Wang, Junye Liu, Wenjuan Yang, Min Niu, Yan Yu and Heping Zhao
Cited by 2 | Viewed by 1989
Abstract
This study explored the proteomic landscape of inflammatory protein dysregulation in ankylosing spondylitis (AS), a chronic inflammatory disorder primarily affecting the axial skeleton and strongly associated with the HLA-B27 allele, particularly the HLA-B2705 and HLA-B2704 subtypes prevalent in Chinese populations. Blood samples from
[...] Read more.
This study explored the proteomic landscape of inflammatory protein dysregulation in ankylosing spondylitis (AS), a chronic inflammatory disorder primarily affecting the axial skeleton and strongly associated with the HLA-B27 allele, particularly the HLA-B2705 and HLA-B2704 subtypes prevalent in Chinese populations. Blood samples from HLA-B27-positive AS patients and normal controls (NC) were analyzed using the Olink Target 96 inflammation panel to profile 92 inflammatory proteins. HLA-B27 subtyping was performed via PCR-SSP. To identify key proteins and stratify AS subtypes, we employed machine learning classifiers, including LightGBM models coupled with SHAP value interpretation, alongside traditional statistical analyses. The proteomic analysis revealed significant dysregulation of pro-inflammatory cytokines, such as IL-6 and IL-17A, in AS patients compared to NC, with CXCL9 and NRTN identified as potential biomarkers associated with disease activity. The combination of LightGBM classifiers and traditional statistical methods demonstrated high accuracy in distinguishing AS from NC and effectively stratifying subtypes. These findings provide valuable insights into the inflammatory mechanisms underlying AS pathogenesis and highlight potential biomarkers and therapeutic targets for improving diagnosis and treatment strategies. Future studies with larger and more diverse cohorts, as well as longitudinal designs, are warranted to validate these biomarkers and elucidate their dynamic changes during disease progression.
Full article
►▼
Show Figures
Open AccessReview
A Potential Role of EFR3A in Human Disease States
by
Karolina Marek-Bukowiec, Magdalena Trybus, Anita Hryniewicz-Jankowska, Aleksander Czogalla and Aleksander F. Sikorski
Viewed by 1769
Abstract
EFR3A is a conserved peripheral membrane protein required for the plasma membrane localization of the phosphatidylinositol-4 kinase (PI4KIIIα/PI4KA) complex and for regulating the responsiveness of G-protein-coupled receptors. Additionally, it was implicated in several other potentially unrelated physiological functions. In metazoan organisms,
EFR3A is
[...] Read more.
EFR3A is a conserved peripheral membrane protein required for the plasma membrane localization of the phosphatidylinositol-4 kinase (PI4KIIIα/PI4KA) complex and for regulating the responsiveness of G-protein-coupled receptors. Additionally, it was implicated in several other potentially unrelated physiological functions. In metazoan organisms,
EFR3A is ubiquitously co-expressed with its paralog
EFR3B which shares similar biological roles. This brief review summarizes the current knowledge regarding the potential roles of EFR3A in human disease states, including neurological and cardiovascular disorders, as well as various neoplasia-based diseases.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceArticle
Tau Oligomers Resist Phase Separation
by
Lathan Lucas, Phoebe S. Tsoi, Josephine C. Ferreon and Allan Chris M. Ferreon
Cited by 3 | Viewed by 2818
Abstract
Tau is a microtubule-associated protein that undergoes liquid–liquid phase separation (LLPS) to form condensates under physiological conditions, facilitating microtubule stabilization and intracellular transport. LLPS has also been implicated in pathological Tau aggregation, which contributes to tauopathies such as Alzheimer’s disease. While LLPS is
[...] Read more.
Tau is a microtubule-associated protein that undergoes liquid–liquid phase separation (LLPS) to form condensates under physiological conditions, facilitating microtubule stabilization and intracellular transport. LLPS has also been implicated in pathological Tau aggregation, which contributes to tauopathies such as Alzheimer’s disease. While LLPS is known to promote Tau aggregation, the relationship between Tau’s structural states and its phase separation behavior remains poorly defined. Here, we examine how oligomerization modulates Tau LLPS and uncover key distinctions between monomeric, oligomeric, and amyloidogenic Tau species. Using dynamic light scattering and fluorescence microscopy, we monitored oligomer formation over time and assessed oligomeric Tau’s ability to undergo LLPS. We found that Tau monomers readily phase separate and form condensates. As oligomerization progresses, Tau’s propensity to undergo LLPS diminishes, with oligomers still being able to phase separate, albeit with reduced efficiency. Interestingly, oligomeric Tau is recruited into condensates formed with 0-day-aged Tau, with this recruitment depending on the oligomer state of maturation. Early-stage, Thioflavin T (ThT)-negative oligomers co-localize with 0-day-aged Tau condensates, whereas ThT-positive oligomers resist condensate recruitment entirely. This study highlights a dynamic interplay between Tau LLPS and aggregation, providing insight into how Tau’s structural and oligomeric states influence its pathological and functional roles. These findings underscore the need to further explore LLPS as a likely modulator of Tau pathogenesis and distinct pathogenic oligomers as viable therapeutic targets in tauopathies.
Full article
►▼
Show Figures
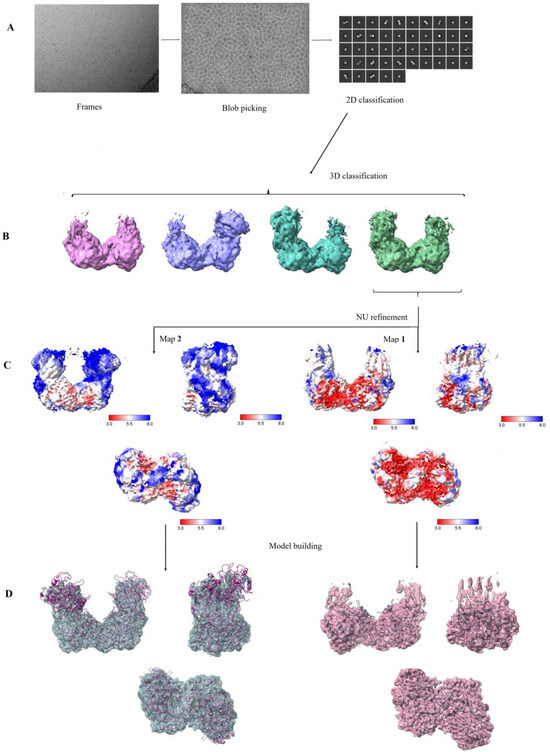
Open AccessArticle
A Bifunctional Phosphoglucomutase/Phosphomannomutase from Thermococcus kodakarensis: Biophysical Analysis and Cryo-EM Structure
by
Zahra Naz, Ishan Rathore, Muhammad Saleem, Moazur Rahman, Alexander Wlodawer and Naeem Rashid
Cited by 2 | Viewed by 2013
Abstract
Phosphoglucomutase (EC 5.4.2.2., PGM), a key enzyme of glycogenolysis and glycogenesis, catalyzes the interconversion of glucose 1-phosphate and glucose 6-phosphate, whereas phosphomannomutase (EC 5.4.2.8., PMM) transfers the phosphate group from the 1′ to the 6′, or from the 6′ to the 1′ position
[...] Read more.
Phosphoglucomutase (EC 5.4.2.2., PGM), a key enzyme of glycogenolysis and glycogenesis, catalyzes the interconversion of glucose 1-phosphate and glucose 6-phosphate, whereas phosphomannomutase (EC 5.4.2.8., PMM) transfers the phosphate group from the 1′ to the 6′, or from the 6′ to the 1′ position in mannose phosphate. However, in the hyperthermophilic archaeon
Thermococcus kodakarensis, a single gene,
Tk1108, encodes a protein with both PGM and PMM activities. Here, we report biophysical analysis and the 2.45 Å resolution cryo-EM structure of this novel enzyme. Our results demonstrate a specific arrangement of the four subunits in the quaternary structure, displaying a distinct catalytic cleft required for the bifunctional activity at extremely high temperatures. To the best of our knowledge, this is the first biophysical characterization and cryo-EM structure elucidation of a thermostable, bifunctional PGM/PMM.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceArticle
Dissecting Cytophagalysin: Structural and Biochemical Studies of a Bacterial Pappalysin-Family Metallopeptidase
by
Eva Estevan-Morió, Juan Sebastián Ramírez-Larrota, Enkela Bushi and Ulrich Eckhard
Cited by 2 | Viewed by 1825
Abstract
Cytophaga is a genus of Gram-negative bacteria occurring in soil and the gut microbiome. It is closely related to pathogenic
Flavobacterium spp. that cause severe diseases in fish. Cytophaga strain L43-1 secretes cytophagalysin (CPL1), a 137 kDa peptidase with reported collagenolytic and gelatinolytic
[...] Read more.
Cytophaga is a genus of Gram-negative bacteria occurring in soil and the gut microbiome. It is closely related to pathogenic
Flavobacterium spp. that cause severe diseases in fish. Cytophaga strain L43-1 secretes cytophagalysin (CPL1), a 137 kDa peptidase with reported collagenolytic and gelatinolytic activity. We performed highly-confident structure prediction calculations for CPL1, which identified 11 segments and domains, including a signal peptide for secretion, a prosegment (PS) for latency, a metallopeptidase (MP)-like catalytic domain (CD), and eight immunoglobulin (Ig)-like domains (D3–D10). In addition, two short linkers were found at the D8–D9 and D9–D10 junctions, and the structure would be crosslinked by four disulfide bonds. The CPL1 CD was found closest to ulilysin from
Methanosarcina acetivorans, which assigns CPL1 to the lower-pappalysin family within the metzincin clan of MPs. Based on the structure predictions, we aimed to produce constructs spanning the full-length enzyme, as well as PS+CD, PS+CD+D3, and PS+CD+D3+D4. However, we were successful only with the latter three constructs. We could activate recombinant CPL1 by PS removal employing trypsin, and found that both zymogen and mature CPL1 were active in gelatin zymography and against a fluorogenic gelatin variant. This activity was ablated in a mutant, in which the catalytic glutamate described for lower pappalyins and other metzincins was replaced by alanine, and by a broad-spectrum metal chelator. Overall, these results proved that our recombinant CPL1 is a functional active MP, thus supporting the conclusions derived from the structure predictions.
Full article
►▼
Show Figures
Open AccessArticle
Mutations of Key Functional Residues in CRM1/XPO1 Differently Alter Its Intranuclear Localization and the Nuclear Export of Endogenous Cargos
by
Miren Josu Omaetxebarria, Maria Sendino, Liher Arrizabalaga, Irune Mota, Ana Maria Zubiaga and José Antonio Rodríguez
Viewed by 2383
Abstract
CRM1 (XPO1) has been well-characterized as a shuttling receptor that mediates the export of protein and RNA cargos to the cytoplasm, and previous analyses have pinpointed several key residues (A541, F572, K568, S1055, and Q742) that modulate CRM1 export activity. CRM1 also has
[...] Read more.
CRM1 (XPO1) has been well-characterized as a shuttling receptor that mediates the export of protein and RNA cargos to the cytoplasm, and previous analyses have pinpointed several key residues (A541, F572, K568, S1055, and Q742) that modulate CRM1 export activity. CRM1 also has a less studied nuclear function in RNA biogenesis, which is reflected by its localization to the Cajal body and the nucleolus. Here, we have investigated how the mutation of these key residues affects the intranuclear localization of CRM1 and its ability to mediate export of endogenous cargos. We identify A541K as a separation-of-function mutant that reveals the independent nature of the Cajal body and nucleolar localizations of CRM1. We also show that the F572A mutation may have strikingly opposite effects on the export of specific cargos. Importantly, and in contrast to previous claims, our findings indicate that S1055 phosphorylation is not generally required for CRM1 function and that the Q742 is not a function-defining residue in human CRM1. Collectively, our findings provide new insights into an understudied aspect of CRM1 biology and highlight several important issues related to CRM1 function and regulation that need to be re-evaluated and addressed in more detail.
Full article
►▼
Show Figures
Open AccessArticle
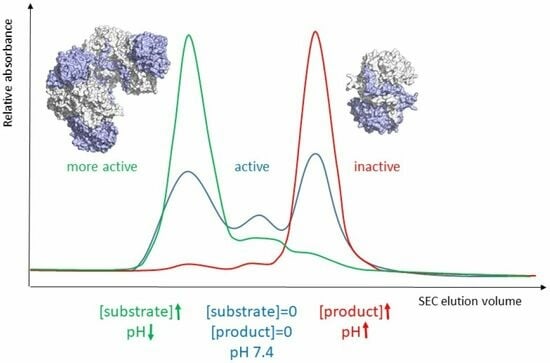
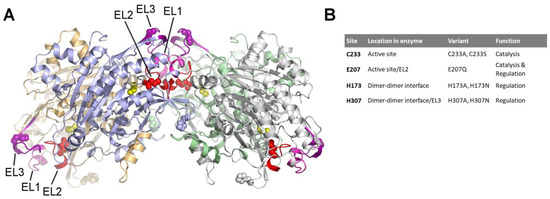
The Allosteric Regulation of Β-Ureidopropionase Depends on Fine-Tuned Stability of Active-Site Loops and Subunit Interfaces
by
Daniela Cederfelt, Dilip Badgujar, Ayan Au Musse, Bernhard Lohkamp, U. Helena Danielson and Doreen Dobritzsch
Cited by 3 | Viewed by 2300
Abstract
The activity of β-ureidopropionase, which catalyses the last step in the degradation of uracil, thymine, and analogous antimetabolites, is cooperatively regulated by the substrate and product of the reaction. This involves shifts in the equilibrium of the oligomeric states of the enzyme, but
[...] Read more.
The activity of β-ureidopropionase, which catalyses the last step in the degradation of uracil, thymine, and analogous antimetabolites, is cooperatively regulated by the substrate and product of the reaction. This involves shifts in the equilibrium of the oligomeric states of the enzyme, but how these are achieved and result in changes in enzyme catalytic competence has yet to be determined. Here, the regulation of human β-ureidopropionase was further explored via site-directed mutagenesis, inhibition studies, and cryo-electron microscopy. The active-site residue E207, as well as H173 and H307 located at the dimer–dimer interface, are shown to play crucial roles in enzyme activation. Dimer association to larger assemblies requires closure of active-site loops, which positions the catalytically crucial E207 stably in the active site. H173 and H307 likely respond to ligand-induced changes in their environment with changes in their protonation states, which fine-tunes the active-site loop stability and the strength of dimer–dimer interfaces and explains the previously observed pH influence on the oligomer equilibrium. The correlation between substrate analogue structure and effect on enzyme assembly suggests that the ability to favourably interact with F205 may distinguish activators from inhibitors. The cryo-EM structure of human β-ureidopropionase assembly obtained at low pH provides first insights into the architecture of its activated state. and validates our current model of the allosteric regulation mechanism. Closed entrance loop conformations and dimer–dimer interfaces are highly conserved between human and fruit fly enzymes.
Full article
►▼
Show Figures
Open AccessArticle
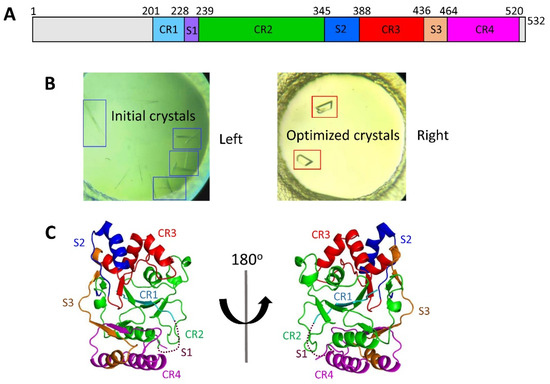
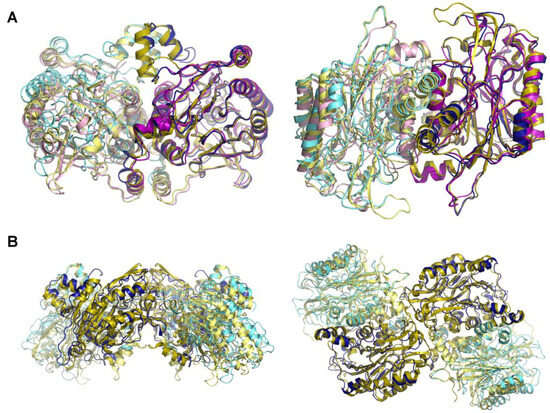
Structural and Functional Insights into the Stealth Protein CpsY of Mycobacterium tuberculosis
by
Dafeng Liu, Cai Yuan, Chenyun Guo, Mingdong Huang and Donghai Lin
Cited by 7 | Viewed by 2588
Abstract
Mycobacterium tuberculosis (
Mtb) is an important and harmful intracellular pathogen that is responsible for the cause of tuberculosis (TB).
Mtb capsular polysaccharides can misdirect the host’s immune response pathways, resulting in additional challenges in TB treatment. These capsule polysaccharides are biosynthesized
[...] Read more.
Mycobacterium tuberculosis (
Mtb) is an important and harmful intracellular pathogen that is responsible for the cause of tuberculosis (TB).
Mtb capsular polysaccharides can misdirect the host’s immune response pathways, resulting in additional challenges in TB treatment. These capsule polysaccharides are biosynthesized by stealth proteins, including CpsY. The structure and functional mechanism of
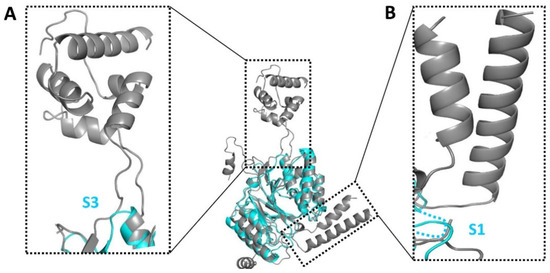
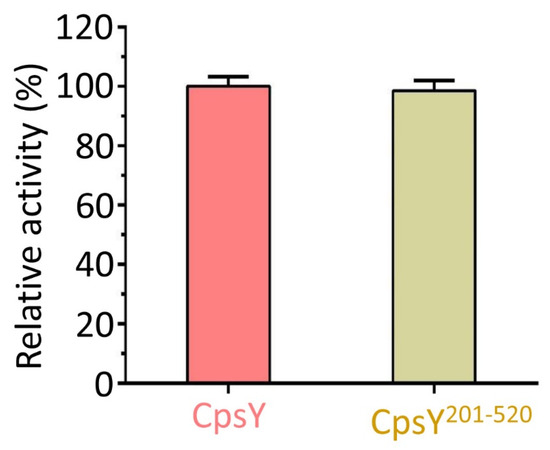
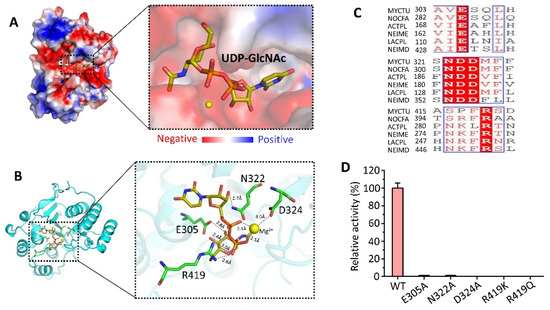
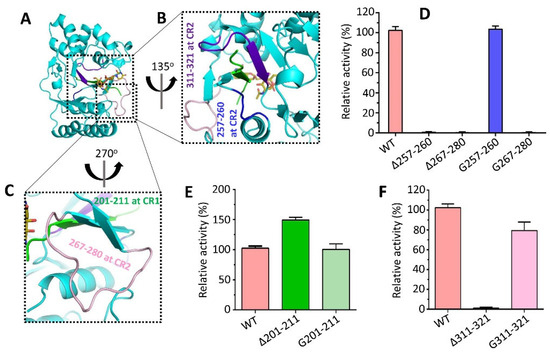
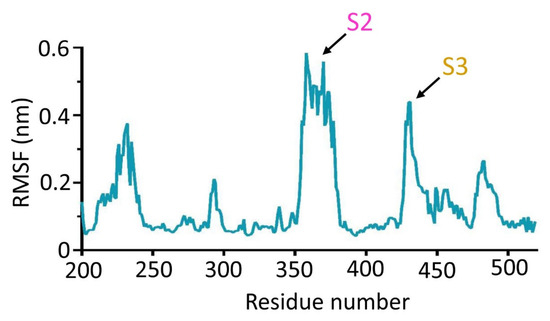
Mtb CpsY are not completely delineated. Here, we reported the crystal structure of CpsY
201−520 at 1.64 Å. CpsY
201−520 comprises three β-sheets with five α-helices on one side and three on the other. Four conserved regions (CR1–CR4) are located near and at the base of its catalytic cavity, and three spacer segments (S1–S3) surround the catalytic cavity. Site-directed mutagenesis demonstrated the strict conservation of R419 at CR3 and S1–S3 in regulating the phosphotransferase activity of CpsY
201−520. In addition, deletion of S2 or S3 (∆S2 or ∆S3) dramatically increased the activity compared to the wild-type (WT) CpsY
201−520. Results from molecular dynamics (MD) simulations showed that S2 and S3 are highly flexible. Our study provides new insights for the development of new vaccines and targeted immunotherapy against
Mtb.
Full article
►▼
Show Figures
Open AccessArticle
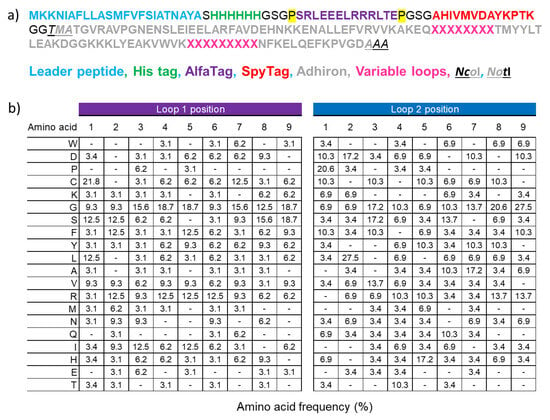
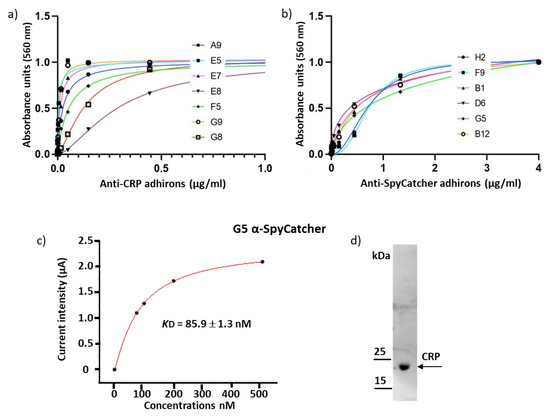
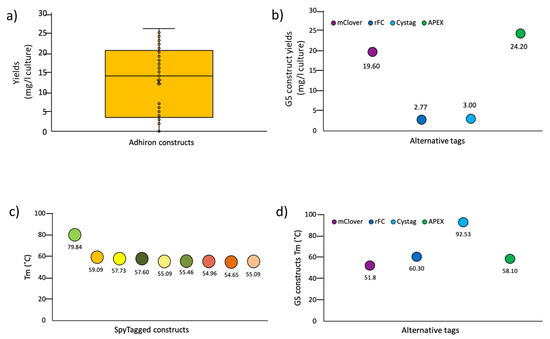
Biological Applications of Synthetic Binders Isolated from a Conceptually New Adhiron Library
by
Claudia D’Ercole, Matteo De March, Gianluca Veggiani, Sandra Oloketuyi, Rossella Svigelj and Ario de Marco
Cited by 3 | Viewed by 3418
Abstract
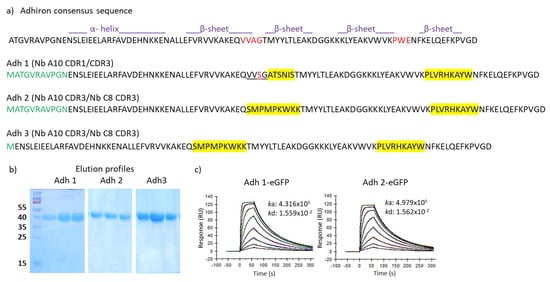
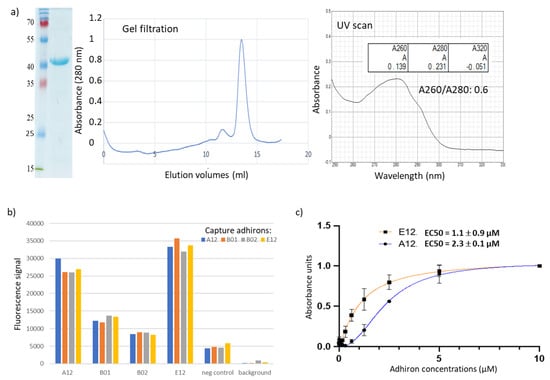
Background: Adhirons are small (10 kDa) synthetic ligands that might represent an alternative to antibody fragments and to alternative scaffolds such as DARPins or affibodies. Methods: We prepared a conceptionally new adhiron phage display library that allows the presence of cysteines in the
[...] Read more.
Background: Adhirons are small (10 kDa) synthetic ligands that might represent an alternative to antibody fragments and to alternative scaffolds such as DARPins or affibodies. Methods: We prepared a conceptionally new adhiron phage display library that allows the presence of cysteines in the hypervariable loops and successfully panned it against antigens possessing different characteristics. Results: We recovered binders specific for membrane epitopes of plant cells by panning the library directly against pea protoplasts and against soluble C-Reactive Protein and SpyCatcher, a small protein domain for which we failed to isolate binders using pre-immune nanobody libraries. The best binders had a binding constant in the low nM range, were produced easily in bacteria (average yields of 15 mg/L of culture) in combination with different tags, were stable, and had minimal aggregation propensity, independent of the presence or absence of cysteine residues in their loops. Discussion: The isolated adhirons were significantly stronger than those isolated previously from other libraries and as good as nanobodies recovered from a naïve library of comparable theoretical diversity. Moreover, they proved to be suitable reagents for ELISA, flow cytometry, the western blot, and also as capture elements in electrochemical biosensors.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceArticle
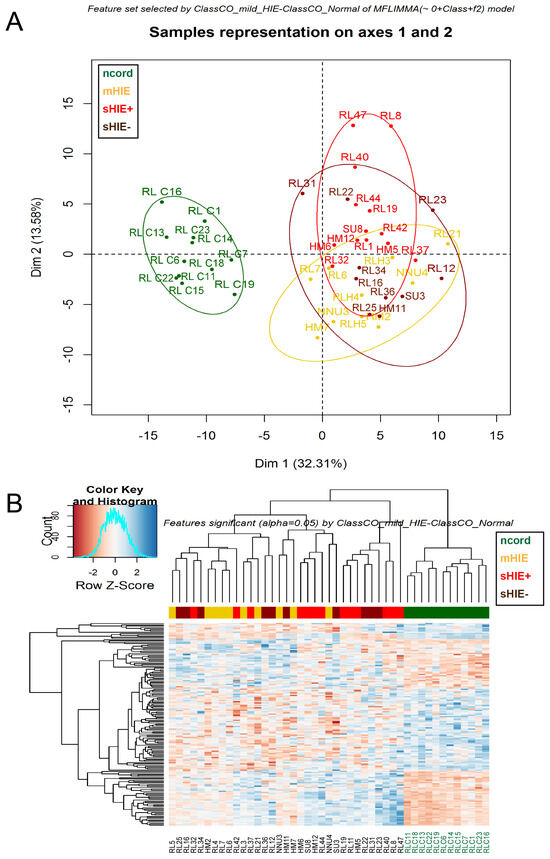
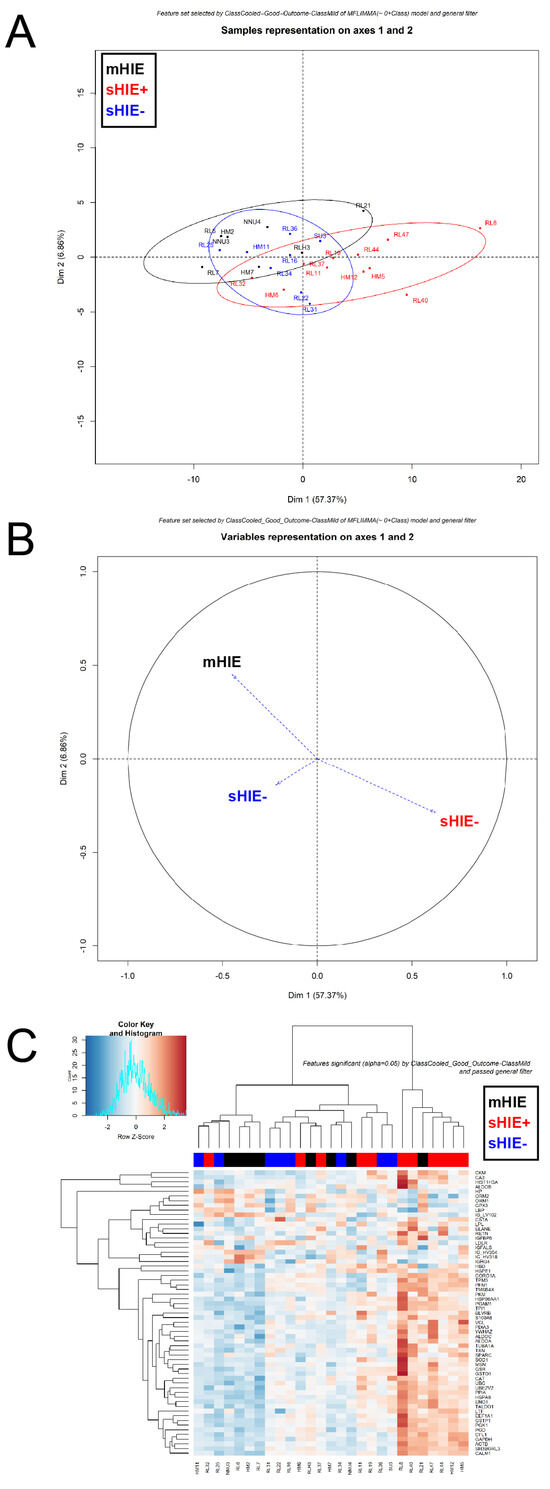
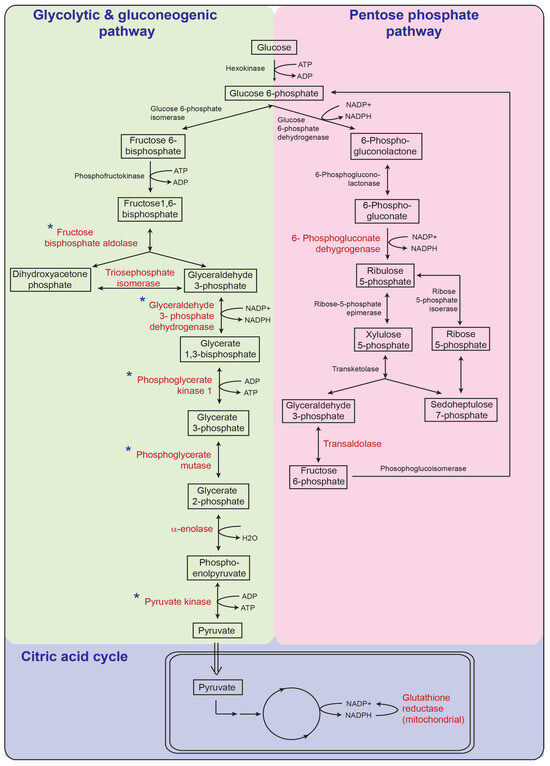
Newborns with Favourable Outcomes after Perinatal Asphyxia Have Upregulated Glucose Metabolism-Related Proteins in Plasma
by
Ping K. Yip, Michael Bremang, Ian Pike, Vennila Ponnusamy, Adina T. Michael-Titus and Divyen K. Shah
Cited by 5 | Viewed by 2712
Abstract
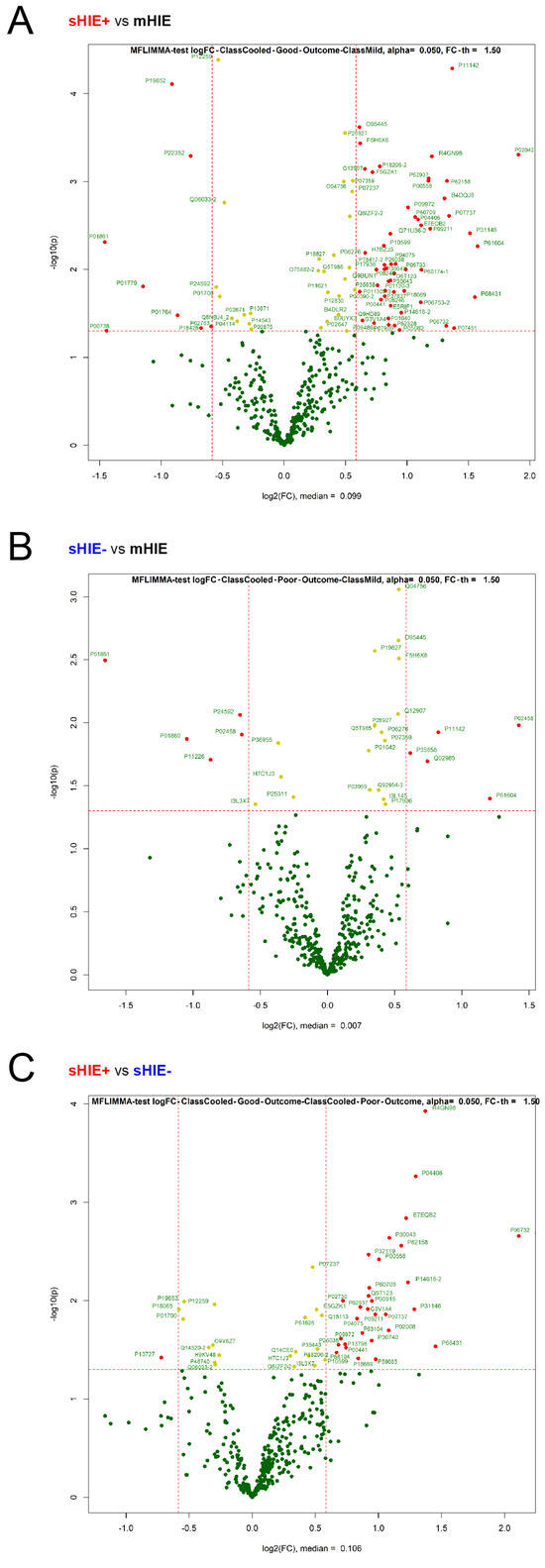
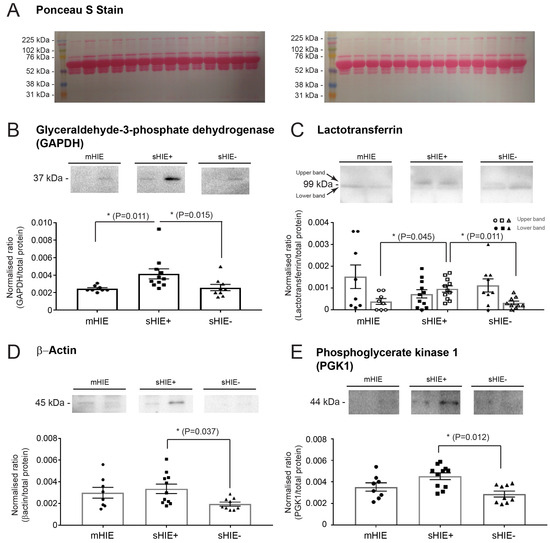
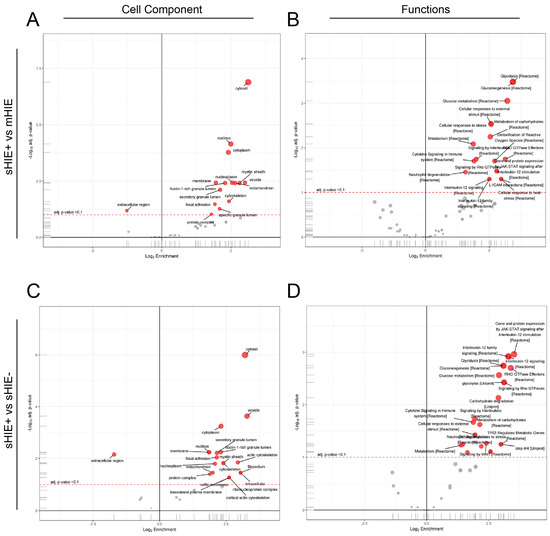
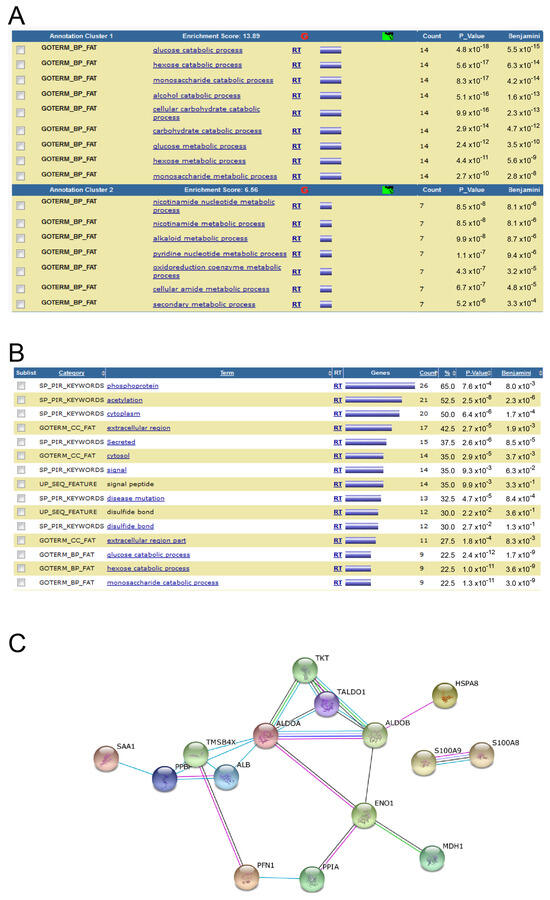
Hypoxic-ischaemic encephalopathy (HIE) is an important cause of morbidity and mortality globally. Although mild therapeutic hypothermia (TH) may improve outcomes in selected babies, the mechanism of action is not fully understood. A proteomics discovery study was carried out to analyse proteins in the
[...] Read more.
Hypoxic-ischaemic encephalopathy (HIE) is an important cause of morbidity and mortality globally. Although mild therapeutic hypothermia (TH) may improve outcomes in selected babies, the mechanism of action is not fully understood. A proteomics discovery study was carried out to analyse proteins in the plasma of newborns with HIE. Proteomic analysis of plasma from 22 newborns with moderate-severe HIE that had initially undergone TH, and relative controls including 10 newborns with mild HIE who did not warrant TH and also cord blood from 10 normal births (non-HIE) were carried out using the isobaric Tandem Mass Tag (TMT
®) 10plex
TM labelling with tandem mass spectrometry. A total of 7818 unique peptides were identified in all TMT10plex
TM samples, translating to 3457 peptides representing 405 proteins, after applying stringent filter criteria. Apart from the unique protein signature from normal cord blood, unsupervised analysis revealed several significantly regulated proteins in the TH-treated moderate-severe HIE group. GO annotation and functional clustering revealed various proteins associated with glucose metabolism: the enzymes fructose-bisphosphate aldolase A, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate mutase 1, phosphoglycerate kinase 1, and pyruvate kinase PKM were upregulated in newborns with favourable (sHIE+) outcomes compared to newborns with unfavourable (sHIE−) outcomes. Those with favourable outcomes had normal MR imaging or mild abnormalities not predictive of adverse outcomes. However, in comparison to mild HIE and the sHIE− groups, the sHIE+ group had the additional glucose metabolism-related enzymes upregulated, including triosephosphate isomerase, α-enolase, 6-phosphogluconate dehydrogenase, transaldolase, and mitochondrial glutathione reductase. In conclusion, our plasma proteomic study demonstrates that TH-treated newborns with favourable outcomes have an upregulation in glucose metabolism. These findings may open new avenues for more effective neuroprotective therapy.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
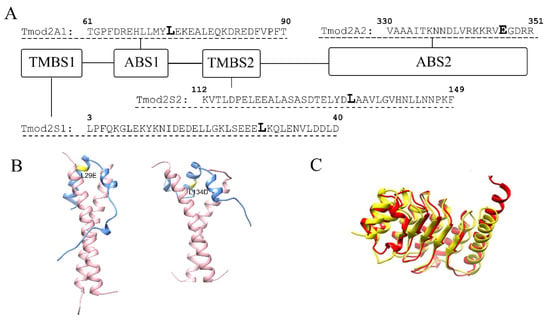
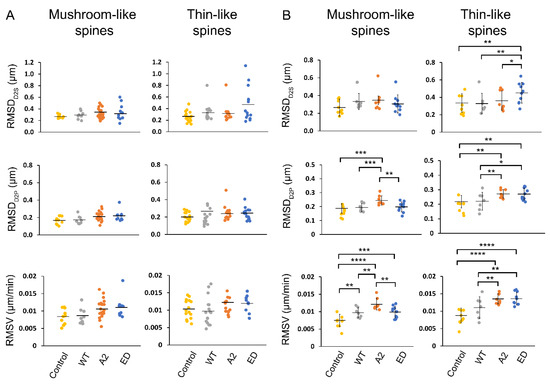
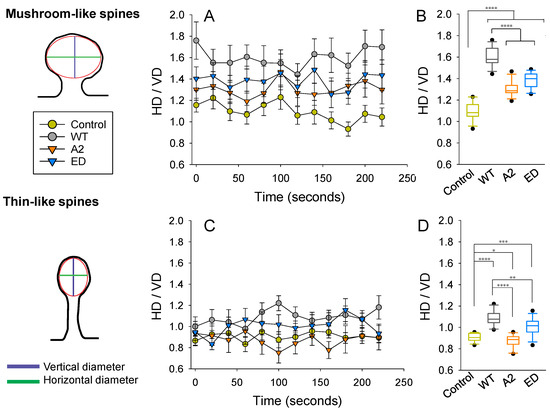
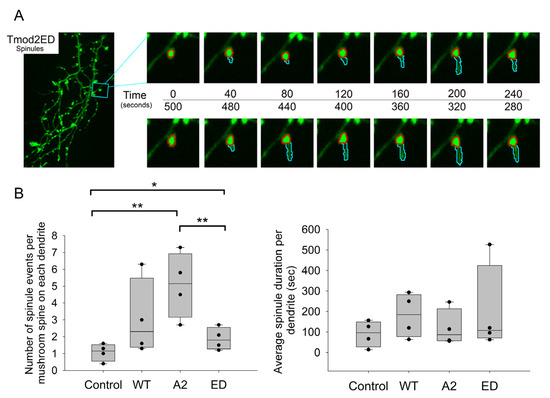
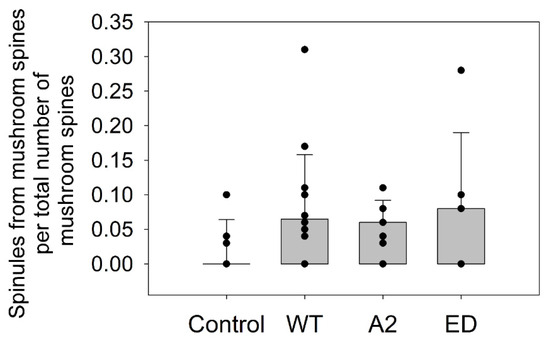
Effects of Tropomodulin 2 on Dendritic Spine Reorganization and Dynamics
by
Balaganesh Kuruba, Nickolas Starks, Mary Rose Josten, Ori Naveh, Gary Wayman, Marina Mikhaylova and Alla S. Kostyukova
Cited by 1 | Viewed by 2790
Abstract
Dendritic spines are actin-rich protrusions that receive a signal from the axon at the synapse. Remodeling of cytoskeletal actin is tightly connected to dendritic spine morphology-mediated synaptic plasticity of the neuron. Remodeling of cytoskeletal actin is required for the formation, development, maturation, and
[...] Read more.
Dendritic spines are actin-rich protrusions that receive a signal from the axon at the synapse. Remodeling of cytoskeletal actin is tightly connected to dendritic spine morphology-mediated synaptic plasticity of the neuron. Remodeling of cytoskeletal actin is required for the formation, development, maturation, and reorganization of dendritic spines. Actin filaments are highly dynamic structures with slow-growing/pointed and fast-growing/barbed ends. Very few studies have been conducted on the role of pointed-end binding proteins in the regulation of dendritic spine morphology. In this study, we evaluated the role played by tropomodulin 2 (Tmod2)—a brain-specific isoform, on the dendritic spine re-organization. Tmod2 regulates actin nucleation and polymerization by binding to the pointed end via actin and tropomyosin (Tpm) binding sites. We studied the effects of Tmod2 overexpression in primary hippocampal neurons on spine morphology using confocal microscopy and image analysis. Tmod2 overexpression decreased the spine number and increased spine length. Destroying Tpm-binding ability increased the number of shaft synapses and thin spine motility. Eliminating the actin-binding abilities of Tmod2 increased the number of mushroom spines. Tpm-mediated pointed-end binding decreased F-actin depolymerization, which may positively affect spine stabilization; the nucleation ability of Tmod2 appeared to increase shaft synapses.
Full article
►▼
Show Figures











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}