
Biological Applications of Synthetic Binders Isolated from a Conceptually New Adhiron Library
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Library Preparation
2.2. Protoplast Preparation
2.3. Panning and Screening of Adhiron Phage Display Library on Whole Protoplasts
2.4. Panning on Soluble Antigens
2.5. Adhiron Expression, Purification, and Characterization
2.6. Binding Affinity Determination
2.7. Differential Scanning Fluorimetry (DSF) Assays
2.8. Biosensor Preparation
3. Results and Discussion
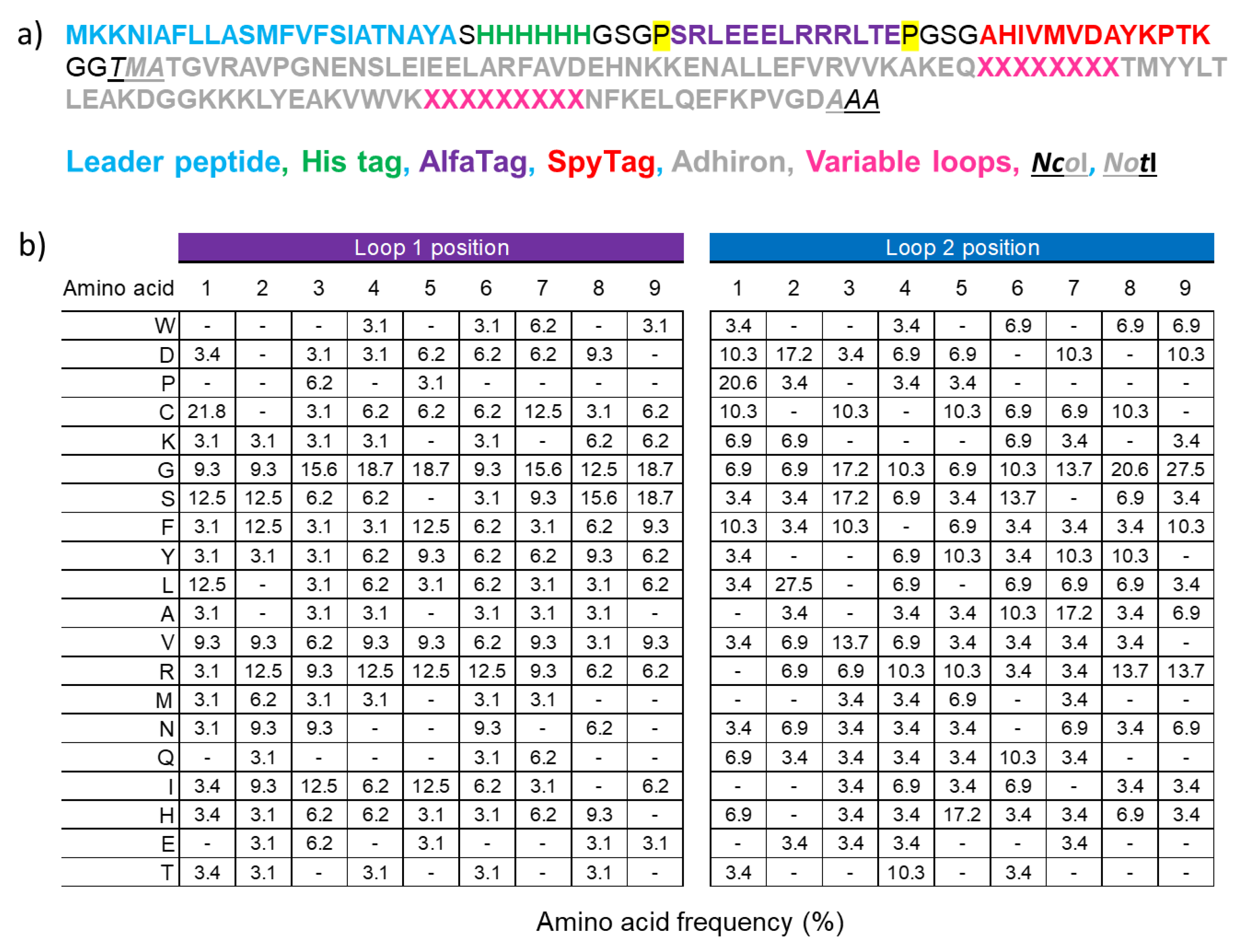
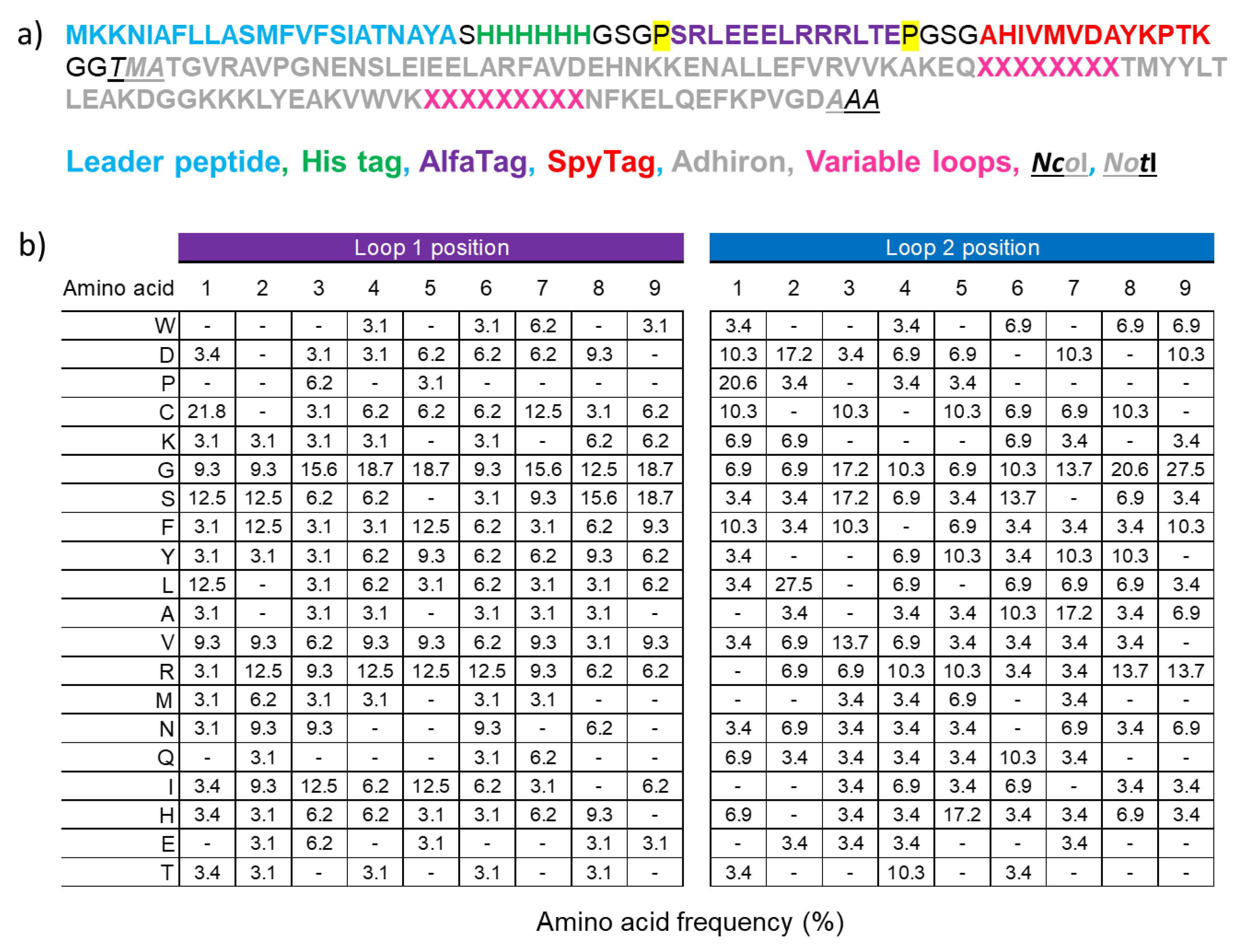
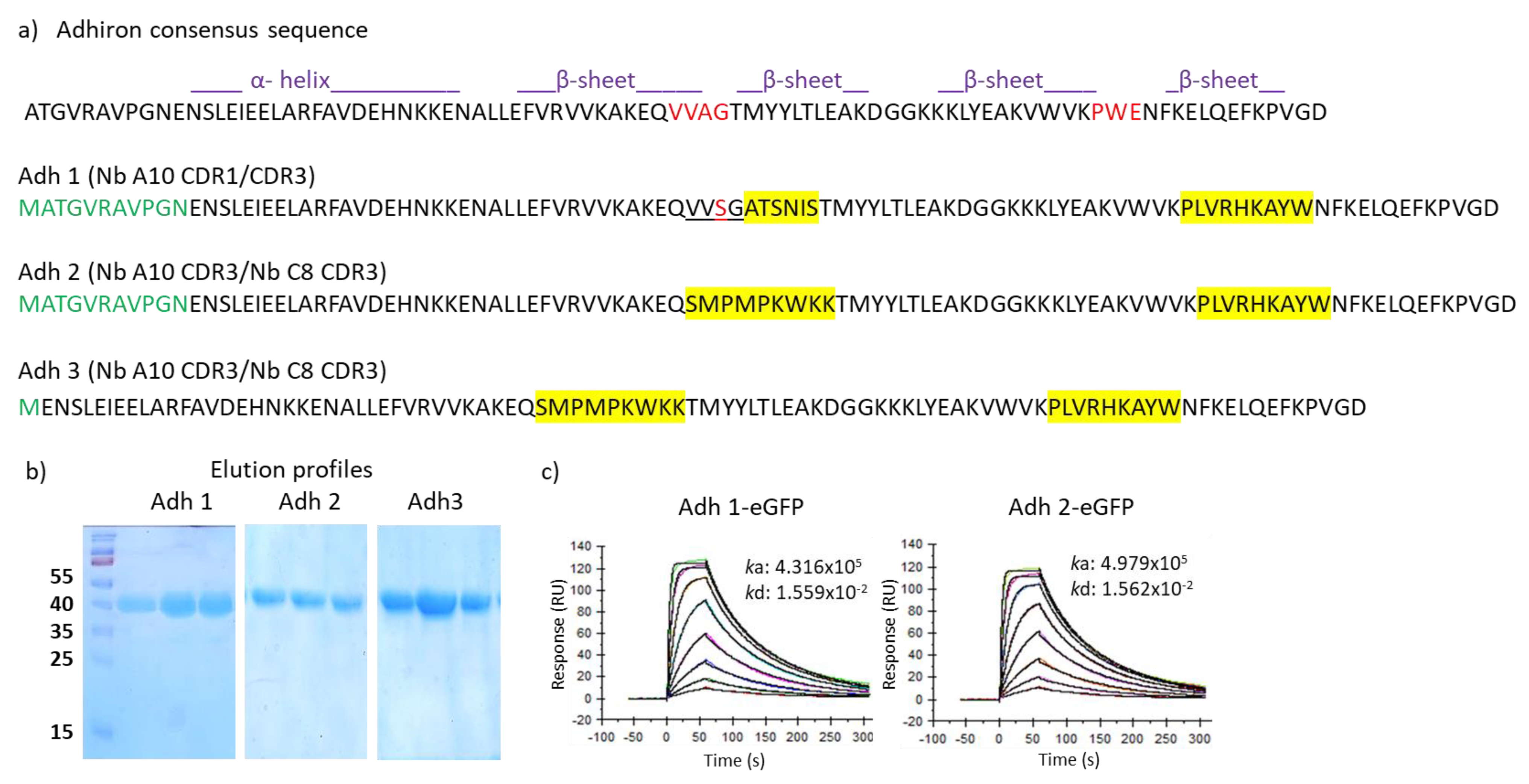
3.1. Design of the Adhiron Phage Display Library
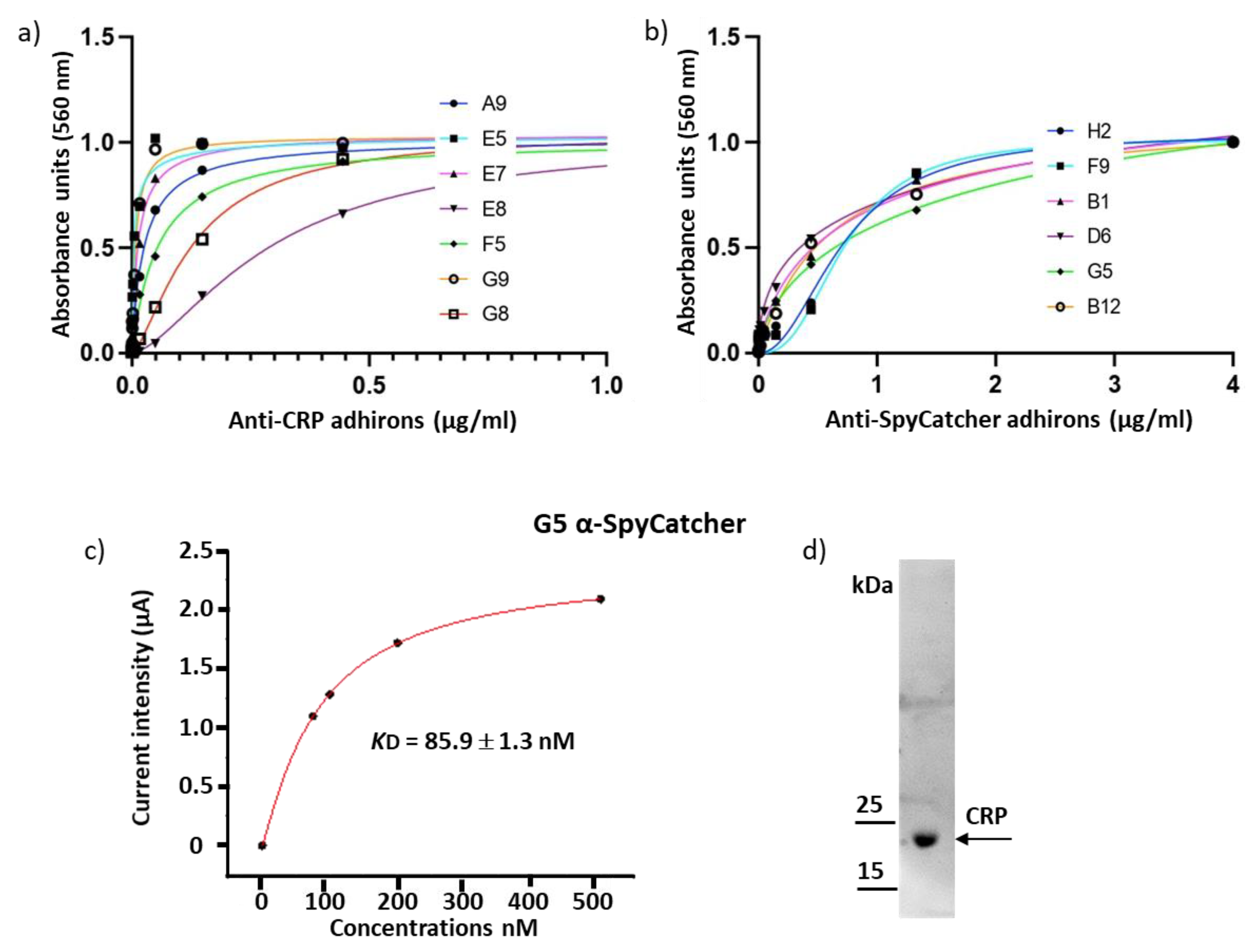
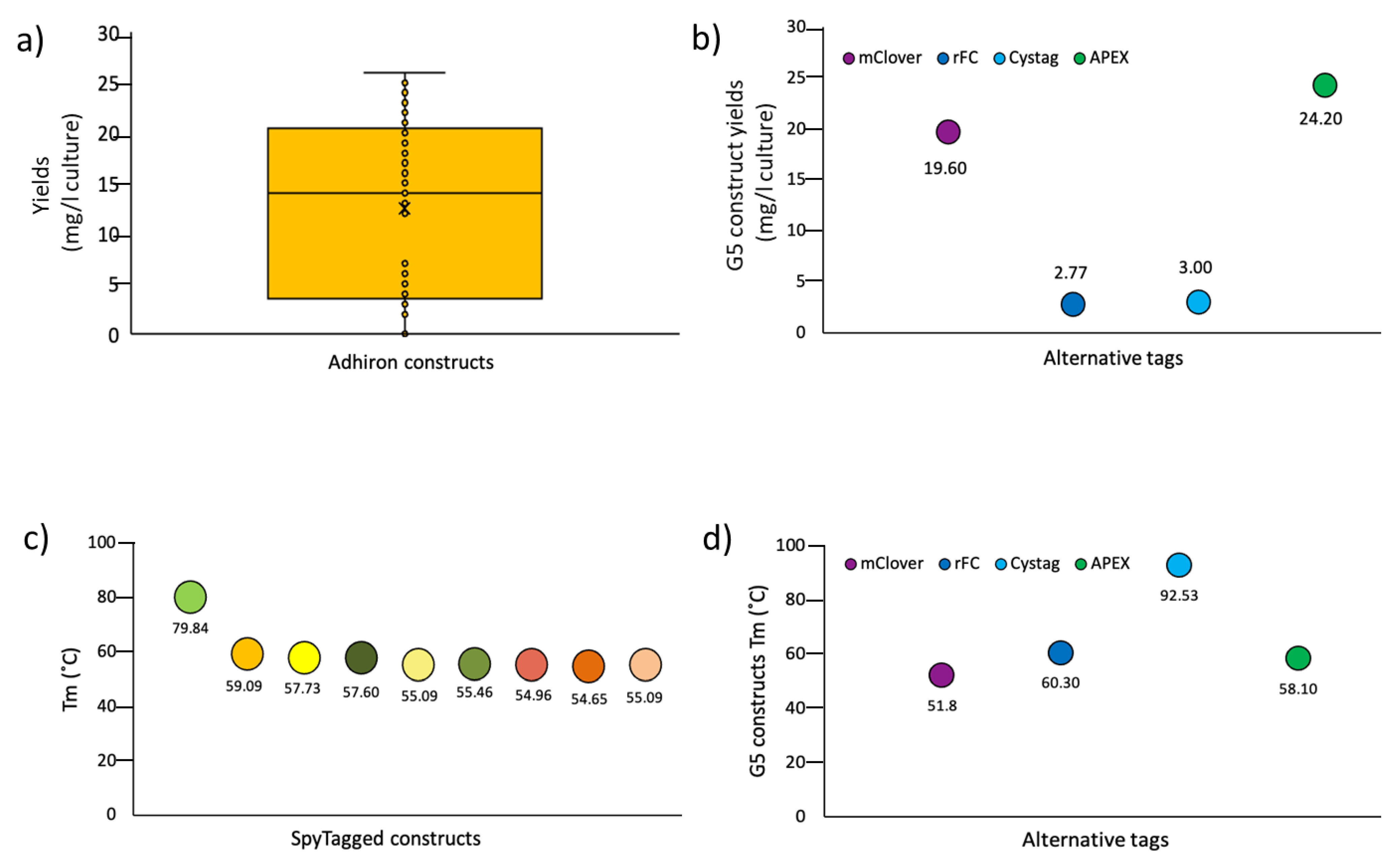
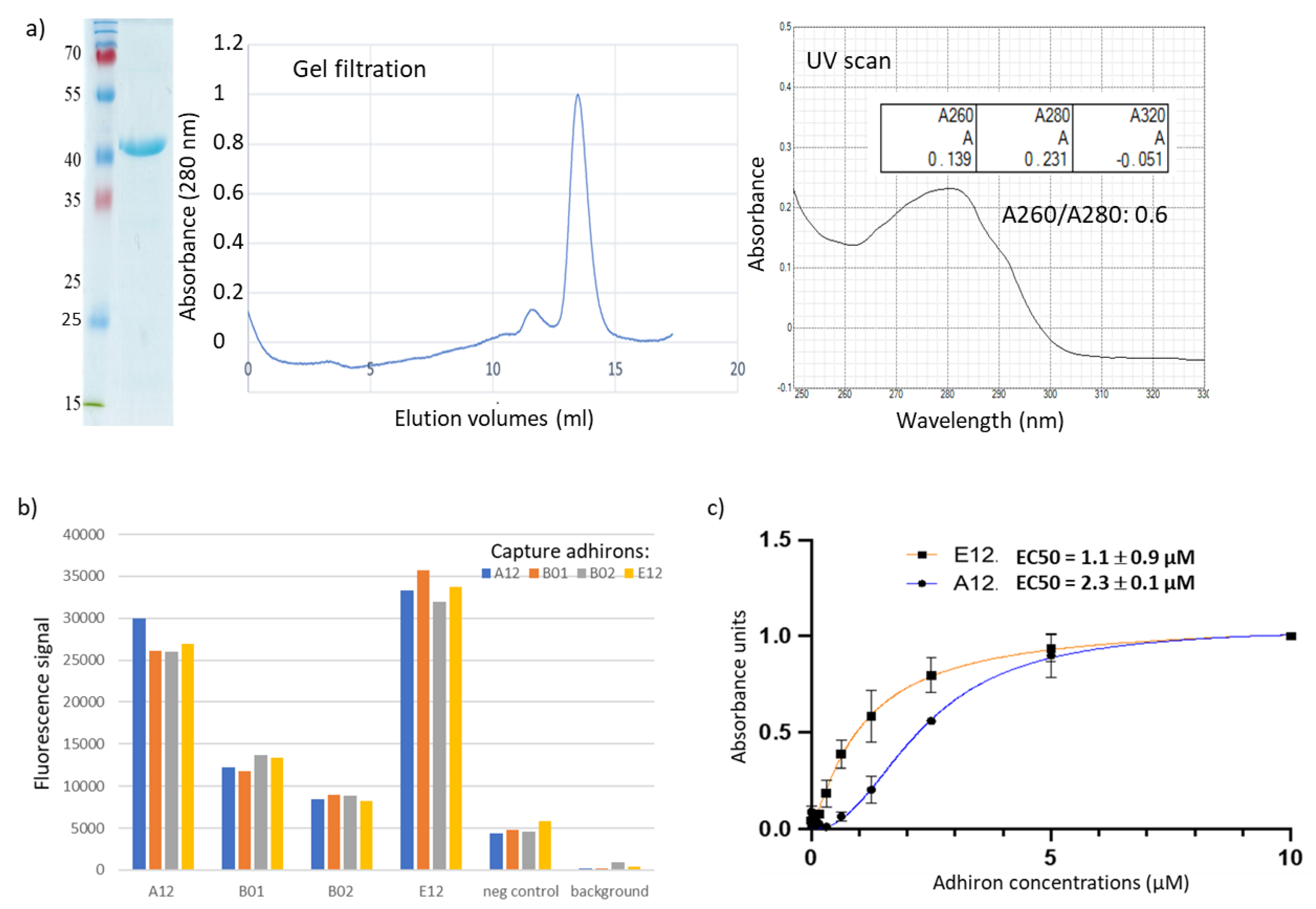
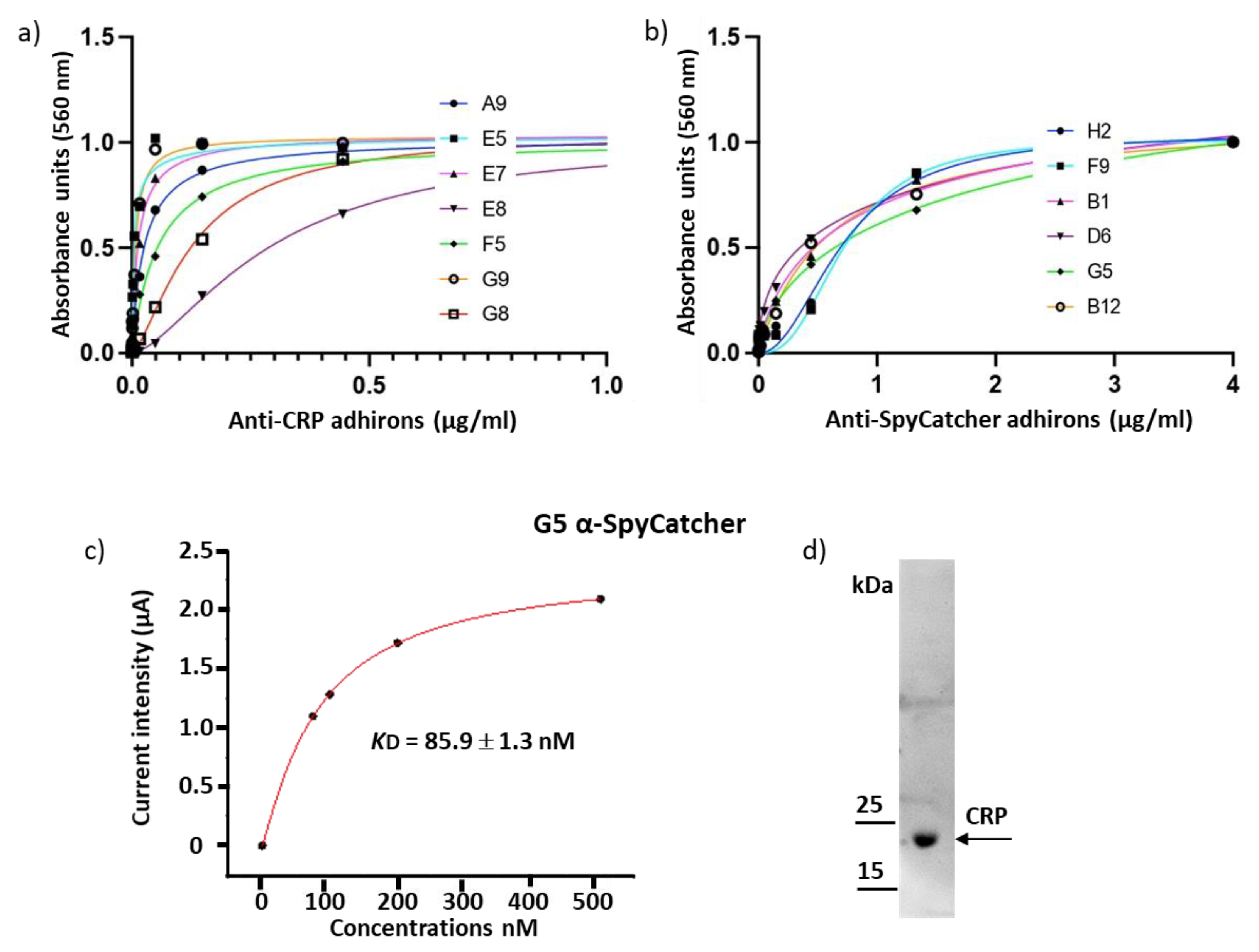
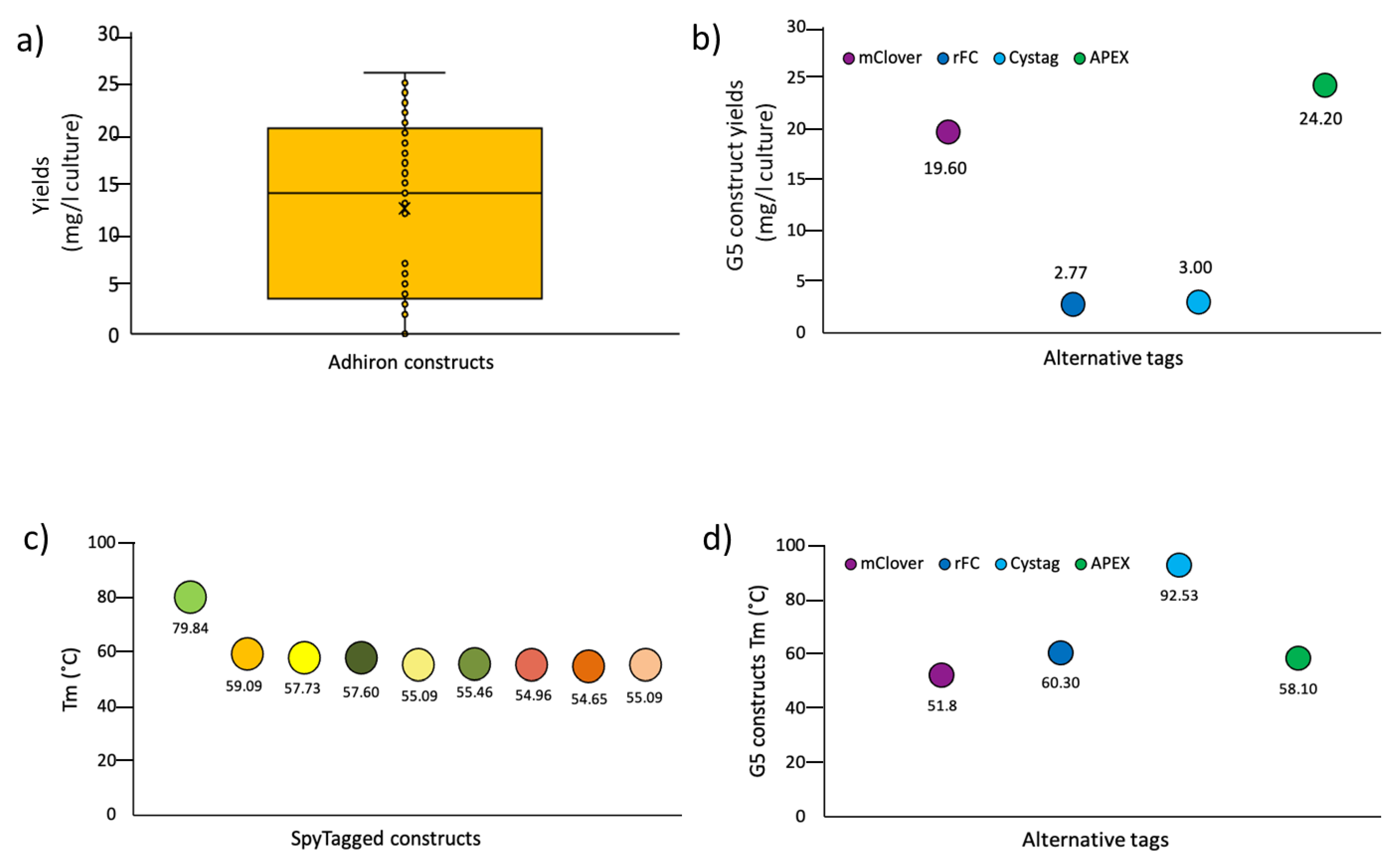
3.2. Panning and Binder Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Traenkle, B.; Rothbauer, U. Under the Microscope: Single-Domain Antibodies for Live-Cell Imaging and Super-Resolution Microscopy. Front. Immunol. 2017, 24, 1030. [Google Scholar] [CrossRef] [PubMed]
- Nieves, D.J.; Pike, J.A.; Levet, F.; Williamson, D.J.; Baragilly, M.; Oloketuyi, S.; de Marco, A.; Griffié, J.; Sage, D.; Cohen, E.A.K.; et al. A framework for evaluating the performance of SMLM cluster analysis algorithms. Nat. Methods 2023, 20, 259–267. [Google Scholar] [CrossRef]
- Ambrosetti, E.; Bernardinelli, G.; Hoffecker, I.; Hartmanis, L.; Kiriako, G.; de Marco, A.; Sandberg, R.; Högberg, B.; Teixeira, A.I. A DNA-nanoassembly-based approach to map membrane protein nanoenvironments. Nat. Nanotechnol. 2021, 16, 85–95. [Google Scholar] [CrossRef]
- Monegal, A.; Ami, D.; Martinelli, C.; Huang, H.; Aliprandi, M.; Capasso, P.; Francavilla, C.; Ossolengo, G.; de Marco, A. Immunological applications of single-domain llama recombinant antibodies isolated from a naïve library. Protein Eng. Des. Sel. 2009, 22, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Moutel, S.; Bery, N.; Bernard, V.; Keller, L.; Lemesre, E.; de Marco, A.; Ligat, L.; Rain, J.C.; Favre, G.; Olichon, A.; et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. Elife 2016, 5, e16228. [Google Scholar] [CrossRef]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.; Arnold, F.M.; Stohler, P.; Bocquet, N.; Hug, M.N.; Huber, S.; Siegrist, M.; Hetemann, L.; et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. Elife 2018, 7, e34317. [Google Scholar] [CrossRef]
- Xiang, Y.; Sang, Z.; Bitton, L.; Xu, J.; Liu, Y.; Schneidman-Duhovny, D.; Shi, Y. Integrative proteomics identifies thousands of distinct, multi-epitope, and high-affinity nanobodies. Cell Syst. 2021, 12, 220–234.e9. [Google Scholar] [CrossRef]
- Chen, X.; Gentili, M.; Hacohen, N.; Regev, A. A cell-free nanobody engineering platform rapidly generates SARS-CoV-2 neutralizing nanobodies. Nat. Commun. 2021, 12, 5506. [Google Scholar] [CrossRef]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015, 33, 408–418. [Google Scholar] [CrossRef]
- Richards, D.A. Exploring alternative antibody scaffolds: Antibody fragments and antibody mimics for targeted drug delivery. Drug Discov. Today Technol. 2018, 30, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Nord, K.; Gunneriusson, E.; Ringdahl, J.; Ståhl, S.; Uhlén, M.; Nygren, P.A. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat. Biotechnol. 1997, 15, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Plückthun, A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Tiede, C.; Tang, A.A.; Deacon, S.E.; Mandal, U.; Nettleship, J.E.; Owen, R.L.; George, S.E.; Harrison, D.J.; Owens, R.J.; Tomlinson, D.C.; et al. Adhiron: A stable and versatile peptide display scaffold for molecular recognition applications. Protein Eng. Des. Sel. 2014, 27, 145–155. [Google Scholar] [CrossRef]
- Woodman, R.; Yeh, J.T.; Laurenson, S.; Ko Ferrigno, P. Design and validation of a neutral protein scaffold for the presentation of peptide aptamers. J. Mol. Biol. 2005, 352, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; Song, Q.; Ko Ferrigno, P.; Bueno, P.R.; Davis, J.J. Sensitive affimer and antibody based impedimetric label-free assays for C-reactive protein. Anal. Chem. 2012, 84, 6553–6560. [Google Scholar] [CrossRef] [PubMed]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. Elife 2017, 6, e24903. [Google Scholar] [CrossRef]
- Kearney, K.J.; Pechlivani, N.; King, R.; Tiede, C.; Phoenix, F.; Cheah, R.; Macrae, F.L.; Simmons, K.J.; Manfield, I.W.; Smith, K.A.; et al. Affimer proteins as a tool to modulate fibrinolysis, stabilize the blood clot, and reduce bleeding complications. Blood 2019, 133, 1233–1244. [Google Scholar] [CrossRef]
- Haza, K.Z.; Martin, H.L.; Rao, A.; Turner, A.L.; Saunders, S.E.; Petersen, B.; Tiede, C.; Tipping, K.; Tang, A.A.; Ajayi, M.; et al. RAS-inhibiting biologics identify and probe druggable pockets including an SII-α3 allosteric site. Nat. Commun. 2021, 12, 4045. [Google Scholar] [CrossRef]
- Tang, A.A.S.; Tiede, C.; McPherson, M.J.; Tomlinson, D.C. Isolation of Artificial Binding Proteins (Affimer Reagents) for Use in Molecular and Cellular Biology. Methods Mol. Biol. 2021, 2247, 105–121. [Google Scholar]
- Carrington, G.; Tomlinson, D.; Peckham, M. Exploiting nanobodies and Affimers for superresolution imaging in light microscopy. Mol. Biol. Cell 2019, 30, 2737–2740. [Google Scholar] [CrossRef] [PubMed]
- de Marco, A. User-friendly expression plasmids enable the fusion of VHHs to application-specific tags. Methods Mol. Biol. 2012, 911, 507–522. [Google Scholar] [PubMed]
- Djender, S.; Schneider, A.; Beugnet, A.; Crepin, R.; Desrumeaux, K.E.; Romani, C.; Moutel, S.; Perez, F.; de Marco, A. Bacterial cytoplasm as an effective cell compartment for producing functional VHH-based affinity reagents and Camelidae IgG-like recombinant antibodies. Microb Cell Fact. 2014, 13, 140. [Google Scholar] [CrossRef]
- Oloketuyi, S.; Bernedo, R.; Christmann, A.; Borkowska, J.; Cazzaniga, G.; Schuchmann, H.W.; Niedziółka-Jönsson, J.; Szot-Karpińska, K.; Kolmar, H.; de Marco, A. Native llama nanobody library panning performed by phage and yeast display provides binders suitable for C-Reactive Protein detection. Biosensors 2021, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Crépin, R.; Gentien, D.; Duché, A.; Rapinat, A.; Reyes, C.; Némati, F.; Massonnet, G.; Decaudin, D.; Djender, S.; Moutel, S.; et al. Nanobodies against surface biomarkers enable the analysis of tumor genetic heterogeneity in uveal melanoma patient-derived xenografts. Pigment. Cell Melanoma Res. 2017, 30, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Keeble, A.H.; Banerjee, A.; Ferla, M.P.; Reddington, S.C.; Anuar, I.N.A.K.; Howarth, M. Evolving accelerated amidation by SpyTag/SpyCatcher to analyze membrane dynamics. Angew. Chem. Int. Ed. Engl. 2017, 56, 16521–16525. [Google Scholar] [CrossRef]
- Oloketuyi, S.; Mazzega, E.; Zavašnik, J.; Pungjunun, K.; Kalcher, K.; de Marco, A.; Mehmeti, E. Electrochemical immunosensor functionalized with nanobodies for the detection of the toxic microalgae Alexandrium minutum using glassy carbon electrode modified with gold nanoparticles. Biosens. Bioelectron. 2020, 154, 112052. [Google Scholar] [CrossRef]
- De March, M.; Terdoslavich, M.; Polez, S.; Guarnaccia, C.; Poggianella, M.; Marcello, A.; Skoko, N.; de Marco, A. Expression, purification and characterization of SARS-CoV-2 spike RBD in ExpiCHO cells. Protein Expr. Purif. 2022, 194, 106071. [Google Scholar] [CrossRef]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.; Wohlkönig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.; Kobilka, B.K.; Steyaert, J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef]
- Sherwood, L.J.; Hayhurst, A. Visualizing Filoviral Nucleoproteins Using Nanobodies Fused to the Ascorbate Peroxidase Derivatives APEX2 and dEAPX. Methods Mol. Biol. 2022, 2446, 427–449. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the Expasy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Veggiani, G.; de Marco, A. Improved quantitative and qualitative production of single-domain intrabodies mediated by the co-expression of Erv1p sulfhydryl oxidase. Protein Expr. Purif. 2011, 79, 111–114. [Google Scholar] [CrossRef] [PubMed]
- de Marco, A.; Casatta, E.; Savaresi, S.; Geerlof, A. Recombinant proteins fused to thermostable partners can be purified by heat incubation. J. Biotechnol. 2004, 107, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Götzke, H.; Kilisch, M.; Martínez-Carranza, M.; Sograte-Idrissi, S.; Rajavel, A.; Schlichthaerle, T.; Engels, N.; Jungmann, R.; Stenmark, P.; Opazo, F.; et al. The ALFA-tag is a highly versatile tool for nanobody-based bioscience applications. Nat. Commun. 2019, 10, 4403. [Google Scholar] [CrossRef] [PubMed]
- Reddington, S.C.; Howarth, M. Secrets of a covalent interaction for biomaterials and biotechnology: SpyTag and SpyCatcher. Curr. Opin. Chem. Biol. 2015, 29, 94–99. [Google Scholar] [CrossRef]
- Prabakaran, P.; Chowdhury, P.S. Landscape of non-canonical cysteines in human VH repertoire revealed by immunogenetic analysis. Cell Rep. 2020, 13, 107831. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.E.; Choy, E.H. C-reactive protein and implications in rheumatoid arthritis and associated comorbidities. Semin. Arthritis Rheum. 2021, 51, 219–229. [Google Scholar] [CrossRef]
- Amelung, S.; Nerlich, A.; Rohde, M.; Spellerberg, B.; Cole, J.N.; Nizet, V.; Chhatwal, G.S.; Talay, S.R. The FbaB-type fibronectin-binding protein of Streptococcus pyogenes promotes specific invasion into endothelial cells. Cell Microbiol. 2011, 13, 1200–1211. [Google Scholar] [CrossRef]
- Li, J.; Kang, G.; Wang, J.; Yuan, H.; Wu, Y.; Meng, S.; Wang, P.; Zhang, M.; Wang, Y.; Feng, Y.; et al. Affinity maturation of antibody fragments: A review encompassing the development from random approaches to computational rational optimization. Int. J. Biol. Macromol. 2023, 247, 125733. [Google Scholar] [CrossRef]
- Yang, D.; Singh, A.; Wu, H.; Kroe-Barrett, R. Comparison of biosensor platforms in the evaluation of high affinity antibody-antigen binding kinetics. Anal. Biochem. 2016, 508, 78–96. [Google Scholar] [CrossRef]
- de Veer, S.J.; Kan, M.W.; Craik, D.J. Cyclotides: From Structure to Function. Chem. Rev. 2019, 119, 12375–12421. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-CRP | |||||||
|---|---|---|---|---|---|---|---|
| A9 | E5 | E7 | E8 | F5 | G8 | G9 | |
| EC50 (nM) | 25.9 ± 3.0 | 3.6 ± 0.2 | 12.9 ± 3.0 | 295 ± 39.0 | 55.3 ± 39.0 | 132 ± 54.0 | 7.4 ± 0.4 |
| Anti-SpyCatcher002 | ||||||
|---|---|---|---|---|---|---|
| B1 | B12 | D6 | F9 | G5 | H2 | |
| EC50 (nM) | 577 ± 109 | 611 ± 70 | 356 ± 106 | 727 ± 117 | 739 ± 263 | 719 ± 91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ercole, C.; De March, M.; Veggiani, G.; Oloketuyi, S.; Svigelj, R.; de Marco, A. Biological Applications of Synthetic Binders Isolated from a Conceptually New Adhiron Library. Biomolecules 2023, 13, 1533. https://doi.org/10.3390/biom13101533
D’Ercole C, De March M, Veggiani G, Oloketuyi S, Svigelj R, de Marco A. Biological Applications of Synthetic Binders Isolated from a Conceptually New Adhiron Library. Biomolecules. 2023; 13(10):1533. https://doi.org/10.3390/biom13101533
Chicago/Turabian StyleD’Ercole, Claudia, Matteo De March, Gianluca Veggiani, Sandra Oloketuyi, Rossella Svigelj, and Ario de Marco. 2023. "Biological Applications of Synthetic Binders Isolated from a Conceptually New Adhiron Library" Biomolecules 13, no. 10: 1533. https://doi.org/10.3390/biom13101533
APA StyleD’Ercole, C., De March, M., Veggiani, G., Oloketuyi, S., Svigelj, R., & de Marco, A. (2023). Biological Applications of Synthetic Binders Isolated from a Conceptually New Adhiron Library. Biomolecules, 13(10), 1533. https://doi.org/10.3390/biom13101533