
Mutations of Key Functional Residues in CRM1/XPO1 Differently Alter Its Intranuclear Localization and the Nuclear Export of Endogenous Cargos
 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
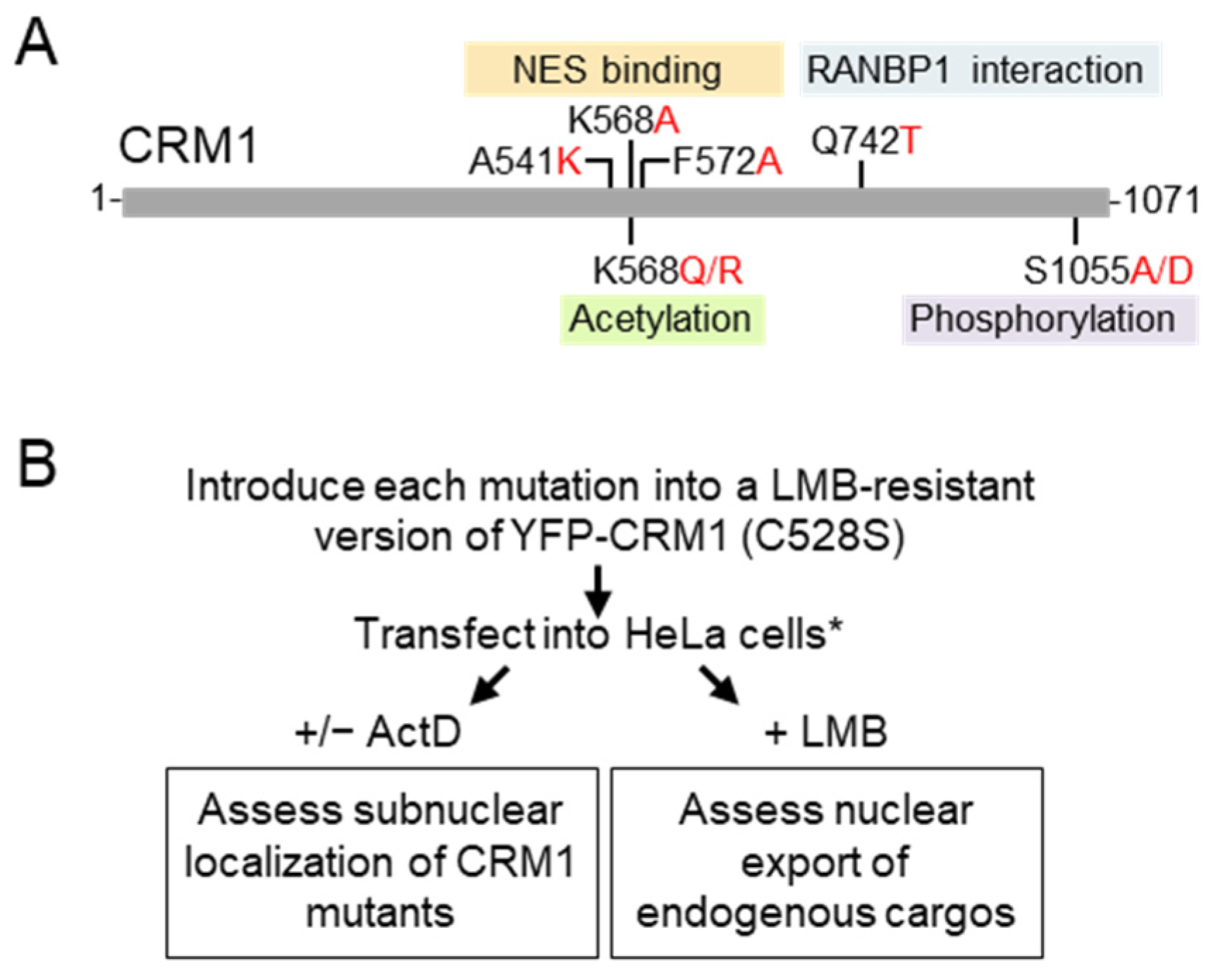
2.1. Plasmids and Site-Directed Mutagenesis
2.2. Cell Culture, Plasmid Transfection, and Drug Treatments
2.3. Immunofluorescence and Microscopy Analysis
2.4. Image Analysis
2.5. Statistical Analysis
3. Results
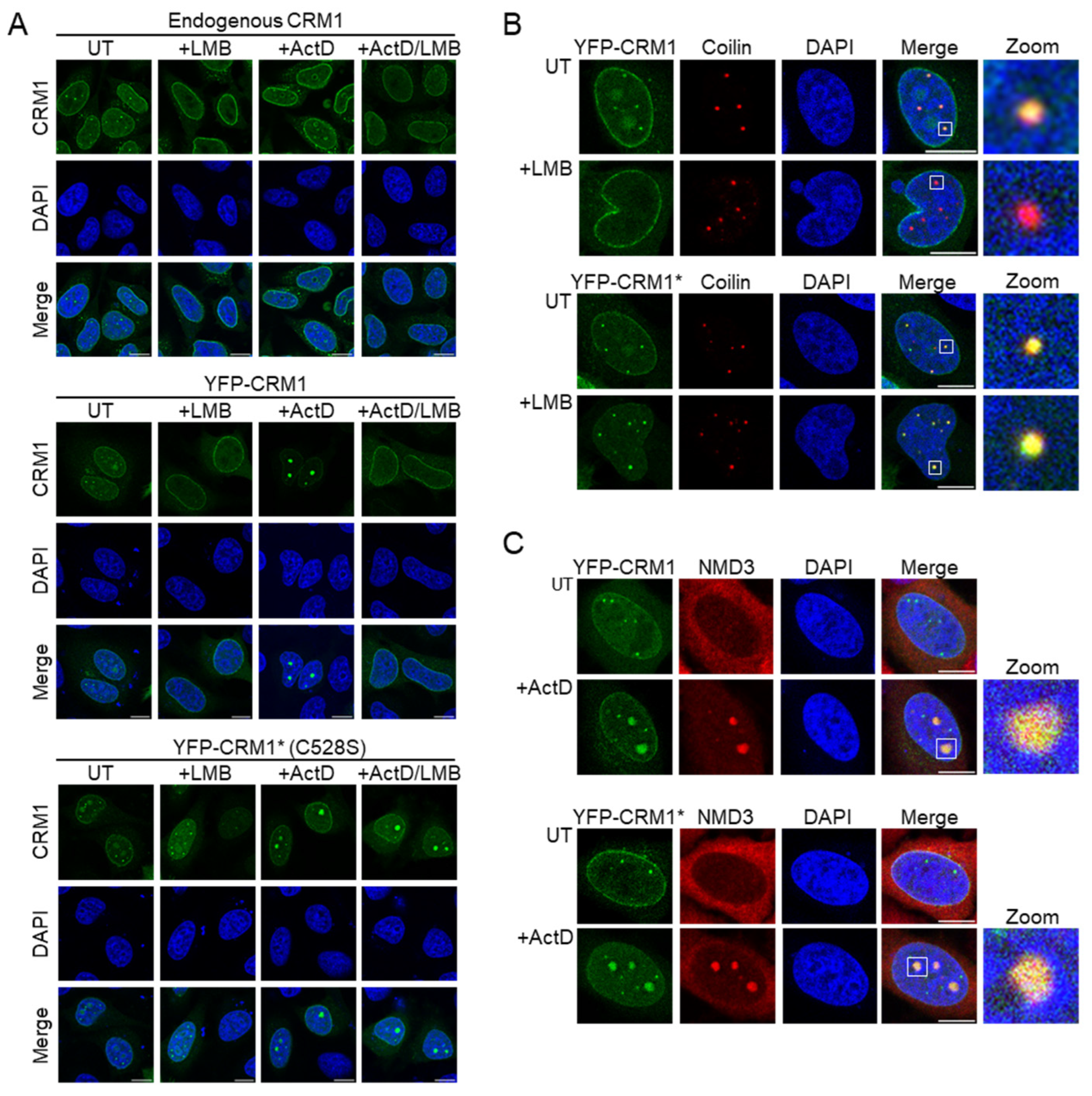
3.1. An YFP-Tagged, LMB Resistant Version of CRM1 Recapitulates the Intranuclear Localization of Endogenous CRM1 in HeLa Cells
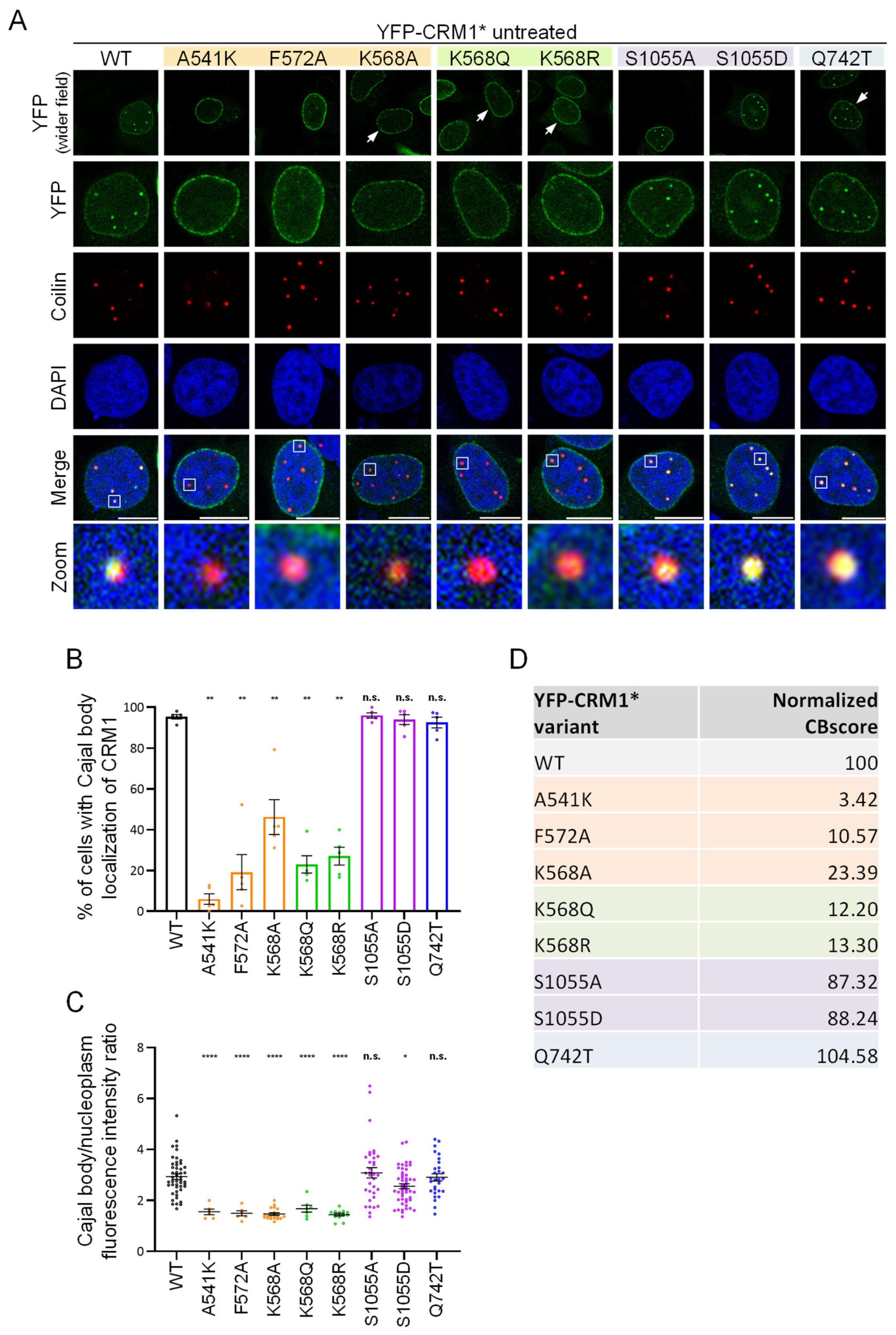
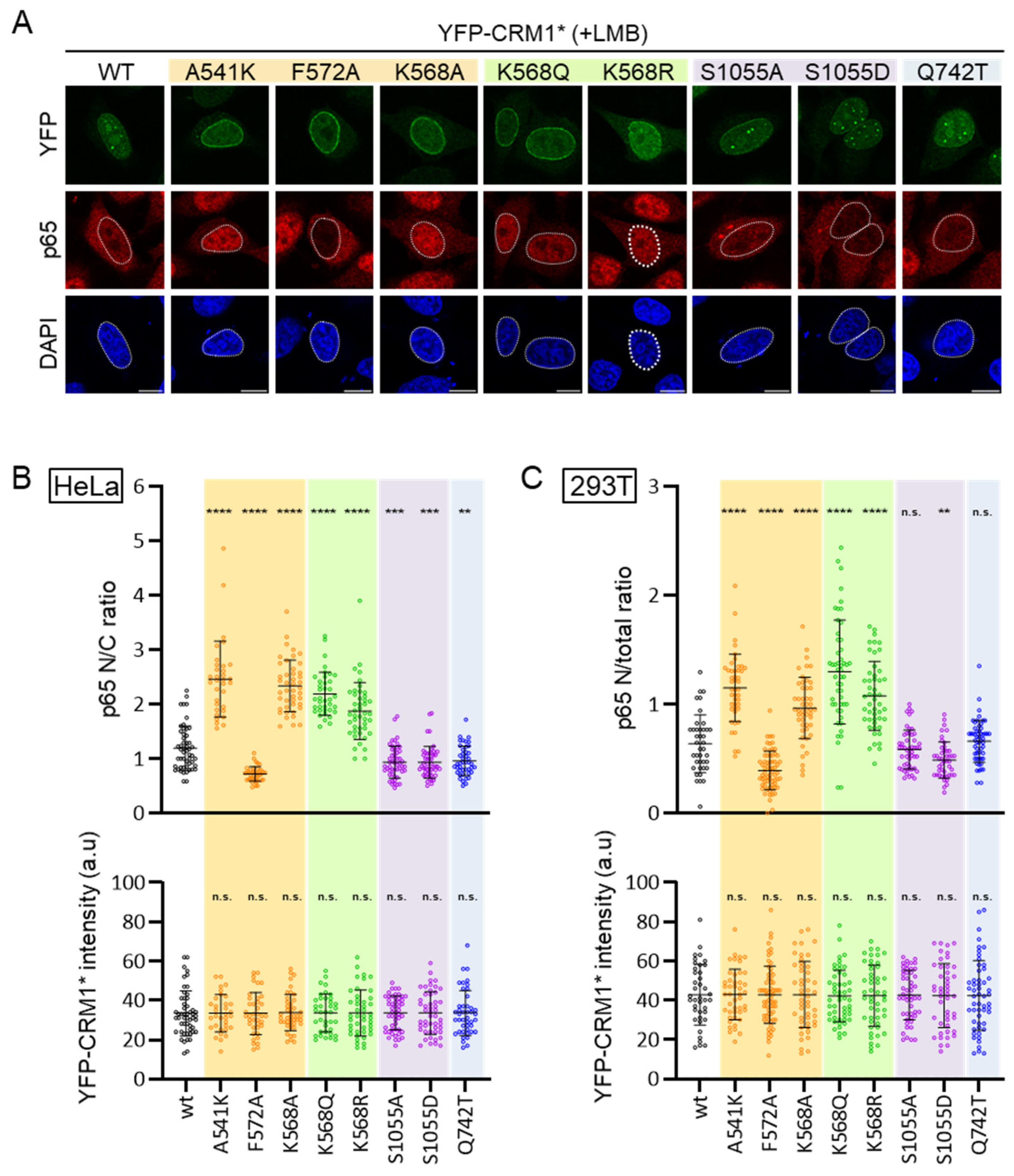
3.2. CRM1 Mutations Differently Alter the Intranuclear Localization of the Receptor
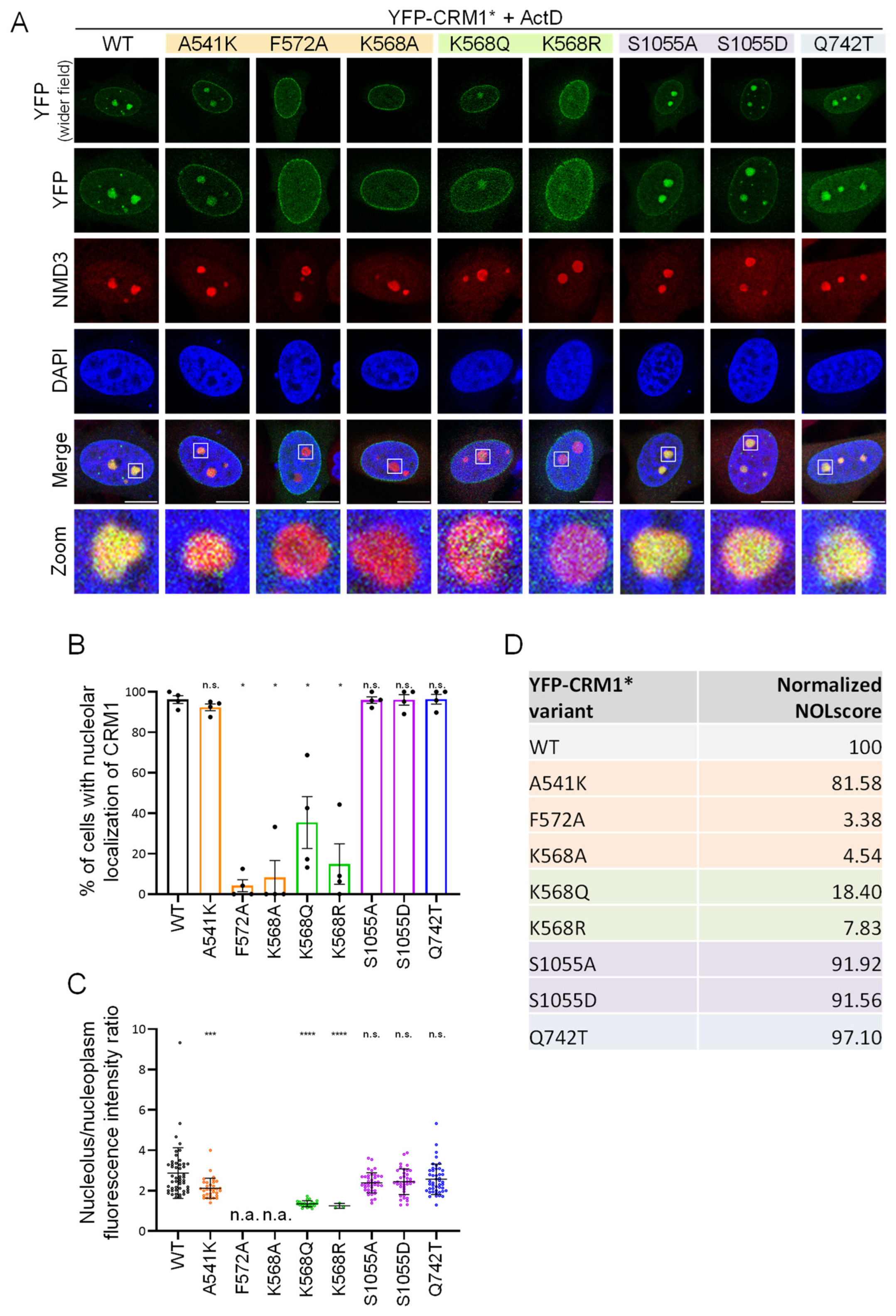
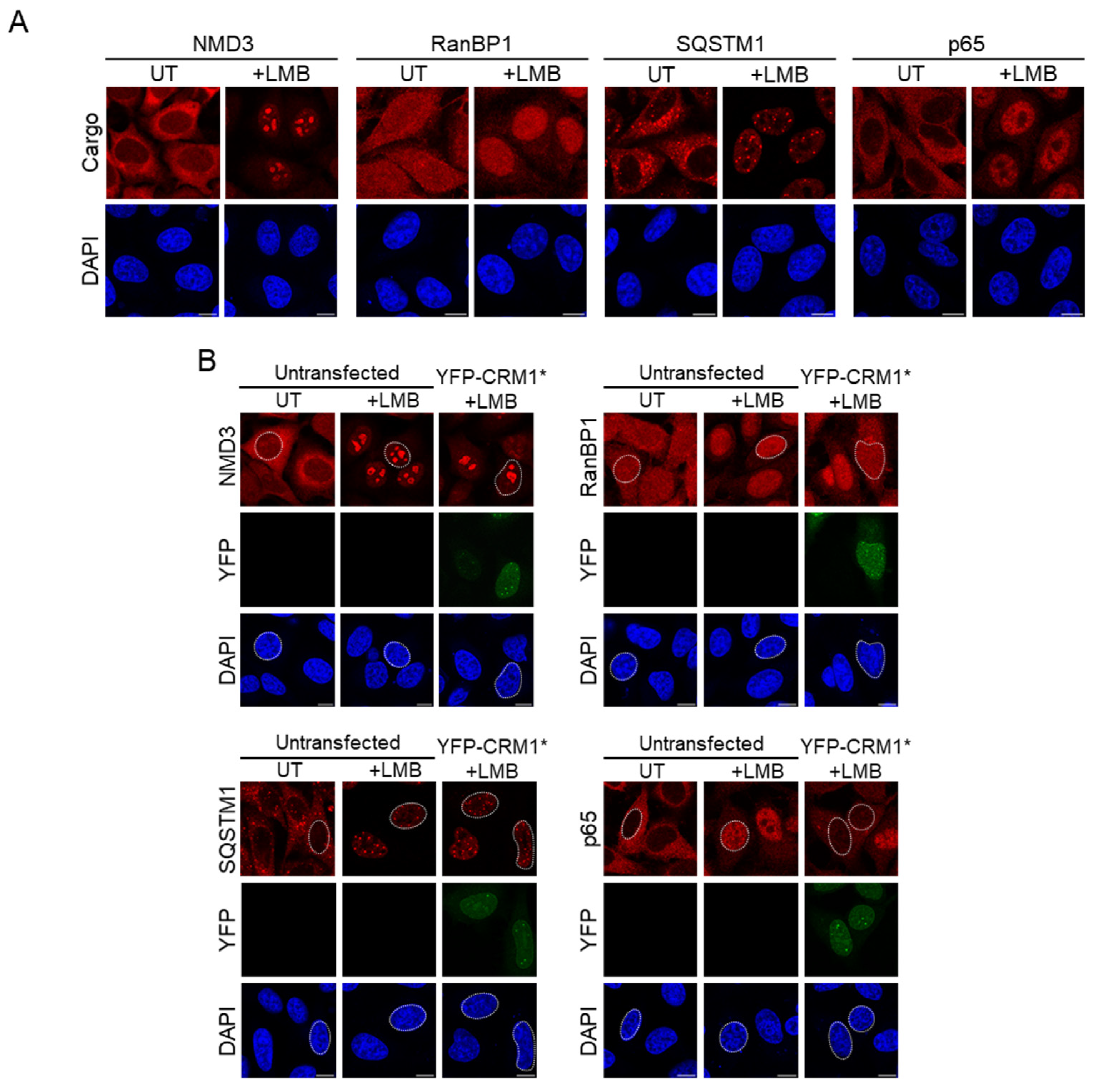
3.3. Selection of Endogenous Cargos to Evaluate the Nuclear Export Activity of CRM1 Mutants
3.4. CRM1 Mutations Differently Alter Nuclear Export of RanBP1 and p65
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sendino, M.; Omaetxebarria, M.J.; Rodriguez, J.A. Hitting a moving target: Inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer. Cancer Drug Resist. 2018, 1, 139–163. [Google Scholar] [CrossRef]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1—From biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Wolff, B.; Sekimoto, T.; Schreiner, E.P.; Yoneda, Y.; Yanagida, M.; Horinouchi, S.; Yoshida, M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 1998, 242, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Selinexor: First Global Approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef]
- Kasamon, Y.L.; Price, L.S.L.; Okusanya, O.O.; Richardson, N.C.; Li, R.-J.; Lian, M.L.; Wu, Y.-T.; Theoret, M.; Pazdur, R.; Gormley, N.J. FDA Approval Summary: Selinexor for Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Oncologist 2021, 26, 879–886. [Google Scholar] [CrossRef]
- Hutten, S.; Kehlenbach, R.H. CRM1-mediated nuclear export: To the pore and beyond. Trends Cell Biol. 2007, 17, 193–201. [Google Scholar] [CrossRef]
- Petosa, C.; Schoehn, G.; Askjaer, P.; Bauer, U.; Moulin, M.; Steuerwald, U.; Soler-López, M.; Baudin, F.; Mattaj, I.W.; Müller, C.W. Architecture of CRM1/Exportin1 suggests how cooperativity is achieved during formation of a nuclear export complex. Mol. Cell. 2004, 16, 761–775. [Google Scholar] [CrossRef]
- Monecke, T.; Güttler, T.; Neumann, P.; Dickmanns, A.; Görlich, D.; Ficner, R. Crystal structure of the nuclear export receptor CRM1 in complex with snurportin1 and RanGTP. Science 2009, 324, 1087–1091. [Google Scholar] [CrossRef]
- Dong, X.; Biswas, A.; Süel, K.E.; Jackson, L.K.; Martinez, R.; Gu, H.; Chook, Y.M. Structural basis for leucine-rich nuclear recognition by CRM1. Nature 2009, 458, 1136–1141. [Google Scholar] [CrossRef]
- Güttler, T.; Madl, T.; Neumann, P.; Deichsel, D.; Corsini, L.; Monecke, T.; Ficner, R.; Sattler, M.; Görlich, D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat. Struct. Mol. Biol. 2010, 17, 1367–1377. [Google Scholar] [CrossRef]
- Dong, X.; Biswas, A.; Chook, Y.M. Structural basis for assembly and disassembly of the CRM1 nuclear export complex. Nat. Struct. Mol. Biol. 2009, 16, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Matsuura, Y. An allosteric mechanism to displace nuclear export cargo from CRM1 and RanGTP by RanBP1. EMBO J. 2010, 29, 2002–2013. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.Y.; Chook, Y.M. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin. Cancer Biol. 2014, 27, 52–61. [Google Scholar] [CrossRef] [PubMed]
- García-Santisteban, I.; Arregi, I.; Alonso-Mariño, M.; Urbaneja, M.A.; García-Vallejo, J.J.; Bañuelos, S.; Rodríguez, J.A. A cellular reporter to evaluate CRM1 nuclear export activity: Functional analysis of the cancer-related mutant E571K. Cell. Mol. Life Sci. 2016, 73, 4685–4699. [Google Scholar] [CrossRef] [PubMed]
- Sendino, M.; Omaetxebarria, M.J.; Prieto, G.; Rodriguez, J.A. Using a simple cellular assay to map NES motifs in cancer-related proteins, gain insight into CRM1-mediated NES export, and search for NES-harboring micropeptides. Int. J. Mol. Sci. 2020, 21, 6341. [Google Scholar] [CrossRef]
- Fung, H.Y.J.; Fu, S.C.; Chook, Y.M. Nuclear export receptor CRM1 recognizes diverse conformations in nuclear export signals. eLife 2017, 6, e23961. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, J.; Min, S.; Zhang, Y.; Zhang, Y.; Zhou, Q.; Shen, X.; Jia, D.; Han, J.; Sun, Q. Distinct RanBP1 nuclear export and cargo dissociation mechanisms between fungi and animals. eLife 2019, 8, e41331. [Google Scholar] [CrossRef]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef]
- Kang, S.S.; Shin, S.H. Phosphorylation of human chromosome maintenance 1 mediates association with 14-3-3 proteins. Anim. Cells Syst. 2013, 17, 186–195. [Google Scholar] [CrossRef]
- Martin, A.P.; Jacquemyn, M.; Lipecka, J.; Chhuon, C.; Aushev, V.N.; Meunier, B.; Singh, M.K.; Carpi, N.; Piel, M.; Codogno, P.; et al. STK38 kinase acts as XPO1 gatekeeper regulating the nuclear export of autophagy proteins and other cargoes. EMBO Rep. 2019, 20, e48150. [Google Scholar] [CrossRef]
- Martin, A.P.; Aushev, V.N.; Zalcman, G.; Camonis, J.H. The STK38-XPO1 axis, a new actor in physiology and cancer. Cell Mol. Life Sci. 2021, 78, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Boulon, S.; Verheggen, C.; Jady, B.E.; Girard, C.; Pescia, C.; Paul, C.; Ospina, J.K.; Kiss, T.; Matera, A.G.; Bordonné, R.; et al. PHAX and CRM1 are required sequentially to transport U3 snoRNA to nucleoli. Mol. Cell. 2004, 16, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, J. A regulatory role for CRM1 in the multi-directional trafficking of splicing snRNPs in the mammalian nucleus. J. Cell Sci. 2007, 120, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Pradet-Balade, B.; Girard, C.; Boulon, S.; Paul, C.; Azzag, K.; Bordonné, R.; Bertrand, E.; Verheggen, C. CRM1 controls the composition of nucleoplasmic pre-snoRNA complexes to licence them for nucleolar transport. EMBO J. 2011, 30, 2205–2218. [Google Scholar] [CrossRef]
- Massenet, S.; Bertrand, E.; Verheggen, C. Assembly and trafficking of box C/D and H/ACA snoRNPs. RNA Biol. 2017, 14, 680–692. [Google Scholar] [CrossRef]
- Hebert, M.D.; Poole, A.R. Towards an understanding of regulating Cajal body activity by protein modification. RNA Biol. 2017, 14, 761–778. [Google Scholar] [CrossRef]
- Staněk, D. Cajal bodies and snRNPs—Friends with benefits. RNA Biol. 2017, 14, 671–679. [Google Scholar] [CrossRef]
- Ernoult-Lange, M.; Wilczynska, A.; Harper, M.; Aigueperse, C.; Dautry, F.; Kress, M.; Weil, D. Nucleocytoplasmic traffic of CPEB1 and accumulation in Crm1 nucleolar bodies. Mol. Biol. Cell. 2009, 20, 176–187. [Google Scholar] [CrossRef]
- Bai, B.; Moore, H.M.; Laiho, M. CRM1 and its ribosome export adaptor NMD3 localize to the nucleolus and affect rRNA synthesis. Nucleus 2013, 4, 315–325. [Google Scholar] [CrossRef]
- Rodríguez, J.A.; Henderson, B.R. Identification of a functional nuclear export sequence in BRCA1. J. Biol. Chem. 2000, 275, 38589–38596. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.E.; Chan, E.K.; Raska, I.; Peebles, C.L.; Roos, G.; Tan, E.M. Human autoantibody to a novel protein of the nuclear coiled body: Immunological characterization and cDNA cloning of p80-coilin. J. Exp. Med. 1991, 173, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Ospina, J.K.; Gonsalvez, G.B.; Bednenko, J.; Darzynkiewicz, E.; Gerace, L.; Matera, A.G. Cross-talk between snurportin1 subdomains. Mol. Biol. Cell. 2005, 16, 4660–4671. [Google Scholar] [CrossRef]
- Lindström, M.S.; Klangby, U.; Inoue, R.; Pisa, P.; Wiman, K.G.; Asker, C.E. Immunolocalization of human p14(ARF) to the granular component of the interphase nucleolus. Exp. Cell Res. 2000, 256, 400–410. [Google Scholar] [CrossRef]
- Thomas, F.; Kutay, U. Biogenesis and nuclear export of ribosomal subunits in higher eukaryotes depend on the CRM1 export pathway. J. Cell Sci. 2003, 116, 2409–2419. [Google Scholar] [CrossRef]
- Trotta, C.R.; Lund, E.; Kahan, L.; Johnson, A.W.; Dahlberg, J.E. Coordinated nuclear export of 60S ribosomal subunits and NMD3 in vertebrates. EMBO J. 2003, 22, 2841–2851. [Google Scholar] [CrossRef]
- Plafker, K.; Macara, I.G. Facilitated nucleocytoplasmic shuttling of the Ran binding protein RanBP1. Mol. Cell Biol. 2000, 20, 3510–3521. [Google Scholar] [CrossRef]
- Guarguaglini, G.; Renzi, L.; D’Ottavio, F.; Di Fiore, B.; Casenghi, M.; Cundari, E.; Lavia, P. Regulated Ran-binding protein 1 activity is required for organization and function of the mitotic spindle in mammalian cells in vivo. Cell Growth Differ. 2000, 11, 455–465. [Google Scholar]
- Pankiv, S.; Lamark, T.; Bruun, J.A.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar] [CrossRef]
- Johnson, C.; Van Antwerp, D.; Hope, T.J. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J. 1999, 18, 6682–6693. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Kudo, N.; Yoshida, M.; Miyamoto, S. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc. Natl. Acad. Sci. USA 2000, 97, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Sun, S.C. Regulation of RelA subcellular localization by a putative nuclear export signal and p50. Mol. Cell Biol. 1999, 19, 7088–7095. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Kamieniarz, K.; Schneider, R. Tools to tackle protein acetylation. Chem. Biol. 2009, 16, 1027–1029. [Google Scholar] [CrossRef]
- Peng, T.; Das, T.; Ding, K.; Hang, H.C. Functional analysis of protein post-translational modifications using genetic codon expansion. Protein Sci. 2023, 32, e4618. [Google Scholar] [CrossRef]
- Rodriguez, J.A. The “STK38-XPO1 axis”: Its general relevance and mechanistic underpinnings remain to be further characterized. Cell Mol. Life Sci. 2021, 78, 4451–4452. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omaetxebarria, M.J.; Sendino, M.; Arrizabalaga, L.; Mota, I.; Zubiaga, A.M.; Rodríguez, J.A. Mutations of Key Functional Residues in CRM1/XPO1 Differently Alter Its Intranuclear Localization and the Nuclear Export of Endogenous Cargos. Biomolecules 2024, 14, 1578. https://doi.org/10.3390/biom14121578
Omaetxebarria MJ, Sendino M, Arrizabalaga L, Mota I, Zubiaga AM, Rodríguez JA. Mutations of Key Functional Residues in CRM1/XPO1 Differently Alter Its Intranuclear Localization and the Nuclear Export of Endogenous Cargos. Biomolecules. 2024; 14(12):1578. https://doi.org/10.3390/biom14121578
Chicago/Turabian StyleOmaetxebarria, Miren Josu, Maria Sendino, Liher Arrizabalaga, Irune Mota, Ana Maria Zubiaga, and José Antonio Rodríguez. 2024. "Mutations of Key Functional Residues in CRM1/XPO1 Differently Alter Its Intranuclear Localization and the Nuclear Export of Endogenous Cargos" Biomolecules 14, no. 12: 1578. https://doi.org/10.3390/biom14121578
APA StyleOmaetxebarria, M. J., Sendino, M., Arrizabalaga, L., Mota, I., Zubiaga, A. M., & Rodríguez, J. A. (2024). Mutations of Key Functional Residues in CRM1/XPO1 Differently Alter Its Intranuclear Localization and the Nuclear Export of Endogenous Cargos. Biomolecules, 14(12), 1578. https://doi.org/10.3390/biom14121578