Abstract
Research on autism spectrum disorders (ASDs) has so far focused primarily on the gut–brain axis, whereas the role of the gut–liver axis remains insufficiently explored. The aim of this study was to evaluate whether microbiota derived from girls with ASD induces dysbiotic changes in gut microbiota composition and leads to alterations in gut–liver axis processes in pseudo-germ-free (PGF) BALB/c mice. We also examined whether these processes could be modulated by altering the gut microbiota using the probiotic strains Lactiplantibacillus plantarum CCM 7512 and Limosilactobacillus reuteri CCM 8617 in combination with ground flaxseed (Linum usitatissimum L.) as a source of omega-3 polyunsaturated fatty acids and fermentable fiber. Colonization with fecal microbiota derived from girls with ASD resulted in dysbiotic changes in the composition of the cecal microbiota, characterized by an increased relative abundance of Escherichia–Shigella, Fusobacterium, Alistipes, and the Ruminococcus gnavus group. These changes were accompanied by impaired intestinal mucosal integrity, altered metabolomic pathways related mainly to aromatic amino acids and lipid metabolism, increased hepatic immunoreactivity of iNOS and COX-2, and elevated activity of the liver-specific LDH-5 isoenzyme. These results suggest that synbiotic intervention contributed to remodeling of the cecal microbiota composition, restoration of intestinal epithelial integrity, and modulation of metabolomic pathways, which was reflected by reduced immunoreactivity of iNOS and COX-2 in liver tissue and decreased activity of the LDH-5 isoenzyme. These findings support the role of microbiota-mediated metabolic processes in communication between the gut and the liver within the gut–liver axis.
1. Introduction
Autism spectrum disorder (ASD) is a multifactorial, highly heritable neurodevelopmental disorder characterized by impairments in social interaction, communication, stereotyped behaviors, restricted interests, and food selectivity [1,2]. Individuals with ASD frequently present with gastrointestinal comorbidities such as constipation, diarrhea, or abdominal discomfort, which are often associated with alterations in the composition and abundance of the gut microbiota. Although dysbiosis has been repeatedly reported in patients with ASD, a definitive microbial signature of the disorder has not yet been identified [3,4].
The gut microbiota plays an important role in the regulation of metabolic and immunological processes of the host, as demonstrated by numerous experimental and clinical studies [5,6]. Microorganisms colonizing the intestine participate in the fermentation of indigestible dietary components, the production of short-chain fatty acids (SCFAs), vitamin synthesis, and the modulation of host immune responses. Due to the broad range of these metabolic and regulatory functions, the gut microbiota is often described as a metabolically active organ that substantially contributes to maintaining physiological homeostasis of the host [7]. Metabolites and other bioactive molecules produced by gut microorganisms may enter the systemic circulation and influence the function of distant organs, including the liver [8]. One of the key mechanisms mediating interactions between the intestine and other organs is the gut–liver axis, which represents a functional connection between the intestine, its microbiota, and the liver. This bidirectional communication is primarily mediated by the portal circulation, through which metabolites, microbial products, and other molecules originating from the intestine are transported directly to the liver. At the same time, the liver influences the intestinal environment and shapes the composition of the gut microbiota through the secretion of bile and immunologically active molecules [9,10]. An important role in regulating this communication is played by the intestinal mucosal and vascular barrier, which represents a functional and anatomical boundary controlling the translocation of microorganisms, toxins, and metabolites into the systemic circulation. The integrity of this barrier is essential for maintaining the homeostasis of the gut–liver axis, whereas its disruption may lead to increased translocation of microbial products, such as lipopolysaccharides (LPSs) or other microbial metabolites, into the portal circulation and the subsequent activation of inflammatory processes in the liver [6,8,9].
In the context of ASD, increasing attention has therefore been devoted to the modulation of the gut microbiota as a potential approach to influencing gastrointestinal disorders frequently reported in patients with ASD. Among the investigated strategies is the use of probiotic microorganisms and nutritional components with prebiotic effects, which may contribute to modifying the composition of the gut microbiota and its metabolic activity. Our previous experimental studies demonstrated that selected probiotic strains of the genus Lactobacillus, particularly Limosilactobacillus reuteri CCM 8617, in combination with nutritional components possessing prebiotic effects, can effectively modulate the composition of the gut microbiota and the metabolic activity of the intestinal environment [11,12]. Experimental findings have also indicated that supplementation with flaxseed, as a source of omega-3 polyunsaturated fatty acids and dietary fiber, may influence fatty acid metabolism, the composition of the gut microbiota, and selected biochemical indicators of tissue damage [13].
Despite the growing interest in the role of the gut microbiota in the pathophysiology of ASD, most studies to date have focused primarily on interactions within the microbiota–gut–brain axis and their influence on the neurobehavioral manifestations of the disorder. These mechanisms have been investigated mainly in various experimental models of ASD, including genetic and environmentally induced mouse models [14,15]. In recent years, experimental approaches based on fecal microbiota transplantation (FMT) have also been used in this context, allowing the transfer of microbiota from patients with ASD to an animal host and enabling the evaluation of its effects on host physiological and behavioral outcomes [16]. Nevertheless, the potential role of the gut–liver axis in the context of gastrointestinal comorbidities accompanying ASD remains insufficiently explored.
The aim of this study was to evaluate in pseudo-germ-free mice (PGF) whether fecal microbiota transplantation from children with ASD induces a dysbiotic gut environment and leads to alterations of the gut–liver axis, including changes in liver tissue. In addition, we aimed to determine whether targeted modulation of the intestinal microbiota using a synbiotic intervention could mitigate these microbiota-induced alterations.
2. Materials and Methods
2.1. Experimental Design and Animal Husbandry
The in vivo experiments were performed using 106 specific pathogen-free (SPF) female BALB/c mice (6 weeks old), obtained from the breeding facility Velaz s.r.o. (Prague, Czech Republic). Animals were transported by air in dedicated transport containers to the accredited Laboratory of Gnotobiology at the Department of Microbiology and Immunology, University of Veterinary Medicine and Pharmacy in Kosice, Slovakia (SK U 10021). Upon arrival, mice were housed in a gnotobiotic animal facility equipped with EHRET THF 3271IE 101/97 isolators (EHRET Labor- und Pharmatechnik GmbH & Co. KG, Emmendingen, Germany) and two-part glove isolators (CBC Ltd., Madison, WI, USA). Animals were maintained under barrier conditions and had ad libitum access to an irradiated complete pelleted diet formulated for laboratory mice (Ssniff Spezialdiäten GmbH, Soest, Germany). The diet contained 22% crude protein, 3.9% crude fiber, 4.5% crude fat, 6.7% ash, 0.7% calcium, and 0.5% phosphorus, and was supplemented with trace elements (iron 100 mg/kg, zinc 100 mg/kg, manganese 30 mg/kg, selenium 0.10 mg/kg, copper 5 mg/kg) and vitamins (vitamin A 28,000 IU/kg, vitamin D3 2200 IU/kg, vitamin E 100 mg/kg). Autoclaved drinking water was supplied in glass bottles without restriction. Sterilized bedding material (Lignocel 3-4S, JRS, Rosenberg, Germany) treated by irradiation was used throughout this study. Environmental conditions within the gnotobiotic units were strictly controlled. The temperature inside the isolators was maintained between 20 and 24 °C via air exchange with a centrally heated gnotobiotic room. Relative humidity was kept within the range of 45–65% by regular bedding replacement and continuous HEPA-based air filtration (CBC Ltd., Madison, WI, USA). The isolator filtration systems ensured 10–15 air exchanges per hour under positive pressure (50–70 kPa), with an airflow rate of 8–30 m3/h.
In the first stage of the procedure, all animals underwent antibiotic-mediated depletion of the gut microbiota to establish a pseudo-germ-free model, as described in Section 2.2. According to the experimental design shown in Figure 1a, Phase I represented the period of antibiotic decontamination, Phase II represented the period following fecal microbiota transplantation (FMT), and Phase III represented the period after synbiotic additive administration. After antibiotic treatment, mice were randomly assigned to four experimental groups: the control group (C; n = 30), NT group (receiving FMT from neurotypical donors; n = 30), ASD group (receiving FMT from ASD donors; n = 23), and ASD+pro/pre group (receiving FMT from ASD donors followed by synbiotic intervention; n = 23). Synbiotic intervention was administered exclusively to the ASD+pro/pre group and included per os administration of probiotic cultures (Lactiplantibacillus plantarum CCM 7512 and Limosilactobacillus reuteri CCM 8617; each inoculum contained 1 × 109 CFU/mL) at a dose of 0.1 mL/strain/mouse/day for 5 days. Concurrently, the feed was supplemented with irradiated ground flaxseed (Linum usitatissimum L.) of the LIBRA cultivar (Osivo, Lučenec, Slovakia) at a concentration of 4%, which was homogeneously mixed into a powdered form of irradiated feed (Ssniff Spezialdiäten GmbH, Soest, Germany). The polyunsaturated fatty acid (PUFA) composition of LIBRA flaxseed was characterized by a high content of omega-3 fatty acids, predominantly α-linolenic acid (ALA; 54.9%), and omega-6 fatty acids, mainly linoleic acid (17.1%).
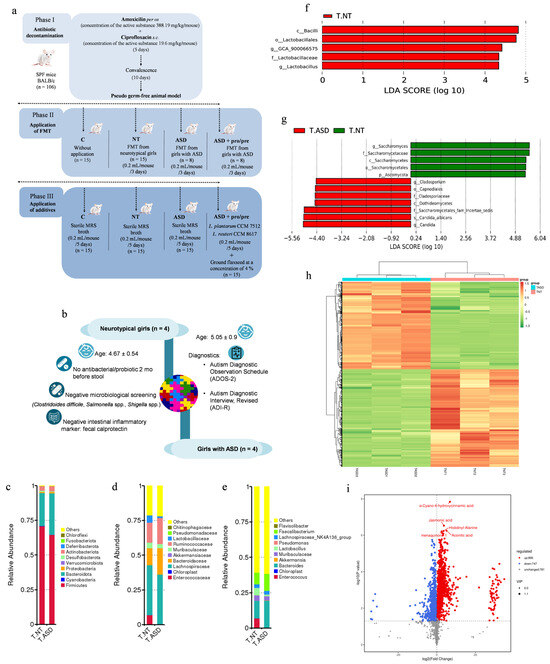
Figure 1.
Experimental design of this study (a), FMT donor selection (b), and composition of fecal microbiota transplants derived from neurotypical girls (T.NT, n = 4) and girls with autism spectrum disorder (T.ASD, n = 4); bacterial microbiota (c–f), gut mycobiome (g), and metabolomic profiles (h,i). (c) Relative abundance of the top 10 bacterial phyla. (d) Relative abundance of the top 10 bacterial families. (e) Relative abundance of the most abundant bacterial genera. (f) Histogram of linear discriminant analysis (LDA) scores identifying bacterial taxa that significantly discriminate between T.ASD and T.NT transplants (red), meeting the LDA significance threshold of log10 > 2. (g) Histogram of linear discriminant analysis (LDA) scores identifying differentially abundant taxa of the gut mycobiome at multiple taxonomic levels between T.ASD (red) and T.NT (green) transplants, meeting the LDA significance threshold of log10 > 2. (h) Heatmap of standardized metabolite abundances (z-scores) across T.NT and T.ASD transplants, each represented by three technical replicates (T.NT1–T.NT3 and T.ASD1–T.ASD3). (i) Volcano plot illustrating differential metabolite abundance between T.NT and T.ASD transplants, where the x-axis represents log2(T.NT/T.ASD) fold change and the y-axis shows −log10(p-value); red dots indicate metabolites significantly increased in T.NT, blue dots represent metabolites significantly increased in T.ASD, and gray dots correspond to non-significant changes; metabolites were considered significantly altered at p < 0.05 and |log2(T.NT/T.ASD)| ≥ 1. Fecal microbiota transplants were generated by pooling samples from four donors per group; three independent pooled biological replicates per group were included in the analyses. log2 (fold change) ≥ 1. Microbial profiling of the FMT inocula was performed using three technical replicates derived from the same pooled inoculum per group.
2.2. Acquisition of the Pseudo-Germ-Free Model
The PGF animal model was obtained by administering broad-spectrum antibiotics to SPF female BALB/c mice according to the application scheme shown in Figure 1a, as previously described by [17]. During PGF induction, amoxicillin–clavulanate (Amoksiklav 2 × 457 mg/5 mL; Sandoz Pharmaceuticals, Ljubljana, Slovenia) was administered orally at a volume of 0.2 mL (387.11 mg/kg body weight), while ciprofloxacin (Ciprinol con infusione 5 × 10 mL/100 mg; Krka d.d., Novo Mesto, Slovenia) was administered subcutaneously at a volume of 0.1 mL (19.60 mg/kg body weight). Both antibiotics were applied every 12 h for five consecutive days.
2.3. Selection and Screening of Fecal Microbiota Donors
Psychological and psychiatric examinations were performed at The Academic Research Center for Autism at the Institute of Physiology, Faculty of Medicine, Comenius University (CU) in Bratislava, Slovakia. Donors for ASD FMT (Figure 1b) were girls diagnosed with ASD using the gold-standard diagnostic tools, including the Autism Diagnostic Observation Schedule, Second Edition (ADOS-2) [18] and the Autism Diagnostic Interview, Revised (ADI-R) [19] with a comparative score greater than seven. Neurotypical donors were recruited from the local population. The mean age of ASD and neurotypical donors was 5.05 ± 0.9 and 4.67 ± 0.54 years, respectively, with no significant difference between groups. Donors had not received any antibiotic or probiotic treatment for at least 2 months, maintained a BMI within the normal range for age, and showed no symptoms of acute gastrointestinal disorders or other infections within the preceding 2 months.
2.4. Fecal Microbiota Collection and Processing
Stool sample collection was performed at home by parents of neurotypical and ASD donors into sterile containers following detailed instructions. The samples were delivered to the laboratory within 2 h, and fecal microbiota transplants were promptly prepared. The filtered material was mixed with glycerol (final concentration 10%), frozen, and stored at −80 °C for up to 6 months [20]. To ensure biosafety, all fecal samples were screened for bacterial, fungal, and parasitic pathogens at the Institute of Microbiology, Faculty of Medicine, (CU) in Bratislava. Additionally, intestinal inflammation was assessed by measuring calprotectin concentrations using the CALPROLAB™ Calprotectin ELISA (ALP) (Calprolab, Lysaker, Norway) according to the manufacturer’s instructions. Fecal calprotectin concentrations below 50 μg/g of feces were considered within the normal range, based on the established criteria [21]. Microbial transplants were prepared by pooling equal volumes of fecal material from four donors (1 mL per donor). Prior to administration, the transplants were slowly thawed for 8 h at 4 °C and subsequently processed under sterile conditions in a class II laminar flow cabinet (Telstar Bio II Advance, Terrassa, Spain), and homogenized. The transplants were administered per os at a volume of 0.2 mL/mouse/day for three consecutive days.
2.5. Preparation of Probiotic Strains for Administration to Mice
Probiotic strains Limosilactobacillus reuteri CCM 8617 and Lactiplantibacillus plantarum CCM 7512 were isolated at the Laboratory of Gnotobiology (Department of Microbiology and Immunology, Kosice, UVMP, Slovakia) and were deposited in the Czech Collection of Microorganisms (CCM, Masaryk University, Brno, Czech Republic). A rifampicin-resistant variant of Limosilactobacillus reuteri CCM 8617 (designated CCM 8617RIF2) was prepared as previously described by [12]. Individual strains were cultivated separately by static incubation in MRS broth (HiMedia Laboratories GmbH, Modautal, Germany) under anaerobic conditions (BBL GasPak™ Plus, BD, Franklin Lakes, NJ, USA) at 37 °C for 24 h. The entire 24 h culture, including the cultivation medium, was used for administration without further processing. The final concentration of each inoculum was 1 × 109 CFU/mL. The inocula were not stored and were administered immediately after cultivation.
2.6. DNA Isolation and Sequencing of Bacterial Microbiota and Fungal Communities
Total microbial DNA was extracted from fecal microbiota transplants derived from neurotypical and ASD donors, as well as from fecal samples collected from experimental mice. Each FMT inoculum was prepared by pooling equal volumes of fecal material from four donors per group (neurotypical and ASD, respectively), as described in Section 2.4. For next-generation sequencing (NGS)-based microbial profiling of the inocula, three technical replicates were generated from the same pooled material per group. Approximately 200 mg of each sample was processed using the Quick-DNA™ Fecal/Soil Microbe Miniprep Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. DNA concentration and purity were determined spectrophotometrically using a NanoDrop™ OneC UV–Vis spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) prior to downstream analyses. The isolated DNA was used for characterization of the bacterial and fungal communities using next-generation sequencing (NGS). Library preparation, sequencing, and primary bioinformatic processing were carried out by Novogene Co., Ltd. (Munich, Germany). Briefly, amplification of the V3–V4 regions of the bacterial 16S rRNA gene was carried out using the universal primer pair 341F (5′-CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACNNGGGTATCTAAT-3′). The resulting libraries were sequenced on an Illumina MiSeq instrument generating 2 × 300 bp paired-end reads. Fungal community profile was assessed based on the ITS1 profiling using primers ITS5-1737F (5′-GGAAGTAAAAGTCGTAACAAGG-3′) and ITS2-2043R (5′-GCTGCGTTCTTCATCGATGC-3′), and sequencing was conducted on the same platform.
2.7. Metabolomic Analysis
Fecal samples were subjected to non-targeted metabolomic profiling performed by Biomarker Technologies (BMKGENE, Münster, Germany), including sample processing, LC-MS and statistical analysis. Briefly, 100 mg of each sample was thawed on ice, then homogenized using bead beating and sonicated in cold organic solvent to precipitate proteins and extract metabolites. The mixtures were centrifuged at 12,000× g for 10 min at 4 °C, after which the supernatants were collected, dried under vacuum, and reconstituted prior to LC–MS analysis. Chromatographic separation was performed on a Waters Acquity UPLC HSS T3 column using a Waters ACQUITY I-Class PLUS UHPLC system coupled to a Waters Xevo G2-XS QTOF mass spectrometer (Waters Corporation, Milford, MA, USA). Data acquisition in both positive and negative electrospray ionization modes was carried out using MassLynx v4.2 (Waters Corporation). Peak detection, alignment, and normalization were carried out in Progenesis QI (version 2.4.6911, Nonlinear Dynamics, Waters). Metabolites were annotated using accurate mass, retention time, and MS/MS spectra matched against several databases, including HMDB [22] (MR1.1), KEGG [23] (MR2.1), LIPID MAPS [24] (MR3.1), METLIN [25] (MR4.1), and the BMKGENE internal library. Quantification was performed using relative peak areas. Quality control samples, prepared by pooling equal portions of all study samples, were analyzed at regular intervals to track instrument performance and reproducibility. Differential metabolites were defined by VIP > 1.0 and p < 0.05. Pathway and functional enrichment analyses were conducted using KEGG to interpret the biological relevance of the identified metabolites.
2.8. Spectrophotometric Analysis of Total Lactate Dehydrogenase
Serum samples (n = 5 per group) were collected for the determination of total lactate dehydrogenase (TLDH) (μkat/L; RX Monaco, Randox, UK). Liver tissues (n = 5 per group) were collected individually at necropsy and immediately stored at −80 °C until analysis. Prior to homogenization, the tissues were sectioned and thoroughly rinsed in isotonic saline to remove residual blood and connective tissue. The samples were then homogenized at a 1:10 (w/v) ratio in ice-cold buffer (0.05 M Tris–HCl, pH 7.3), following the protocol described [26]. Total lactate dehydrogenase (TLDH) activity in the resulting tissue extracts was measured using a commercial assay kit (LD Pyruvate–Lactate [LDH P-L]) on an automated biochemical analyzer (µkat/L; RX Monaco, Randox, UK). Absorbance was recorded at 340 nm (primary wavelength) and 405 nm (secondary wavelength), and net absorbance was calculated as the difference between these readings. Tissue extracts were also analyzed for total protein analysis (g/L; RX Monaco, Randox, UK). Enzyme activity is expressed as international units per gram of protein (IU/g protein).
2.9. Electrophoresis Analysis of Lactate Dehydrogenase Isoenzymes
Hydragel 15 ISO-LDH electrophoresis kits (Sebia Slovakia s.r.o., Bratislava, Slovakia) were used for the separation of individual LDH isoenzymes. Relative proportions of LDH isoenzymes were quantified by densitometric scanning of electrophoretograms at 570 nm using a densitometer and analyzed with PHORESIS software (version 9.20; Sebia, Lisses, France).
2.10. Histological and Immunohistochemical Analysis
Segments of the caudal colon (1–2 cm; n = 6 per group) and liver tissue samples (0.5–1 cm; n = 6 per group) were rinsed with cold physiological saline and fixed in 4% paraformaldehyde. Fixed tissues were dehydrated through a graded ethanol series, cleared in xylene, and embedded in paraffin (Paraplast, Sigma-Aldrich, St. Louis, MO, USA).
For histological evaluation, sections were deparaffinized in xylene, rehydrated through a descending ethanol series, and stained with Harrison’s hematoxylin (Sigma-Aldrich, St. Louis, MO, USA) and eosin Y (Merck, Darmstadt, Germany) to assess overall tissue morphology. Stained sections were subsequently dehydrated, cleared in xylene, mounted using a synthetic mounting medium (Entellan™ New, Merck, Darmstadt, Germany), and coverslipped. Histopathological changes in colonic tissue were evaluated using the Histological Activity Index (HAI). To minimize subjective bias, all analyses were performed in a blinded manner, with evaluators unaware of the experimental group allocation. Each section was independently assessed by two experienced pathologists. Histological scoring was performed in 50 randomly selected microscopic fields per section. The following parameters were evaluated: epithelial erosion (0—morphologically normal; 1—focal destruction; 2—zonal destruction; 3—diffuse and mucosal ulcerations), crypt damage (0—none; 1—some crypt damage, spaces between crypts; 2—large spaces between crypts; 3—large spaces without crypts, surrounded by normal crypts; 4—no crypts), and infiltration of inflammatory cells (0—absence of infiltrate; 1—infiltrate at the subepithelial and lamina propria; 2—infiltrate reaching muscularis mucosae; 3—severe and extensive infiltrate reaching submucosa and involving muscularis propria). Microscopic analysis was performed using an Axiolab light microscope (Carl Zeiss, Jena, Germany), and representative microphotographs were acquired and analyzed using Image C software (version 2.52, Imtronic GmbH, Berlin, Germany).
For immunohistochemical analysis, paraffin-embedded liver tissue samples were cut into 5 μm-thick sections using a Leica RM2255 microtome (Leica Microsystems Nussloch GmbH, Nussloch, Germany). The sections were rehydrated and subjected to heat-induced epitope retrieval in citrate buffer (pH 6.0) by bringing the solution to boiling. Endogenous peroxidase activity was blocked with 1% H2O2 in PBS for 30 min. Non-specific binding was blocked by incubation in 1% bovine serum albumin (BSA; Sigma-Aldrich, MO, USA) in PBST for 1 h. Primary antibodies against cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS; Santa Cruz Biotechnology Inc., Dallas, TX, USA) were applied at dilutions of 1:50 for both antibodies, followed by overnight incubation at 4 °C. After washing in PBST, sections were incubated with secondary antibodies (Dako REAL EnVision/HRP, Rabbit/Mouse [ENV], Dako, Glostrup, Denmark) for 2 h. Following additional washes in PBST and PBS, immunoreactivity was visualized using 3,3′-diaminobenzidine tetrahydrochloride (DAB; Dako REAL DAB+ Chromogen, Dako, Glostrup, Denmark) as the chromogen. Sections were counterstained with hematoxylin and subsequently mounted in Pertex mounting medium (Pertex Mounting Media, Gothenburg, Sweden). Positive immunoreactivity was identified based on gray level (GL) intensity values obtained from microphotographs captured using a light microscope (Optica Microscope B-290TB, Ponteranica, Italy). Quantitative image analysis was performed using ImageJ software (version 1.53t, National Institutes of Health, Bethesda, MD, USA). Further procedures were conducted according to [27].
2.11. Bioinformatic and Statistical Analysis
Statistical analysis of the microbiome and mycobiome data was performed by Novogene Co., Ltd. (Munich, Germany). Briefly, raw paired-end reads were demultiplexed, and trimmed using cutadapt (v3.3) [28]. Forward and reverse reads were then joined with FLASH (v1.2.11) [29] and quality filtering was performed with fastp (v0.23.1) [30]. Next, chimeric sequences were detected and removed using vsearch (v2.16.0) [31], yielding high-quality reads. Denoising and amplicon sequence variant (ASV) inference were conducted with the DADA2 method [32] within QIIME2 (version 2022.2) [33]. Taxonomy was assigned using the SILVA 138 database for bacteria [34] and UNITE 10.0 for fungi [35]. Species abundance tables were generated at multiple taxonomic ranks based on the annotated ASVs. Abundance values were normalized to the sequencing depth of the sample with the fewest reads, and all downstream analyses were performed on this normalized dataset. Group differences at various taxonomic levels were assessed using complementary statistical approaches, including conventional t-tests (p < 0.05) together with microbiome-oriented differential abundance methods such as Metastat [36], and results were visualized in R (v2.15.3) [37]. Differential taxa were additionally identified using the LEfSe algorithm (Linear Discriminant Analysis Effect Size) following the approach of Segata et al. [38]. These complementary analytical approaches were applied to provide an exploratory assessment of differential abundance patterns in the microbiome data. Regarding the NGS-based microbial profiling of the fecal microbiota transplant (FMT) inocula, it is important to note that three technical replicates were generated from a single pooled sample per donor group (T.NT and T.ASD), with each pool comprising equal contributions from four individual donors. These technical replicates were used to characterize and compare the microbial profiles of the pooled inocula and to assess the reproducibility of sequencing-based profiling. However, they were not treated as independent biological observations for inference on donor-level variability. Accordingly, the resulting analyses describe differences between pooled group-level inocula, whereas donor-level variance cannot be estimated directly from these data.
Metabolome data were statistically analyzed by Biomarker Technologies (BMKGENE, Münster, Germany). After normalizing metabolite data by total peak area using Progenesis QI software (version 2.4.6911, Nonlinear Dynamics, Waters), all subsequent analyses were carried out in R. Hierarchical clustering heatmaps were produced with the pheatmap package (v1.0.2) [39] using UV scaling to illustrate overall similarity patterns among samples and metabolites. Group differences were examined using both univariate and multivariate approaches. Univariate analysis relied on fold change (FC) and t-test-based comparisons (p < 0.05), interpreted in an exploratory manner, whereas multivariate analysis employed Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) with Variable Importance in Projection (VIP > 1), implemented in ropls (v1.6.2) [40], and model validity was evaluated through 200-permutation testing. Metabolites were considered differential when meeting the combined thresholds of |FC| > 1, p < 0.05, and VIP > 1. These features were then analyzed for KEGG-based pathway enrichment using a hypergeometric test.
LDH analyses were conducted in R (version 4.4.1) [37]. Permutation t-tests were performed using the MKinfer package (version 1.2) [41] to assess differences in mean parameter values. The Holm correction was applied for multiple comparisons.
Immunohistochemical (IHC) data for COX-2 and iNOS expression, as well as Histological Activity Index (HAI) scores, were evaluated using one-way analysis of variance (one-way ANOVA). When a significant overall effect was observed, Tukey’s multiple comparison test was applied for pairwise comparisons. All data are presented as mean ± SEM. A significance level of p < 0.05 was used for all analyses.
3. Results and Discussion
3.1. Microbiome, Mycobiome and Metabolomic Profiles of Neurotypical and ASD-Derived FMT
3.1.1. Fecal Microbiota
Based on NGS analysis of FMT obtained from girls with ASD (T.ASD) and neurotypical girls (T.NT), distinct differences in microbial composition were observed across multiple taxonomic levels.
At the phylum level (Figure 1c), Firmicutes were the dominant phylum in both transplants. T.NT contained 70.83% Firmicutes and 23.87% Bacteroidota, whereas FMT (T.ASD) consisted of 64.58% Firmicutes and 30.08% Bacteroidota. These results suggest a largely comparable phylum-level structure, with only minor shifts in the Firmicutes/Bacteroidota ratio. In contrast to these findings, several published studies report an increased Firmicutes/Bacteroidota ratio in children with ASD compared to neurotypical controls, which has been interpreted as a potential marker of gut dysbiosis. Strati et al. [42] reported a higher relative abundance of Firmicutes accompanied by a reduction in Bacteroidota in a cohort of children with ASD, whereas the systematic review and meta-analysis by Lewandowska-Pietruszka et al. [43] demonstrated that an increased Firmicutes/Bacteroidota ratio is not a general characteristic of the ASD population, but was observed only in a subset of the analyzed cohorts, with substantial variability across both studies and individuals.
Among the ten most abundant families in both transplants (Figure 1d) were Lachnospiraceae, Ruminococcaceae, Bacteroidaceae, Lactobacillaceae, and Muribaculaceae, representing the core shared microbiota. Despite this similarity, T.ASD exhibited higher relative abundances of the families Lachnospiraceae (35.50%) and Ruminococcaceae (18.82%) compared to the transplant obtained from neurotypical donors. Both families belong to the phylum Firmicutes, and their increased abundance has been repeatedly reported in previous studies of children with ASD as a potential indicator of dysregulated fermentative processes in the gut. These families comprise metabolically heterogeneous taxa whose functional roles depend on the overall composition and interactions within the microbial ecosystem. In this context, their relative enrichment may be consistent with altered fermentative and metabolic activity reported in ASD-associated microbiota [42,44], although such functional interpretation remains tentative in the present dataset.
Analysis at the genus level (Figure 1e) showed that both transplants shared dominant bacterial genera, with differences primarily reflected in their relative abundances. Among the top 10 most abundant genera were Bacteroides, Faecalibacterium, Lactobacillus, and the Lachnospiraceae NK4A136 group. The genera Bacteroides and Faecalibacterium represented dominant components of the microbiota in both transplants, with comparable relative abundances between T.NT and T.ASD. Faecalibacterium is a major butyrate producer with anti-inflammatory properties, which may suggest preservation of some microbiota-associated SCFA-producing potential. Although several studies have reported reduced Faecalibacterium abundance in ASD [44,45,46], this trend was not observed in the analyzed transplants. In contrast, T.ASD showed a higher relative abundance of the Lachnospiraceae NK4A136 group, which is broadly consistent with previous reports describing altered representation of Lachnospiraceae-related genera in ASD [47]. LEfSe analysis (Figure 1f) revealed differences in taxonomic composition between T.NT and T.ASD. Taxa exhibiting statistically significant differences were limited exclusively to members of the class Bacilli, order Lactobacillales, family Lactobacillaceae, and genus Lactobacillus, whose relative abundance was significantly higher in T.NT. The genus Lactobacillus comprises bacteria involved in carbohydrate fermentation and lactic acid production and is frequently associated with the maintenance of gut homeostasis [46,48,49]. In contrast to the findings of Tomova et al. [48], who described increased representation of the genus Lactobacillus in children with ASD, its relative abundance in the present study was significantly higher in T.NT.
In summary, both transplants exhibited a similar core microbiota, while differences were mainly reflected in subtle but biologically relevant shifts at the family and genus levels. ASD-derived microbiota showed patterns consistent with enrichment of Firmicutes-associated taxa, whereas significant differences were limited to Lactobacillus, which was more abundant in T.NT.
3.1.2. Fecal Mycobiome
LDA of the gut mycobiome (Figure 1g) revealed statistically significant differences in the relative abundance of selected fungal taxa between the T.NT and T.ASD transplants. The T.NT transplant showed a higher representation of the genus Saccharomyces and related taxa within the family Saccharomycetaceae, class Saccharomycetes, order Saccharomycetales, and phylum Ascomycota. Members of the genus Saccharomyces are generally considered non-pathogenic components of the gut mycobiome and are frequently detected in association with dietary intake, particularly fermented foods. Their presence in the gut has been associated with a more stable fungal community structure of the intestinal ecosystem and with a potential role in maintaining intestinal barrier integrity and modulating host immune responses [50,51].
In contrast, the T.ASD transplant exhibited an increased presence of environmental fungal taxa belonging to the genus Cladosporium (order Capnodiales, family Cladosporiaceae, class Dothideomycetes), as well as a higher relative abundance of Candida species, including Candida albicans. The genus Cladosporium comprises common environmental fungi that enter the gut mycobiome predominantly through exogenous exposure, and their increased presence in the intestinal environment is often interpreted as an indicator of disrupted fungal community balance [52]. Under conditions of disrupted gut microbial homeostasis, Candida albicans overgrowth may occur, and its increased abundance has been repeatedly associated with inflammatory responses, impairment of intestinal barrier integrity, and increased intestinal permeability [53,54]. Such alterations may represent part of a broader microbial and fungal dysbiosis, which has been increasingly reported in individuals with ASD, particularly in association with gastrointestinal symptoms and immune dysregulation [50,51].
Mycobiome analysis suggested more pronounced differences between the transplants than bacterial microbiota analysis, with enrichment of Candida representing the main distinguishing feature of ASD-derived FMT.
3.1.3. Comparative Metabolomic Profiling of FMTs
The heatmap of the metabolomic profile (Figure 1h), based on hierarchical clustering, revealed a clear separation between T.NT and T.ASD transplants, indicating systematic differences in their overall metabolic signatures. Similar separation patterns have been reported in previous studies comparing neurotypical individuals and individuals with ASD across different biological matrices, including fecal samples and serum [44,55]. Two major metabolite clusters were identified showing opposite abundance patterns between T.NT and T.ASD. This coordinated shift suggests systematic alterations in microbial metabolic activity rather than random interindividual donor variability. Such metabolite clustering is a characteristic feature of distinct functional states of the microbiota and has been repeatedly observed in ASD-related metabolomic studies [55,56,57]. Importantly, the pronounced separation between T.NT and T.ASD suggests that the metabolomic profiling may capture functional differences more sensitively than taxonomic composition alone, which showed a largely shared core microbiota. This observation is consistent with accumulating evidence that the metabolomic profile of the gut microbiota can differ substantially even when overall taxonomic composition appears relatively similar [58,59,60]. From a biological perspective, these differences may reflect coordinated alterations in fermentative pathways, substrate utilization, and microbiota–host interactions within the gut–brain axis. Building on the global separation observed in the heat map (Figure 1h), volcano analysis (Figure 1i) enabled a more detailed identification of specific metabolites contributing to the differentiation between T.NT and T.ASD.
Metabolites exhibiting significant differences in abundance point to alterations across multiple microbially derived metabolic pathways, further supporting the possibility of differences in the functional metabolic activity of the transplants [61,62]. Among metabolites enriched in T.NT, α-cyano-4-hydroxycinnamic acid may reflect increased presence or microbial processing of dietary phenolic compounds. Phenolic derivatives are frequently associated with antioxidant and regulatory effects in the intestinal environment and are considered indicators of active microbial polyphenol metabolism [55]. Jasmonic acid, a plant-derived lipid compound, has been reported to exert immunomodulatory and anti-inflammatory effects in experimental systems.
Its higher abundance in T.NT may indicate differences in the handling of plant-derived substrates and overall microbial metabolic activity, features commonly linked to a more favorable intestinal environment [56,58]. Elevated histidinyl-alanine levels in T.NT suggest distinct amino acid and proteolytic metabolism of the gut microbiota, consistent with the literature describing balanced amino acid metabolism as a feature of intestinal homeostasis [63,64]. In contrast, metabolites enriched in T.ASD were primarily related to alterations in energy metabolism. Increased aconitic acid may indicate differences in tricarboxylic acid cycle activity, which may reflect altered microbial energy dynamics and substrate availability in the gut ecosystem [44,58]. Elevated levels of menaquinone (vitamin K2) may reflect differences in the abundance of bacteria capable of its biosynthesis, suggesting shifts in microbial metabolic capacity and functional composition [65,66].
These findings suggest that T.NT and T.ASD transplants differ not only in taxonomic composition but, more importantly, in their metabolic output, highlighting metabolomics as a sensitive indicator of microbiota functionality [61].
3.2. Engraftment of Fecal Microbiota from Neurotypical and ASD Donors in PGF Mice
Antibiotic decontamination of the gastrointestinal tract of SPF mice resulted in a pronounced reduction in the original gut microbiota (Figure 2a), reflected by the dominance of a single bacterial genus, Enterococcus (66.28%). Such a microbial profile is characteristic of antibiotic-treated or PGF animal models and facilitates the engraftment of transplanted microbiota [17,67]. These results are consistent with the successful establishment of a PGF animal model using our previously described protocol [68,69].
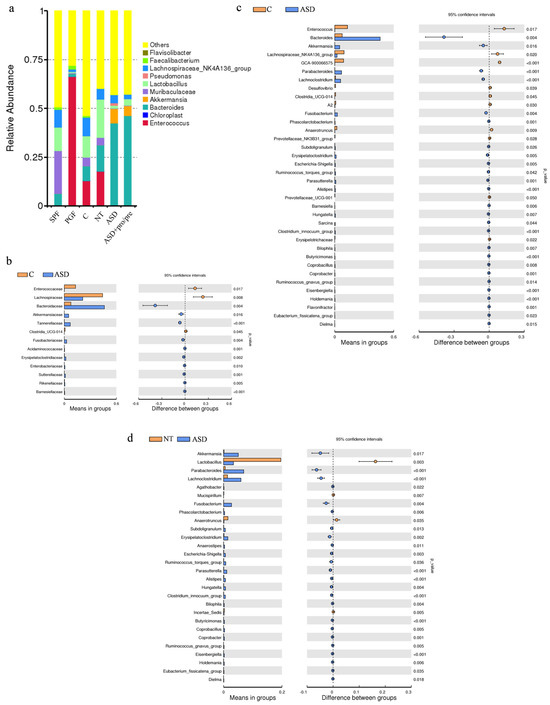
Figure 2.
Gut microbiota composition and differential taxa in PGF mice following fecal microbiota transplantation from neurotypical and ASD donors. (a) Relative abundance of the top 10 bacterial genera in specific pathogen-free animals (SPF; n = 4), pseudo-germ-free animals (PGF; n = 4), the control group (C; without FMT application; n = 4), the neurotypical group (NT; FMT from neurotypical donors; n = 4), and autism spectrum disorder groups (ASD and ASD+pro/pre; FMT from ASD; n = 4 per group). (b) Differential relative abundance analysis at the family level between C and ASD groups. (c) Differential relative abundance analysis at the genus level between C and ASD groups. (d) Differential relative abundance analysis at the genus level between NT and ASD groups. Statistical comparisons were performed using an unpaired two-tailed t-test (p < 0.05; p < 0.01; p < 0.001).
Following FMT administration, differences in microbial community composition were observed across experimental groups, as demonstrated by beta diversity analysis, which showed distinct clustering of fecal microbiota. At the level of bacterial families (Figure 2b), the group receiving T.ASD exhibited a significantly higher relative abundance of the Bacteroidaceae (p = 0.004), Tannerellaceae (p < 0.001), Fusobacteriaceae (p = 0.004), Enterobacteriaceae (p = 0.010), and Sutterellaceae (p = 0.001) compared with the control group. These changes across multiple taxonomic ranks suggest a broader reorganization of the intestinal microbial community following transplantation of ASD-derived microbiota. This pattern is consistent with observations from GF mouse models, where human FMT induced shifts across broader phylogenetic groups [56], as well as with antibiotic-treated models, where engraftment effects are most evident at the family and genus levels [14].
At the genus level (Figure 2a), pronounced differences in microbial composition were observed depending on donor origin. In mice colonized with T.NT, higher relative abundances of Lactobacillus (19.62%) and Enterococcus (17.73%) were observed among the dominant genera, along with contributions from Bacteroides (13.43%), Lachnospiraceae NK_4A136 (5.50%), and Muribaculaceae (3.78%). In contrast, mice colonized with T.ASD (ASD and ASD+pro/pre, Figure 2a) showed a pronounced dominance of Bacteroides (42.15% and 46.22%), while the Lachnospiraceae NK_4A136 (4.22% and 2.33%) and Lactobacillus (1.51% and 3.37%) were present at lower relative abundances. This taxonomic shift toward Bacteroides dominance indicates selective engraftment of the donor microbiota and may reflect differences in the colonization capacity of specific bacterial taxa, as reported in previous human FMT studies [56,70,71].
The increased relative abundance of Escherichia-Shigella (p = 0.005), together with the significant presence of Fusobacterium (p = 0.004), Alistipes (p < 0.001), Ruminococcus gnavus_group (p = 0.005) and Parabacteroides (p < 0.001) in the ASD group (Figure 2c), suggests changes in the structure of the intestinal ecosystem following transplantation of ASD-derived microbiota. These taxa are known to thrive under modified physicochemical conditions including altered redox potential or tolerance to a more neutral pH, which selectively favor facultative anaerobes and microorganisms adapted to dynamic intestinal environments [72,73]. Although amplicon-based sequencing does not allow definitive confirmation at the species level, the observed enrichment of these taxa is consistent with features of dysbiotic microbial profiles associated with ASD. The presence of Fusobacterium is particularly notable, while its representative species, Fusobacterium mortiferum, has been associated with gastrointestinal diseases and its documented capacity to translocate into the systemic circulation under certain conditions [74,75]. Similarly, increased abundance of Alistipes, along with members of the Escherichia-Shigella genus and Ruminococcus gnavus_group, has been described in association with altered microbiota in various diseases [76,77]. In addition, a significantly higher relative abundance of Bacteroides (p = 0.004) was observed in the ASD group. Members of the genus Bacteroides are characterized by an extensive repertoire of enzymes involved in the degradation of complex polysaccharides, including mucin, enabling efficient utilization of host-derived substrates and modification of the structure of the mucus layer under conditions of microbial restructuring [78]. Sequences assigned to Bacteroides fragilis have previously been reported in association with inflammatory conditions of the intestinal mucosa [79].
In contrast, animals colonized with T.NT exhibited higher relative abundances associated with intestinal homeostasis, reflected by a significantly increased abundance of the genus Lactobacillus (p = 0.003). Concurrently, these animals showed lower relative abundances of taxa commonly associated with gut dysbiosis and inflammatory states, including Escherichia–Shigella, Fusobacterium [74,75], Ruminococcus gnavus group, Alistipes [76,77], Parabacteroides, Erysipelatoclostridium (p = 0.002), and Bilophila (p = 0.004). Compared to the ASD group, differences in microbial composition were not pronounced in terms of taxonomic structure, with changes primarily reflected at the level of relative abundances of individual taxa.
In summary, analysis of fecal microbiota in PGF mice suggested that the ASD group was characterized by enrichment of taxa associated with dysbiosis and inflammatory processes, whereas the NT group did not show a significant increase in taxa commonly associated with inflammatory processes.
3.3. Effects of Synbiotic Intervention on Gut Microbiota and Metabolic Profiles
3.3.1. Synbiotic-Induced Remodeling of Gut Microbiota
Probiotics and prebiotics influence gut microbiota structure and function through interconnected metabolic and immunoregulatory mechanisms. Their combined application as synbiotics can simultaneously modulate microbial composition and enhance fermentative activity, including SCFA production, thereby supporting epithelial integrity and immune homeostasis [80,81,82].
The following synbiotic intervention differences in gut microbiota composition between experimental groups were assessed using kernel-based association testing (Metastat). Compared to the ASD group without intervention, the ASD+pro/pre group showed a statistically significant reduction in the genus Bacteroides (Figure 3a,c,e,f). Concurrently, the ASD+pro/pre group exhibited an increased relative abundance of the genera Akkermansia, Phascolarctobacterium and Blautia (Figure 3b,g,h).
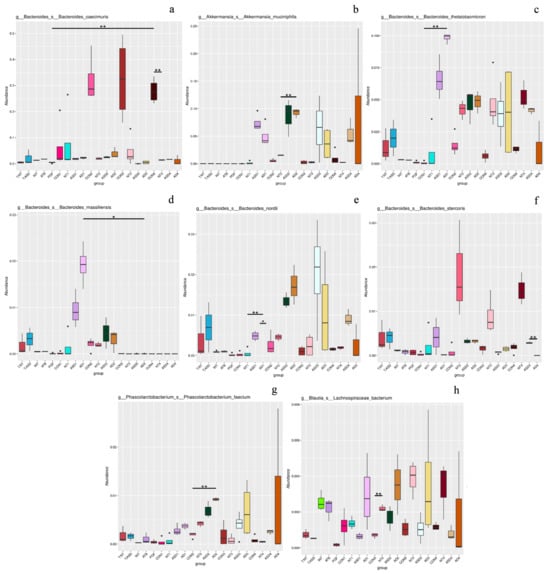
Figure 3.
Synbiotic-driven remodeling of gut microbiota and metabolomic profiles in ASD-colonized PGF mice. Relative abundance of bacterial taxa at the genus levels in control (C), neurotypical (NT), ASD, and ASD+pro/pre (AD) groups. Additives administered exclusively to the ASD+pro/pre group included Lactiplantibacillus plantarum CCM 7512, Limosilactobacillus reuteri CCM 8617, and flaxseed. Statistical analyses were performed on samples collected at the final time point (day 9 post intervention). Statistical comparisons were performed using kernel-based association testing (Metastat). Asterisks denote statistical significance (* p < 0.05; ** p < 0.01). (a,c–f) Bacteroides; (b) Akkermansia; (g) Phascolarctobacterium; (h) Blautia.
In experimental ASD models based on the transfer of ASD-associated microbiota into an animal host, persistence of a specific gut microbiota composition is often observed following intestinal colonization [56]. Subsequent modulation with probiotic strains or alterations in substrate availability may lead to selective changes in the relative abundance of specific bacterial groups without substantially altering the overall gut microbiota composition. In the maternal immune activation (MIA) model, administration of Limosilactobacillus reuteri resulted in partial modulation of gut microbiota composition [83], and similarly, application of Lactiplantibacillus plantarum modified the abundance of selected bacterial groups without markedly changing the overall structure of the gut microbiota [84].
The reduction in the relative abundance of the genus Bacteroides in the ASD+pro/pre group is relevant in this context, as the abundance of this genus is influenced by the availability of fermentable substrates and competitive interactions within the gut microbiota [85]. Members of the genus Bacteroides are metabolically flexible and capable of utilizing a wide range of complex polysaccharides. The observed decline may therefore reflect a shift within this genus rather than its overall suppression, likely due to a redistribution of metabolic processes following the synbiotic intervention. Concurrently, the ASD+pro/pre group exhibited an increased relative abundance of Akkermansia, Phascolarctobacterium and Blautia, indicating a shift in gut microbiota composition toward a fermentation-oriented profile. The genus Phascolarctobacterium comprises succinate-utilizing bacteria involved in the propionate pathway, whereas several members of the family Lachnospiraceae are linked to butyrate-associated metabolic pathways.
The increased relative abundance of these groups is therefore consistent with a potential shift in fermentative activity and may indicate a change in short-chain fatty acid production [82,86].
Compared to the ASD group without intervention, synbiotic modulation in the ASD+pro/pre group was accompanied by a reduction in the relative abundance of the genus Bacteroides and an increase in taxa associated with fermentative activity, including members of the family Lachnospiraceae and the genus Akkermansia. This pattern of changes suggests a gut microbiota profile characterized by a higher representation of bacteria involved in SCFA production and mucin metabolism, processes linked to epithelial integrity and functional stability of the intestinal environment.
3.3.2. Metabolomic Shifts Following Synbiotic Intervention
To assess whether synbiotic modulation of dysbiotic microbiota was reflected at the metabolic level (Figure 4a), the metabolomic profiles of the ASD and ASD+pro/pre groups were analyzed using principal component analysis (PCA). The analysis demonstrated a clear separation between groups, with no overlap, indicating a distinct shift in the metabolomic profiles following synbiotic intervention.
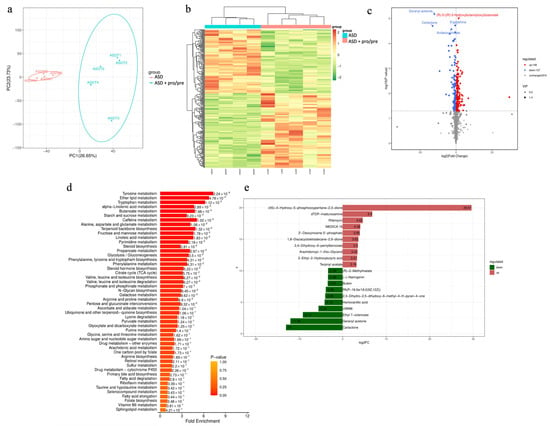
Figure 4.
Synbiotic-driven remodeling of metabolomic profiles in ASD-colonized PGF mice. (a) Principal component analysis (PCA) of metabolomic profiles in the ASD and ASD+pro/pre groups. Points represent individual animals (ASDM1–ASDM4 for ASD; ASDT1–ASDT4 for ASD+pro/pre). PC1 and PC2 explain 26.65% and 23.73% of the total variance, respectively. (b) Heatmap showing standardized metabolite abundance values (z-scores) across samples from animals in the ASD and ASD+pro/pre groups. Each column represents an individual animal (ASDM1–ASDM4 for ASD; ASDT1–ASDT4 for ASD+pro/pre). Colors range from dark green (lowest metabolite abundance, −2) to dark red (highest metabolite abundance, 2), indicating low to high relative abundance levels. (c) Volcano plot showing differential metabolites between the ASD and ASD+pro/pre groups (n = 4 animals per group). The X-axis represents log2 (fold change) and the Y-axis −log10 (p-value). Red dots indicate metabolites significantly increased in the ASD+pro/pre group, blue dots indicate metabolites increased in the ASD group, and gray dots represent metabolites without significant change. The horizontal dashed line indicates the significance threshold (p < 0.05). (d) Metabolic pathway enrichment plot (MSEA) comparing the ASD and ASD+pro/pre groups (n = 4 animals per group). Bar length represents the fold enrichment of metabolites within each pathway. The color scale (0–1) indicates pathway enrichment p-values, while the values shown next to the bars represent the exact p-values in scientific notation. Pathways are ranked according to fold enrichment. (e) Bar plot showing the top differentially abundant metabolites between the ASD and ASD+pro/pre groups ranked by log2 fold change (log2FC) (n = 4 animals per group). Positive log2FC values (red bars) indicate metabolites increased in the ASD group relative to ASD+pro/pre, whereas negative log2FC values (green bars) indicate metabolites increased in the ASD+pro/pre group. Values shown on the bars correspond to log2FC.
This separation was further supported by hierarchical clustering (Figure 4b), which showed consistent grouping of samples according to experimental condition. While minor interindividual variability was observed within the ASD+pro/pre group, differences between groups were more pronounced than within-group variation, suggesting a systematic reorganization of the metabolome.
Volcano plot analysis (Figure 4c) identified 148 metabolites with significantly higher and 127 metabolites with significantly lower levels in the ASD+pro/pre group compared with the ASD group, while 810 metabolites remained unchanged. This balanced distribution suggests that synbiotic intervention induced a coordinated metabolic shift rather than a uniform increase or decrease in metabolic activity.
To evaluate whether these changes extended to metabolic pathways, metabolite set enrichment analysis (MSEA) was performed (Figure 4d). Statistical significance was assessed after correction for multiple testing using the Benjamini–Hochberg method (FDR). After correction (FDR < 0.05), pathways related primarily to aromatic amino acid metabolism and lipid metabolism remained significantly enriched. The strongest enrichment was observed for tyrosine metabolism (6/24 metabolites; FDR = 1.37 × 10−7; 7.32-fold enrichment), followed by ether lipid metabolism (4/16 metabolites; FDR = 0.0146; 6.74-fold enrichment) and tryptophan metabolism (4/18 metabolites; FDR = 0.0228; 6.19-fold enrichment). Significant enrichment was also detected for α-linolenic acid metabolism (3/14 metabolites; FDR = 0.0441; 4.68-fold enrichment).
The predominance of pathways related to aromatic amino acid metabolism suggests that the differences between the ASD and ASD+pro/pre groups substantially involved tyrosine- and tryptophan-related metabolic circuits. Tryptophan is a key precursor of serotonin, and its metabolism is strongly modulated by the gut microbiota [87]. As alterations in aromatic amino acid metabolism have been repeatedly reported in autism spectrum disorders, including changes in the tryptophan and tyrosine axes [55], these pathways may represent an important link between the gut microbiota and host metabolism.
The butanoate metabolism pathway showed increased enrichment (3/13 metabolites; 4.79-fold enrichment) but did not reach statistical significance after correction for multiple testing (FDR = 0.0808). Although this pathway cannot be classified among the principal metabolic circuits distinguishing the groups, its representation is biologically meaningful, as butyrate and other SCFAs play important roles in maintaining intestinal barrier integrity and regulating immune responses [63,88]. The concurrent increase in bacteria associated with fermentative and SCFA-related pathways (Akkermansia, Phascolarctobacterium and Blautia) is consistent with the observed metabolic alterations and may indicate a functional shift in the gut environment, although its precise significance remains to be further elucidated. In addition to changes in aromatic amino acid metabolism, differences in lipid metabolic pathways were also observed between the groups. Alterations in lipid metabolism have been repeatedly reported in metabolomic studies of autism spectrum disorders, where differences between ASD and control groups were identified in the absence of nutritional intervention [55]. When interpreting our findings, it is important to consider that flaxseed administered as part of the synbiotic intervention represented a direct source of α-linolenic acid, and therefore some of the observed changes may reflect a dietary contribution. However, the presence of differences across broader lipid metabolic pathways suggests that the shift between groups was likely not limited to a direct nutritional effect.
At the level of individual metabolites (Figure 4e), molecules belonging to aromatic amino acid metabolism were also represented. In the ASD+pro/pre group, a higher level of homovanillic acid (log2FC = −4.14) was detected. Homovanillic acid represents the end product of dopamine degradation within the tyrosine–catecholamine metabolic axis. This difference may indicate a shift in tyrosine metabolism between the groups. Aromatic amino acid metabolism is partially modulated by the gut microbiota [89,90], and the observed difference may therefore be related to changes in the microbial environment induced by the synbiotic intervention. Although our data do not allow determination of the exact mechanism underlying this shift, the parallel changes in microbial profile and metabolic pathways suggest that the probiotic and prebiotic intervention was associated with modulation of interconnected microbiota–metabolic processes. Differences identified at the level of lipid pathways were also reflected in the statistically significant differential representation of specific lipid compounds between the groups. In the ASD+pro/pre group, significantly higher levels of selected phospholipid derivatives were observed, including PA(P–16:0e/18:2(9Z,12Z)), whereas in the ASD group, persistently elevated concentrations of certain lipid mediators were detected, including arachidonoyl-1-thio-glycerol (log2FC = 3.42). This pattern may indicate both quantitative and qualitative differences in the lipid spectrum between the groups. At the level of individual molecules, the changes were not limited to a single compound but involved multiple lipid classes, suggesting a broader reorganization of the lipid metabolic profile between the groups.
In addition to metabolites belonging to significantly enriched pathways, other differentially represented compounds were identified that point to a broader metabolic shift between the studied groups. In the ASD+pro/pre group, increased levels of phenolic compounds such as naringenin and butein were confirmed. Polyphenols and their derivatives undergo metabolism mediated by the gut microbiota, and their concentrations may reflect interactions between dietary components and the intestinal microbiota [91]. Given the presence of a prebiotic component in the intervention and the parallel changes observed in gut microbial composition, it is likely that the increased representation of these molecules is associated with modified microbial activity.
In contrast, in the ASD group without probiotic and prebiotic administration, certain nucleotide derivatives were more prominently represented, including 2′-deoxyinosine 5′-phosphate (log2FC = 3.95). Alterations in purine and nucleotide metabolism have been reported in association with ASD [55,92], suggesting that the difference observed in our study may fit within the broader spectrum of metabolic changes linked to this disorder. Although purine pathways were not significantly enriched in the MSEA, the differential representation of individual nucleotide metabolites indicates a broader range of metabolic differences between the groups.
The results suggest that the studied groups differed not only in individual metabolites but also in broader segments of the metabolic profile. The ASD group was characterized by a higher representation of certain nucleotide derivatives and specific lipid mediators. In contrast, the ASD+pro/pre group showed a more pronounced representation of pathways related to aromatic amino acid metabolism, along with alterations in selected lipid metabolites. These differences likely reflect that modulation of the dysbiotic gut microbiota following synbiotic intervention was accompanied by a systematic shift in the metabolic profile.
3.4. Effects of Synbiotic Intervention on Colon Histopathology
3.4.1. Colonic Histopathology and Histological Activity Index (HAI) Following FMT
Following identification of ASD-associated alterations in gut microbiota composition, histological analysis of the colon was subsequently performed to evaluate whether these alterations were accompanied by disruption of intestinal mucosal integrity and the development of histopathological alterations in the colon.
Marked reduction in the intestinal microbiota in PGF animals of the control group C (Figure 5(a1–3)) was associated with morphological characteristics typical of an environment with minimal microbial stimulation, resembling findings described in germ-free and pseudo-germ-free models [68,69]. The histological appearance of the mucosa was characterized by preserved intestinal wall architecture, lower cellularity of the lamina propria mucosae, and shallower intestinal crypts compared with animals with conventional microbiological status [68,69]. Semiquantitative evaluation using the Histological Activity Index (HAI; Figure 5e), based on the scoring of epithelial erosion, inflammatory infiltrate, and intestinal crypt damage [69], confirmed only a mild degree of mucosal alterations in the control group. HAI scores remained significantly lower across all evaluated parameters compared with the NT, ASD, and ASD+pro/pre groups (p ˂ 0.001; Figure 5e).
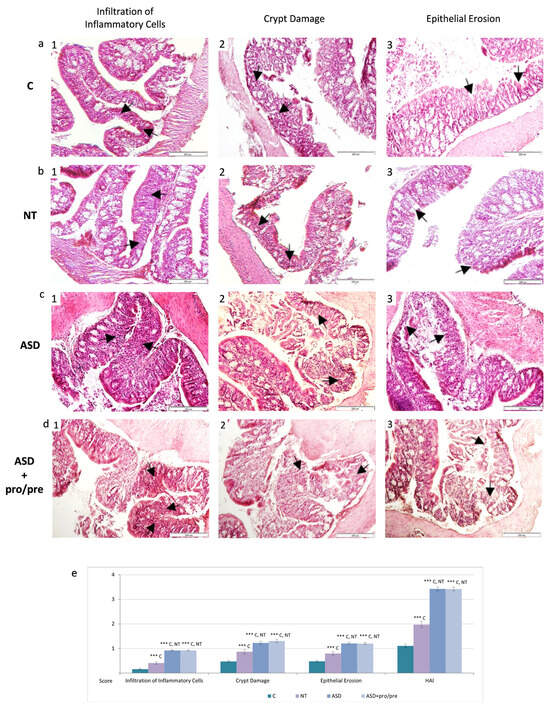
Figure 5.
Representative histological photomicrographs of colonic tissue following fecal microbiota transplantation. (a) (1–3) control group C (n = 5). (b) (1–3) NT group (n = 5). (c) (1–3) ASD group (n = 5). (d) (1–3) ASD+pro/pre group (n = 5). Histopathological alterations included inflammatory cell infiltration, crypt architectural damage, and epithelial erosion (arrows, hematoxylin–eosin staining; ×100). (e) Semiquantitative evaluation of histopathological alterations using the Histological Activity Index (HAI). Results are presented as mean ± SEM. *** p < 0.001 (between groups).
In contrast to the control group, animals colonized with transplanted microbiota exhibited more pronounced histopathological alterations of the intestinal mucosa. The most prominent alterations were observed in the ASD and ASD+pro/pre groups, where marked disruption of the histological architecture of the mucosa and submucosa was confirmed (Figure 5(c1–3,d1–3)), characterized by the presence of localized inflammatory infiltrates within the tunica mucosa and submucosa (Figure 5(c1,d1)). The inflammatory infiltrates were accompanied by destructive alterations of the superficial layers of the colonic epithelial lining (Figure 5(c3,d3)). Disruption of the integrity of the tunica mucosa was associated with deformation of the intestinal crypts, including cryptitis and irregular crypt architecture (Figure 5(c2,d2)). The extent of histopathological alterations was confirmed by significantly increased HAI scores in both groups with ASD-associated microbiota across all evaluated parameters compared with the control group C and the NT group (p ˂ 0.001; Figure 5e).
These results suggest that ASD-associated alterations in gut microbiota were linked to marked disruption of intestinal mucosal integrity and histopathological changes in the colon.
3.4.2. Histopathological Changes Following Synbiotic Intervention
Histological sections of the colon in ASD+pro/pre animals revealed reduced inflammatory cells infiltration following synbiotic administration, which was confirmed by significantly lower inflammatory infiltration scores compared with the ASD group following application of dysbiotic microbiota (Figure 6(d1,e), p < 0.001). Similarly, a significant reduction in crypt damage score was observed after the intervention (Figure 6(d2,e), p < 0.001). The 5-day synbiotic administration also appeared to promote restoration of the colonic epithelial lining (Figure 6(d3)), reflected by significantly lower epithelial damage scores not only compared with the ASD group following FMT (Figure 6e, p < 0.001), but also compared with the group transplanted with microbiota from neurotypical girls (NT, p < 0.05).
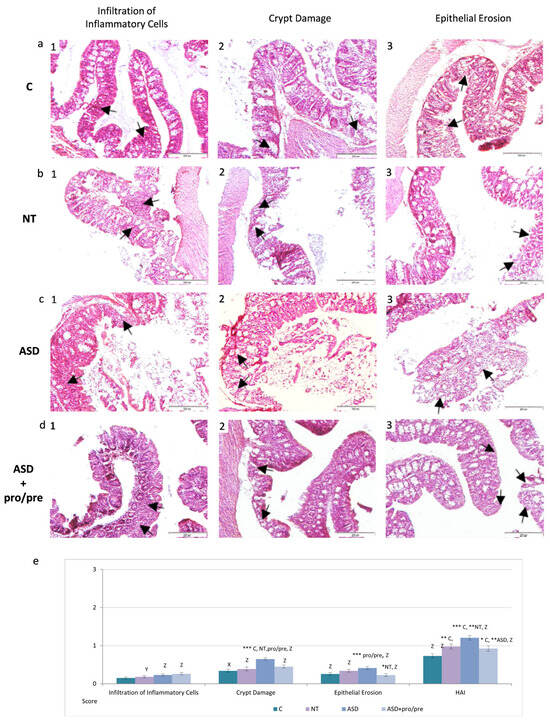
Figure 6.
Representative histological photomicrographs of colonic tissue following synbiotic intervention: (a) (1–3) control group C (n = 5). (b) (1–3) NT group (n = 5). (c) (1–3) ASD group (n = 5). (d) (1–3) ASD+pro/pre group (n = 5). Histopathological alterations included inflammatory cell infiltration, crypt architectural damage, and epithelial erosion (arrows, hematoxylin–eosin staining; ×100). (e) Semiquantitative evaluation of histopathological alterations using the Histological Activity Index (HAI). Results are presented as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001 (between groups). X p < 0.05, Y p < 0.01, Z p < 0.001 (between periods: after FMT vs. after synbiotic intervention within the same group).
The beneficial effect of selective probiotic–prebiotic modulation of ASD-associated dysbiotic microbiota was further supported by a significantly lower overall HAI in the ASD+pro/pre group compared with the untreated ASD group (Figure 6e, p < 0.01). Moreover, the HAI score in the ASD+pro/pre group remained lower, although without statistical significance, also in comparison with the NT group.
These histopathological changes were paralleled by shifts in gut microbiota composition and metabolic pathways following the intervention. Collectively, these findings may suggest that targeted modulation of ASD-associated microbiota may contribute to improved intestinal homeostasis, preservation of epithelial integrity, and attenuation of inflammatory infiltration and epithelial damage in the colon.
3.5. Effect of FMT on Hepatic Inflammatory Markers and LDH Activity
3.5.1. Hepatic Immunoreactivity of iNOS and COX-2 Following FMT
Analysis of gut microbiota composition suggested that transplantation of microbiota derived from donors with autism spectrum disorder led to the development of a dysbiotic microbial profile, which was observed in the ASD and ASD+pro/pre groups. Histological analysis of the colon in these groups additionally confirmed marked disruption of intestinal mucosal integrity accompanied by mucosal histopathological alterations. Increased immunoreactivity of the inflammatory markers iNOS and COX-2 has been repeatedly described in both experimental and clinical models of intestinal inflammation, including ulcerative colitis and dextran sulfate sodium (DSS)-induced colitis models [60,68,69]. Based on the potential association between intestinal dysbiosis, disruption of intestinal mucosal integrity and alterations in the host inflammatory response, we therefore further analyzed the immunoreactivity of inflammatory activation markers—specifically inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2)—in the liver tissue of the individual experimental groups.
Inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) are inducible enzymes involved in the regulation of the inflammatory response. They contribute to this process through the production of different mediators. iNOS is one of the three main isoforms of nitric oxide synthase, together with the neuronal (nNOS) and endothelial (eNOS) isoforms. Activation of iNOS leads to increased production of nitric oxide (NO), which participates in the regulation of immune responses and inflammatory processes and, when produced in excess, may contribute to nitrosative and oxidative stress [93,94,95]. In contrast, COX-2 catalyzes the conversion of arachidonic acid into prostaglandins, which represent important mediators of the inflammatory response [96].
Immunohistochemical evaluation of the inflammatory markers iNOS and COX-2 (Figure 7) during the period of FMT administration revealed differences among the experimental groups. Quantification of relative optical density (ROD) showed that in the groups receiving transplants from children with ASD, a more pronounced inflammatory response was observed, confirmed by significantly higher values of both iNOS and COX-2 compared with the NT group (p < 0.001; Figure 7a,b; ASD II, ASD+pro/pre II). The negative effect of dysbiotic FMT was also observed in the ASD and ASD+pro/pre groups when compared with the control group C, where the ROD values of both markers were likewise significantly higher (p < 0.001 and p < 0.01, respectively; Figure 7a,b; ASD II, ASD+pro/pre II). In contrast, the NT group showed lower ROD values of iNOS and COX-2 compared with the control group C (p < 0.05 and p < 0.01, respectively; Figure 7a,b; NT II).
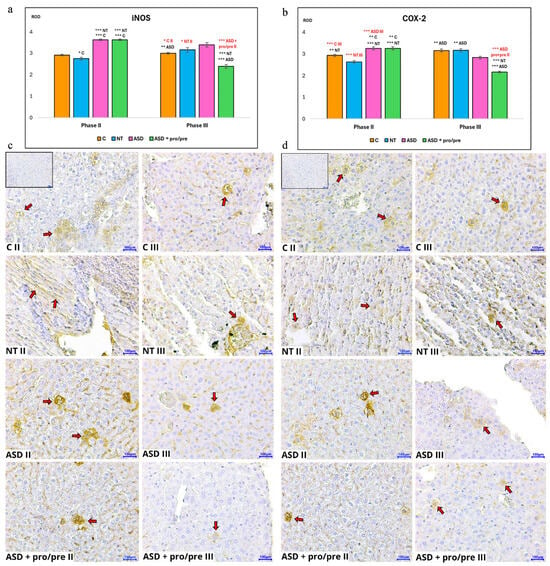
Figure 7.
Relative optical density (ROD) of (a) inducible nitric oxide synthase (iNOS) and (b) cyclooxygenase-2 (COX-2) in liver tissue in Phase II after fecal microbiota transplantation (FMT) and in Phase III after additive administration. Bar graphs show ROD values; red labeling indicates comparisons between Phase II and Phase III within the same group. Representative immunohistochemical images of liver tissue for (c) iNOS and (d) COX-2 corresponding to the evaluated groups. Positive immunoreactivity in the hepatocyte cytoplasm (red arrows) is indicated by brown staining. Magnification 400×; scale bar = 100 µm. The inset image (upper left corner of panel C II) represents a negative control (without primary antibody). Experimental groups included the control group (C; without FMT application; n = 6), the neurotypical group (NT; FMT from neurotypical donors; n = 6), and autism spectrum disorder groups (ASD and ASD+pro/pre; FMT from ASD donors; n = 6 per group). In Phase III, additives were administered exclusively to the ASD+pro/pre group: probiotic strains (Lactiplantibacillus plantarum CCM 7512, Limosilactobacillus reuteri CCM 8617) and a prebiotic (flaxseed). Data are presented as mean ± SEM. Statistical comparisons were performed using an unpaired two-tailed t-test (* p < 0.05; ** p < 0.01; *** p < 0.001).
The results of the quantitative evaluation of the relative optical density (ROD) of the inflammatory markers iNOS and COX-2 may indicate an adverse effect of the T.ASD transplant on the hepatic parenchyma. Increased immunoreactivity of both markers in the ASD and ASD+pro/pre groups suggests a higher degree of inflammatory activation in the liver following FMT derived from girls with autism spectrum disorder. This finding is consistent with the possibility that dysbiotically altered gut microbiota may influence not only the local intestinal environment but also distant organs involved in the gut–liver axis.
One possible mechanism involves disruption of the functional integrity of the intestinal barrier and the subsequent exposure of the liver to microbial products or metabolites derived from the gut microbiota. This assumption is further supported by our histological findings demonstrating marked disruption of intestinal mucosal integrity accompanied by epithelial erosions and crypt architectural alterations in the ASD and ASD+pro/pre groups. Increased intestinal permeability has been repeatedly reported in patients with ASD in association with intestinal dysbiosis, and such alterations in the intestinal environment may facilitate the translocation of bacterial components, including lipopolysaccharides (LPSs), into the systemic and portal circulation [97,98]. Although LPS translocation was not directly assessed in our experiment, the increased immunoreactivity of iNOS and COX-2 in liver tissue may suggest activation of inflammatory mechanisms that may be mediated by microbial signals.
These observations are also supported by changes in the taxonomic composition of the gut microbiota following administration of the T.ASD transplant. In the ASD group, an increased relative abundance of Escherichia–Shigella, Fusobacterium, Alistipes, Parabacteroides, and Bacteroides was observed. Such a microbial community is characteristic of an intestinal environment with disrupted microbial balance, in which facultative anaerobic bacteria capable of adapting to dynamically changing conditions of the intestinal ecosystem are selectively favored [73]. Some of these microorganisms are able to utilize host-derived substrates, including components of the mucin layer, which may contribute to its remodeling and potentially affect the integrity of the intestinal barrier. In this context, the increased abundance of bacteria carrying pro-inflammatory microbial components, including lipopolysaccharides, may represent a potential source of signals activating inflammatory pathways in the liver.
It is well established that bacterial endotoxins can initiate activation of the NF-κB signaling pathway following binding to the TLR4 receptor, which subsequently induces transcription of genes encoding iNOS and COX-2 [99,100]. Increased expression of these markers has been repeatedly observed in various models of inflammatory tissue injury, including experimental models of colitis [57,58,59,60,61,62,63,64,65,66,67,68,69] or following LPS stimulation of macrophages, where it was associated with increased production of prostaglandin E2 and other inflammatory mediators [101,102]. Activation of this signaling pathway therefore may represent one of the possible mechanisms explaining the increased immunoreactivity of iNOS and COX-2 observed in our experiment.
The interpretation of the inflammatory response in the liver is further supported by our metabolomic results. Metabolomic analysis suggested that the changes we observed were mainly associated with the metabolism of aromatic amino acids, particularly tyrosine and tryptophan, which represent metabolic pathways known to be modulated by the gut microbiota. Since metabolites generated within these pathways can influence host immune regulation, these alterations may represent one of the factors contributing to inflammatory activation within the gut–liver axis. In addition to changes in aromatic amino acid metabolism, we also detected differences in lipid metabolic pathways. These alterations are biologically relevant in the context of inflammatory responses, as COX-2 catalyzes the conversion of arachidonic acid into prostaglandins—key mediators of inflammation. Therefore, changes in lipid metabolic pathways may represent an additional factor contributing to the inflammatory activation observed in liver tissue.
Based on our findings, the increased immunoreactivity of iNOS and COX-2 observed in the liver following administration of the T.ASD transplant may reflect the pro-inflammatory effect of dysbiotically altered gut microbiota. This effect may be mediated by the combined action of microbial products and metabolites generated in the intestinal environment together with disruption of intestinal mucosal integrity, which through the gut–liver axis may contribute to the activation of inflammatory mechanisms in liver tissue. However, the precise mechanisms will need to be clarified in further studies focusing on the direct assessment of intestinal permeability, endotoxemia, and signaling pathways involved in inflammatory responses.
3.5.2. Modulation of Hepatic Inflammation by Synbiotic Intervention
Synbiotic intervention led to a statistically significant reduction in both monitored inflammatory markers, iNOS and COX-2, in the ASD+pro/pre group (Figure 7a–d; ASD+pro/pre III).
The iNOS marker in the ASD+pro/pre group (Figure 7a; ASD+pro/pre III) showed a decrease in relative optical density (ROD) compared with all monitored groups—NT and ASD (p < 0.001) as well as the control group C (p < 0.01). A significant decrease was also observed in comparison with the period of FMT administration (p < 0.01; Figure 7a,b; ASD+pro/pre II, ASD+pro/pre III). A similar decrease was observed for the COX-2 marker. In the ASD+pro/pre group (Figure 7b; ASD+pro/pre III), the ROD values were significantly lower compared with the NT and ASD groups (p < 0.001) as well as the control group C (p < 0.01). The same trend was also observed when compared with the period of FMT administration (p < 0.001; Figure 7a,b; ASD+pro/pre II, ASD+pro/pre III).
The observed decrease in the immunoreactivity of the iNOS and COX-2 markers suggests that the probiotic and prebiotic intervention may have contributed to attenuation of the inflammatory response in liver tissue induced by the preceding transplantation of dysbiotic gut microbiota. The modulatory effects of probiotic bacteria of the genus Lactobacillus have also been described in experimental models of autism spectrum disorders. Guo et al. [103] demonstrated that administration of the Lactobacillus plantarum ST-III strain modified gut microbiota composition and was accompanied by changes in the metabolic activity of the intestinal environment. Remodeling of the gut microbiota following a synbiotic intervention based on the combination of Lactobacillus reuteri and the prebiotic inulin was also reported by Wang et al. [104], who additionally observed alterations in metabolic processes mediated by the gut microbiota. The importance of the intestinal barrier in this context was demonstrated by Song et al. [105], who showed that administration of Lactobacillus acidophilus led to improvement in intestinal barrier function and a reduction in intestinal permeability.
It should be noted that in many published studies, probiotic interventions have been tested primarily in genetic or behaviorally characterized models of autism spectrum disorders. In our experiment, the model was based on fecal microbiota transplantation from donors with ASD, which allowed evaluation of the effects of probiotic and prebiotic modulation in an environment of dysbiotically altered gut microbiota. In the studies cited above, probiotic interventions were applied for several weeks (approximately 3–4 weeks), whereas in our experiment the application lasted only five days. Despite this short-term application, we observed changes at multiple levels of the biological response, including modifications in gut microbiota composition, alterations in metabolomic pathways, and a decrease in the immunoreactivity of the inflammatory markers iNOS and COX-2. These findings may indicate a link between modulation of the intestinal environment and attenuation of inflammatory responses in liver tissue within the gut–liver axis.
3.5.3. FMT-Induced Changes in Hepatic LDH Activity in PGF Mice
To complement the immunohistochemical evaluation of inflammatory changes in liver tissue, we analyzed lactate dehydrogenase (LDH) activity as a biochemical indicator of cell integrity (Figure 8a–f). LDH is a cytosolic enzyme involved in anaerobic glycolysis that is localized intracellularly under physiological conditions. Disruption of the cell membrane leads to its release into the extracellular space and subsequently into the circulation; therefore, increased LDH activity represents a sensitive, although relatively non-specific, marker of cellular damage, which may include cytolytic processes in various tissues, including hepatocytes [106,107,108]. During the period of FMT administration, total serum lactate dehydrogenase (TLDH) activity did not show statistically significant differences among the experimental groups (Figure 8a). The highest values were observed in the control group C representing the PGF model without FMT application, whereas groups receiving fecal microbiota transplantation (NT, ASD, and ASD+pro/pre) showed a slight tendency toward lower serum TLDH activity. A different pattern was observed when analyzing total TLDH activity in liver tissue (Figure 8d). Higher values were recorded particularly in the ASD and ASD+pro/pre groups, while C and NT groups showed lower enzymatic activity; however, these differences did not reach statistical significance. The interpretation of total LDH activity is limited by its low organ specificity, as this enzyme is widely distributed across various tissues. Serum and tissue LDH therefore differ not only in their overall activity levels but also in the relative proportions of individual isoenzyme fractions, whose distribution is organ- and species-specific [109]. Isoenzymes containing the H subunit encoded by the LDHB gene are more abundant in blood and cells of the reticuloendothelial system, whereas parenchymal organs, including the liver, exhibit a higher proportion of isoenzymes containing the M subunit [15,110]. For this reason, further analysis focused on LDH-4 and LDH-5 isoenzymes, which are considered more relevant indicators of metabolic and cytolytic changes in the hepatic parenchyma. No significant differences in serum activity of the LDH-4 isoenzyme were observed among the experimental groups (Figure 8b). However, in liver tissue (Figure 8e), the control group C showed significantly lower LDH-4 activity compared with all groups receiving FMT (p < 0.05). The ASD and ASD+pro/pre groups, receiving microbiota transplants derived from girls with ASD, also exhibited significantly higher activity of this isoenzyme compared with the NT group (p < 0.05). Additional information was provided by the analysis of the LDH-5 isoenzyme (Figure 8c,f), which is considered a more sensitive indicator of hepatocyte damage. In the liver parenchyma (Figure 8f), an opposite trend compared with LDH-4 was observed, confirmed by significantly lower LDH-5 activity in the FMT-treated groups compared with the control group C (p < 0.001).
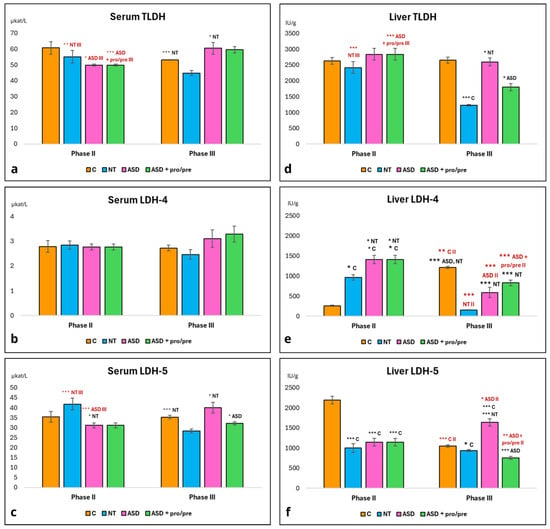
Figure 8.
Activities of lactate dehydrogenase (LDH) and selected LDH isoenzymes in serum and liver. (a) Total LDH activity in serum (TLDH). (b) LDH isoenzyme 4 activity in serum. (c) LDH isoenzyme 5 activity in serum. (d) Total LDH activity in liver (TLDH). (e) LDH isoenzyme 4 activity in liver. (f) LDH isoenzyme 5 activity in liver. Values are shown for Phase II after fecal microbiota transplantation (FMT) and Phase III after additive administration. Red labeling indicates comparisons between Phase II and Phase III within the same group. Experimental groups included the control group (C; without FMT application; n = 6), the neurotypical group (NT; FMT from neurotypical donors; n = 6), and autism spectrum disorder groups (ASD and ASD+pro/pre; FMT from ASD donors; n = 6 per group). In Phase III, additives were administered exclusively to the ASD+pro/pre group: probiotic strains (Lactiplantibacillus plantarum CCM 7512, Limosilactobacillus reuteri CCM 8617) and a prebiotic (flaxseed). Data are presented as mean ± SEM. Statistical comparisons were performed using an unpaired two-tailed t-test (* p < 0.05; ** p < 0.01; *** p < 0.001).
The interpretation of these results should also be considered in the context of the immunohistochemical evaluation of the inflammatory markers iNOS and COX-2. Increased expression of these markers primarily reflects activation of inflammatory signaling in liver tissue and does not constitute direct evidence of cytolytic damage to hepatocytes, as they may be expressed by various hepatic cell populations, including Kupffer cells, endothelial cells, or infiltrating immune cells. The lower activity of the LDH-5 isoenzyme, which is more closely associated with hepatocytes, in the FMT-treated groups therefore may suggest that increased cytolytic damage to hepatocytes did not occur despite the presence of inflammatory activation. In contrast, the higher LDH-5 activity observed in the control group C may be related to the known metabolic and immunological differences characteristic of models with reduced or absent gut microbiota, which exhibit extensive alterations in host metabolic and immune processes [111,112,113]. A potential factor contributing to these differences may also be the microbial metabolites present in the applied fecal microbiota transplants. In the T.NT transplant, we identified metabolites mainly associated with polyphenol and amino acid metabolism, whereas in the T.ASD transplant we detected metabolites indicative of differences in the energy metabolism of the microbial community. These observations suggest that restoration of microbial colonization may contribute to the partial re-establishment of metabolic interactions between the gut microbiota and the host. Compared with models with reduced microbiota, FMT may therefore contribute to the stabilization of metabolic processes within the gut–liver axis, which may also be reflected by a lower degree of cytolytic burden on hepatocytes.
Experimental studies indicate that restoration of gut microbial colonization can significantly influence hepatic metabolic processes through the gut–liver axis. For example, Yang et al. [114] demonstrated in a mouse model of acetaminophen-induced liver injury that orally administered fecal microbiota transplantation led to enrichment of butyrate-producing bacteria from the family Lachnospiraceae, increased production of microbial metabolites, and subsequent attenuation of liver injury. Similarly, Yuan et al. [115], in a model of acute liver failure induced by LPS/D-galactosamine, showed that FMT not only modulated the composition of the gut microbiota but also resulted in changes in the hepatic metabolic profile and a reduction in the inflammatory response. These findings suggest that restoration of the gut microbiota may influence hepatic processes through metabolic signals transmitted via the gut–liver axis.
In our experiment, we likewise observed differences in the composition of the cecal bacterial community between the NT and ASD groups following FMT. These differences were also reflected at the level of the host response in our model, as the ASD group exhibited more pronounced inflammatory activation of the liver parenchyma, as reflected by increased expression of the markers iNOS and COX-2. Despite these inflammatory changes, both FMT-treated groups showed lower activity of the hepatocyte-associated isoenzyme LDH-5 compared with the control group C. This finding suggests that the presence of gut microbiota after FMT—despite differences in bacterial composition—may have been sufficient to maintain basic metabolic interactions within the gut–liver axis, thereby contributing to a lower degree of cytolytic burden on hepatocytes.
3.5.4. Modulation of Hepatic LDH Activity and Isoenzymes by Synbiotic Intervention
Following the observed reduction in inflammatory markers, we further evaluated whether the synbiotic intervention also affected enzymatic parameters in liver tissue, particularly total tissue lactate dehydrogenase activity (TLDH) and the distribution of LDH isoenzymes (Figure 8).
In the ASD+pro/pre group, TLDH (Figure 8d) was significantly lower compared with the ASD group without intervention (p < 0.05), with a more pronounced decrease observed relative to the period following FMT administration (p < 0.001). As TLDH represents the sum of the activities of all LDH isoenzymes (LDH1–LDH5), the observed decrease reflects an overall reduction in LDH activity in liver tissue and may suggest that the beneficial effect of the probiotic–prebiotic intervention may also be manifested at the level of individual LDH isoenzyme fractions. In the ASD+pro/pre group, differential changes in the activities of the monitored LDH isoenzymes were observed following the period of additive administration. The activity of the LDH4 isoenzyme (Figure 8e) was significantly lower compared with the period following FMT administration (p < 0.001); however, it remained non-significantly higher compared with the ASD group. Changes in LDH4 activity may therefore indicate a shift in lactate and pyruvate metabolism and in the intensity of glycolytic metabolism in the given tissue. In contrast, an opposite trend was observed for LDH5, an isoenzyme more closely associated with hepatic tissue (Figure 8f). In the ASD+pro/pre group, its activity significantly decreased not only compared with the period following FMT administration (p < 0.001), but also relative to the other monitored groups, with the most pronounced difference observed in comparison with the ASD group without intervention (p < 0.001).
A similar effect of synbiotic supplementation was also observed in our previous studies using the same probiotic strain Lactobacillus reuteri CCM 8617 in combination with flaxseed. Andrejcakova et al. [13] demonstrated that such an intervention led to a reduction in TLDH activity and several LDH isoenzymes, including LDH-5, in liver tissue, which was accompanied by increased incorporation of n-3 polyunsaturated fatty acids and a decreased n-6/n-3 PUFA ratio. In addition, Sopkova et al. [116] showed that flaxseed supplementation supports the fermentative activity of the intestinal microbiota and increases the production of organic acids, particularly SCFAs such as acetate, propionate, and butyrate. Our current results further extend this interpretation, as the synbiotic intervention was also accompanied by pronounced changes in the composition of the gut microbiota and the metabolic profile. In the ASD+pro/pre group, the abundance of fermentatively active bacteria increased, which are associated with enhanced fermentative metabolism and the production of metabolites involved in maintaining intestinal epithelial integrity and the functional stability of the gut environment. Concurrently, alterations in metabolic pathways were identified, particularly in the metabolism of aromatic amino acids and lipid compounds, including differences in the abundance of certain phospholipid derivatives and mediators derived from arachidonic acid. These changes point to a broader reorganization of microbiota–metabolic processes, which may represent one of the factors contributing to the observed changes in tissue metabolic markers. It should be emphasized that in previous studies, flaxseed supplementation was applied over a longer experimental period (approximately 28–35 days). In the present experiment, however, the synbiotic intervention was administered for only 5 days, yet changes in metabolic and tissue parameters were still observed despite this short duration. These findings may suggest that the combination of the probiotic strain Lactobacillus reuteri CCM 8617 and the prebiotic component is capable of relatively rapidly influencing interconnected microbial and metabolic processes of the host. The fact that these changes were also detected at the level of liver tissue indicates that the synbiotic intervention may quickly affect systemic metabolic processes mediated through the gut–liver axis.
Our study had several limitations. First, fecal microbiota transplants were derived exclusively from female donors, which may limit the generalizability of the findings; future studies should therefore include both sexes. Second, the taxonomic resolution of the cecal microbiota analysis did not allow reliable identification at the species level, limiting a more precise interpretation of microbiota–host relationships in the context of ASD. Third, microbiome analyses were conducted with a modest sample size (n = 4 animals per group), which limits statistical power in a high-dimensional omics setting and increases the risk of type II errors (false negatives). Consequently, microbiome- and pathway-level findings should be considered exploratory and hypothesis-generating and interpreted with caution, and will require validation in larger, appropriately powered studies. In addition, fecal microbiota transplants were generated by pooling donor material, which reduces inter-donor variability but does not allow independent assessment of donor-specific effects. Another limitation is the absence of bacteriophage analysis, as the gut virome may significantly modulate bacterial communities, although its role in health and disease remains insufficiently understood. Furthermore, the experimental design focused on the combined effect of probiotic strains and flaxseed, which precluded distinguishing their individual contributions; future studies should therefore evaluate these components separately. Finally, the analysis did not include standard serum liver parameters (AST, ALT, GGT, ALP), which was constrained by the limited volume of biological material in the small animal model. Future work should also incorporate more detailed histopathological assessment and a broader panel of inflammatory and metabolic markers to provide a more comprehensive characterization of the observed effects. These limitations should be considered when interpreting the results and represent a basis for further research.
4. Conclusions
Analysis of fecal transplants obtained from neurotypical girls and girls with autism spectrum disorder revealed a comparable representation of dominant bacterial taxa, with differences primarily reflected in their relative abundances. More evident differences were observed in the mycobiome—characterized by a higher abundance of the genus Candida in T.ASD and Saccharomyces in T.NT—as well as in the metabolomic profile of the transplants, which showed systematic differences in metabolites associated with polyphenol, amino acid, and energy metabolism. Transplantation of microbiota from girls with ASD was associated with compositional changes in the cecal microbiota, including a higher relative abundance of bacterial groups commonly associated with dysbiotic states. These changes were accompanied by disruption of intestinal mucosal integrity; metabolomic differences, particularly in metabolites related to aromatic amino acid and lipid metabolism; increased immunoreactivity of the inflammatory markers iNOS and COX-2 in liver tissue; and increased activity of the liver-specific LDH-5 isoenzyme, which is consistent with a possible link between microbiota-associated metabolic alterations, impaired intestinal barrier integrity, and hepatic responses within the gut–liver axis. Synbiotic intervention combining Lactiplantibacillus plantarum CCM 7512 and Limosilactobacillus reuteri CCM 8617 with ground flaxseed (Linum usitatissimum L.), a source of omega-3 PUFA and dietary fiber, was associated with remodeling of the gut microbiota, restoration of intestinal epithelial integrity, metabolic changes, reduced immunoreactivity of inflammatory markers in the liver, and decreased activity of the hepatic LDH-5 isoenzyme. These findings support a possible role of the gut microbiota in processes operating within the gut–liver axis; however, the microbiome- and pathway-level observations should be regarded as exploratory and require validation in larger, appropriately powered studies.
Author Contributions
Study concept and design, S.G., V.D., A.T., D.S. (Drahomira Sopkova) and K.S.R.; data acquisition, S.G., S.L., D.S. (Daniela Spisakova), Z.A., V.S., B.R. and K.S.R.; data analysis and interpretation, S.G., V.D., M.R., M.F., V.S., B.R. and K.S.R.; drafting of the manuscript, S.G., K.S.R., V.D. and M.R.; critical revision of the manuscript for important intellectual content, S.G., V.D., D.S. (Drahomira Sopkova), R.V. and Z.A.; statistical analysis, M.R. and M.F.; obtained funding, A.T., S.G., K.S.R. and Z.A.; administrative, technical, or material support, S.G., S.L., A.T., D.S. (Daniela Spisakova), D.S. (Drahomira Sopkova), Z.A., R.V., B.R. and K.S.R.; study supervision, V.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the projects APVV-20-0114, VEGA 1/0414/23 and VEGA 1/0139/25, and funded by the EU NextGenerationEU through the Recovery and Resilience Plan for Slovakia under project No. 09I03-03-V05-00017.
Institutional Review Board Statement
The animal experimental protocol (No. 4356/2022-220) was approved by the State Veterinary and Food Administration of the Slovak Republic. The collection of biological material from healthy children and children with ASD was approved by the Ethics Committee of the Faculty of Medicine, Comenius University, and the University Hospital Bratislava (Old Town Hospital) within the framework of the research project No. 79/2021, approved on 2 August 2021.
Informed Consent Statement
Written informed consent for participation, including data collection and analysis as well as the collection of biological material for FMT preparation, was obtained from the parents of neurotypical and ASD donors prior to inclusion.
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed at the corresponding author.
Acknowledgments
The authors would like to express their sincere gratitude to Radomira Nemcova, for providing the probiotic strains Lactiplantibacillus plantarum CCM 7512 and Limosilactobacillus reuteri CCM 8617 used in this study. The authors also thank J. Kalatova and M. Kozackova for technical assistance and A. Krupa for assistance with the visualization of immunohistochemical and LDH images.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Angrand, L.; Masson, J.-D.; Rubio-Casillas, A.; Nosten-Bertrand, M.; Crépeaux, G. Inflammation and autophagy: A convergent point between autism spectrum disorder (ASD)-related genetic and environmental factors: Focus on aluminum adjuvants. Toxics 2022, 10, 518. [Google Scholar] [CrossRef]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Yang, J.; Fu, X.; Liao, X.; Li, Y. Effects of gut microbial-based treatments on gut microbiota, behavioral symptoms, and gastrointestinal symptoms in children with autism spectrum disorder: A systematic review. Psychiatry Res. 2020, 293, 113471. [Google Scholar] [CrossRef]
- Żebrowska, P.; Łaczmańska, I.; Łaczmański, Ł. Future directions in reducing gastrointestinal disorders in children with ASD using fecal microbiota transplantation. Front. Cell. Infect. Microbiol. 2021, 11, 630052. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of disease: Inflammatory bowel diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef]
- Hsu, C.L.; Schnabl, B. The gut-liver axis and gut microbiota in health and liver disease. Nat. Rev. Microbiol. 2023, 21, 719–733. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Madar, M.; Slizova, M.; Czerwinski, J.; Hrckova, G.; Mudronova, D.; Gancarcikova, S.; Popper, M.; Pistl, J.; Soltys, J.; Nemcova, R. Histo-FISH protocol to detect bacterial compositions and biofilms formation in vivo. Benef. Microbes 2015, 6, 899–907. [Google Scholar] [CrossRef]
- Gancarčíková, S.; Nemcová, R.; Popper, M.; Hrčková, G.; Sciranková, Ľ.; Maďar, M.; Mudroňová, D.; Vilček, Š.; Žitňan, R. The influence of feed-supplementation with probiotic strain Lactobacillus reuteri CCM 8617 and alginite on intestinal microenvironment of SPF mice infected with Salmonella Typhimurium CCM 7205. Probiotics Antimicrob. Proteins 2019, 11, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Andrejčáková, Z.; Sopková, D.; Vlčková, R.; Hertelyová, Z.; Gancarčíková, S.; Nemcová, R. The application of Lactobacillus reuteri CCM 8617 and flaxseed positively improved the health of mice challenged with enterotoxigenic E. coli O149:F4. Probiotics Antimicrob. Proteins 2020, 12, 937–951. [Google Scholar] [CrossRef]
- Park, J.C.; Sim, M.A.; Lee, C.; Park, H.E.; Lee, J.; Choi, S.Y.; Byun, S.; Ko, H.; Lee, H.; Kim, S.W.; et al. Gut microbiota and brain-resident CD4+ T cells shape behavioral outcomes in autism spectrum disorder. Nat. Commun. 2025, 16, 6422. [Google Scholar] [CrossRef]
- Jin, H.; Liu, Q.; Li, J.; Zhao, S.; Tuo, B. Multifaceted roles of lactate dehydrogenase in liver cancer (review). Int. J. Oncol. 2025, 66, 50. [Google Scholar] [CrossRef]
- Prince, N.; Peralta Marzal, L.N.; Roussin, L.; Monnoye, M.; Philippe, C.; Maximin, E.; Ahmed, S.; Salenius, K.; Lin, J.; Autio, R.; et al. Mouse strain-specific responses along the gut-brain axis upon fecal microbiota transplantation from children with autism. Gut Microbes 2025, 17, 2447822. [Google Scholar] [CrossRef]
- Gancarčíková, S.; Popper, M.; Hrčková, G.; Maďar, M.; Mudroňová, D.; Sopková, D.; Nemcová, R. Antibiotic-treated SPF mice as a gnotobiotic model. In Antibiotic Use in Animals; Savic, S., Ed.; InTech: Rijeka, Croatia, 2018; pp. 45–83. [Google Scholar] [CrossRef][Green Version]
- Lord, C.; Rutter, M.; Dilavore, P.C.; Risi, S.; Gotham, K.; Bishop, S. Autism Diagnostic Observation Schedule; Western Psychological Services: Torrance, CA, USA, 2012. [Google Scholar]
- Lord, C.; Pickles, A.; McLennan, J.; Rutter, M.; Bregman, J.; Folstein, S.; Fombonne, E.; Leboyer, M.; Minshew, N. Diagnosing autism: Analyses of data from the Autism Diagnostic Interview. J. Autism Dev. Disord. 1997, 27, 501–517. [Google Scholar] [CrossRef]
- Reygner, J.; Charrueau, C.; Delannoy, J.; Mayeur, C.; Robert, V.; Cuinat, C.; Meylheuc, T.; Mauras, A.; Augustin, J.; Nicolis, I.; et al. Freeze-dried fecal samples are biologically active after long-lasting storage and suited to fecal microbiota transplantation in a preclinical murine model of Clostridioides difficile infection. Gut Microbes 2020, 11, 1405–1422. [Google Scholar] [CrossRef]
- Ikhtaire, S.; Shajib, M.S.; Reinisch, W.; Khan, W.I. Fecal calprotectin: Its scope and utility in the management of inflammatory bowel disease. J. Gastroenterol. 2016, 51, 434–446. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Conroy, M.J.; Andrews, R.M.; Andrews, S.; Cockayne, L.; Dennis, E.A.; Fahy, E.; Gaud, C.; Griffiths, W.J.; Jukes, G.; Kolchin, M.; et al. LIPID MAPS: Update to databases and tools for the lipidomics community. Nucleic Acids Res. 2024, 52, D1677–D1682. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Heinova, D.; Rosival, I.; Avidar, Y.; Bogin, E. Lactate dehydrogenase isoenzyme distribution and patterns in chicken organs. Res. Vet. Sci. 1999, 67, 309–312. [Google Scholar] [CrossRef]
- Andrejčáková, Z.; Vlčková, R.; Sopková, D.; Kozioł, K.; Koziorowski, M.; Fabián, D.; Šefčíková, Z.; Holovská, K.; Almášiová, V.; Sirotkin, A.V. Dietary flaxseed’s protective effects on body tissues of mice after oral exposure to xylene. Saudi J. Biol. Sci. 2021, 28, 3789–3798. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumu-gam, M.; Asnicar, F.; et al. Author correction: Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; May, T.W.; Frøslev, T.G.; Pawlowska, J.; Lindahl, B.; Põldmaa, K.; Truong, C.; et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: Sequences, taxa and classifications reconsidered. Nucleic Acids Res. 2024, 52, D791–D797. [Google Scholar] [CrossRef]
- Paulson, J.N.; Pop, M.; Bravo, H.C. Metastats: An improved statistical method for analysis of metagenomic data. Genome Biol. 2011, 12, P17. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.R-project.org/ (accessed on 12 January 2026).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package 2025, 1.0.13. Available online: https://github.com/raivokolde/pheatmap (accessed on 5 June 2025).
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Kohl, M. MKinfer: Inferential Statistics. R Package Version 1.2. 2024. Available online: https://github.com/stamats/MKinfer (accessed on 12 February 2026).
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Renzi, D.; Calabrò, A.; et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 2017, 5, 24. [Google Scholar] [CrossRef]
- Lewandowska-Pietruszka, Z.; Figlerowicz, M.; Mazur-Melewska, K. Microbiota in autism spectrum disorder: A systematic review. Int. J. Mol. Sci. 2023, 24, 16660. [Google Scholar] [CrossRef]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota transfer therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef]
- Liu, F.; Li, J.; Wu, F.; Zheng, H.; Peng, Q.; Zhou, H. Altered composition and function of intestinal microbiota in autism spectrum disorders: A systematic review. Transl. Psychiatry 2019, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Coretti, L.; Paparo, L.; Riccio, M.P.; Amato, F.; Cuomo, M.; Natale, A.; Borrelli, L.; Corrado, G.; Comegna, M.; Buommino, E.; et al. Gut microbiota features in young children with autism spectrum disorders. Front. Microbiol. 2018, 9, 3146. [Google Scholar] [CrossRef]
- Tomova, A.; Husarova, V.; Lakatosova, S.; Bakos, J.; Vlkova, B.; Babinska, K.; Ostatnikova, D. Gastrointestinal microbiota in children with autism in Slovakia. Physiol. Behav. 2015, 138, 179–187. [Google Scholar] [CrossRef]
- Wang, Y.; Li, N.; Yang, J.J.; Zhao, D.M.; Chen, B.; Zhang, G.Q.; Chen, S.; Cao, R.F.; Yu, H.; Zhao, C.Y.; et al. Probiotics and fructo-oligosaccharide intervention modulate the microbiota-gut brain axis to improve autism spectrum reducing also the hyper-serotonergic state and the dopamine metabolism disorder. Pharmacol. Res. 2020, 157, 104784. [Google Scholar] [CrossRef]
- Retuerto, M.; Al-Shakhshir, H.; Herrada, J.; McCormick, T.S.; Ghannoum, M.A. Analysis of gut bacterial and fungal microbiota in children with autism spectrum disorder and their non-autistic siblings. Nutrients 2024, 16, 3004. [Google Scholar] [CrossRef]
- Nirmalkar, K.; Patel, J.; Kang, D.-W.; Bellinghiere, A.; Bowes, D.A.; Qureshi, F.; Adams, J.B.; Krajmalnik-Brown, R. Bimodal Distribution of Intestinal Candida in Children with Autism and Its Potential Link with Worse ASD Symptoms. Gut Microbes Rep. 2024, 1, 2358324. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, B.S.; Roşu, O.A.; Enache, R.M.; Manciulea Profir, M.; Pavelescu, L.A.; Creţoiu, S.M. Gut Mycobiome: Latest Findings and Current Knowledge Regarding Its Significance in Human Health and Disease. J. Fungi 2025, 11, 333. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Iliev, I.D.; Leonardi, I. Fungal dysbiosis: Immunity and interactions at mucosal barriers. Nat. Rev. Immunol. 2017, 17, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Needham, B.D.; Adame, M.D.; Serena, G.; Rose, D.R.; Preston, G.M.; Conrad, M.C.; Campbell, A.S.; Donabedian, D.H.; Fasano, A.; Ashwood, P.; et al. Plasma and fecal metabolite profiles in autism spectrum disorder. Biol. Psychiatry 2021, 89, 451–462. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 2019, 177, 1600–1618.e17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hua, X.; Yang, T.; Guo, M.; Li, Q.; Xiao, L.; Li, L.; Chen, J.; Li, T. Alterations in gut vitamin and amino acid metabolism are associated with symptoms and neurodevelopment in children with autism spectrum disorder. J. Autism Dev. Disord. 2022, 52, 3116–3128. [Google Scholar] [CrossRef] [PubMed]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef]
- Zheng, R.; Huang, S.; Feng, P.; Liu, S.; Jiang, M.; Li, H.; Zheng, P.; Mi, Y.; Li, E. Comprehensive analysis of gut microbiota and fecal metabolites in patients with autism spectrum disorder. Front. Microbiol. 2025, 16, 1557174. [Google Scholar] [CrossRef]
- Huang, Z.; Wei, A.; Yuan, H.; Huang, S.; Chen, X.; Han, Y.; Li, X. Gut microbiota and urine metabolomics signature in autism spectrum disorder children from Southern China. BMC Pediatr. 2025, 25, 621. [Google Scholar] [CrossRef]
- Kim, S.; Seo, S.U.; Kweon, M.N. Gut microbiota derived metabolites tune host homeostasis fate. Semin. Immunopathol. 2024, 46, 2. [Google Scholar] [CrossRef]
- Keshet, A.; Segal, E. Identification of gut microbiome features associated with host metabolic health in a large population-based cohort. Nat. Commun. 2024, 15, 9358. [Google Scholar] [CrossRef]
- Liu, X.F.; Shao, J.H.; Liao, Y.T.; Wang, L.N.; Jia, Y.; Dong, P.J.; Liu, Z.Z.; He, D.D.; Li, C.; Zhang, X. Regulation of short-chain fatty acids in the immune system. Front. Immunol. 2023, 14, 1186892. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, G.; Wan, L.; Liang, Y.; Liu, X.; Yan, H.; Zhang, B.; Yang, G. Effect of fecal microbiota transplantation in children with autism spectrum disorder: A systematic review. Front. Psychiatry 2023, 14, 1123658. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.L.; Karl, J.P.; Oliverio, A.M.; Fu, X.; Soares, J.W.; Wolfe, B.E.; Hernandez, C.J.; Mason, J.B.; Booth, S.L. Dietary vitamin K is remodeled by gut microbiota and influences community composition. Gut Microbes 2021, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Masatoshi, H.; Ma, Y.; Guo, Y.; Zhang, B. Role of vitamin K in intestinal health. Front. Immunol. 2022, 12, 791565. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Kaiser, T.; Beura, L.K.; Hamilton, M.J.; Weingarden, A.R.; Bobr, A.; Kang, J.; Masopust, D.; Sadowsky, M.J.; Khoruts, A. Stable engraftment of human microbiota into mice with a single oral gavage following antibiotic conditioning. Microbiome 2017, 5, 87. [Google Scholar] [CrossRef]
- Gancarcikova, S.; Lauko, S.; Hrckova, G.; Andrejcakova, Z.; Hajduckova, V.; Madar, M.; Kolesar Fecskeova, L.; Mudronova, D.; Mravcova, K.; Strkolcova, G.; et al. Innovative animal model of DSS-induced ulcerative colitis in pseudo germ-free mice. Cells 2020, 9, 2571. [Google Scholar] [CrossRef]
- Lauko, S.; Gancarcikova, S.; Hrckova, G.; Hajduckova, V.; Andrejcakova, Z.; Fecskeova, L.K.; Bertkova, I.; Hijova, E.; Kamlarova, A.; Janicko, M.; et al. Beneficial effect of faecal microbiota transplantation on mild, moderate and severe dextran sodium sulphate-induced ulcerative colitis in a pseudo germ-free animal model. Biomedicines 2023, 12, 43. [Google Scholar] [CrossRef]
- Xiao, L.; Yan, J.; Yang, T.; Zhu, J.; Li, T.; Wei, H.; Chen, J. Fecal microbiome transplantation from children with autism spectrum disorder modulates tryptophan and serotonergic synapse metabolism and induces altered behaviors in germ-free mice. mSystems 2021, 6, e01343-20. [Google Scholar] [CrossRef]
- Borbélyová, V.; Szabó, J.; Sušienková, P.; Potvin, J.; Belvončíková, P.; Groß, T.; Jančovičová, A.; Bačová, Z.; Rašková, B.; Szadvári, I.; et al. The effect of parental faecal microbiome transplantation from children with autism spectrum disorder on behavior and gastrointestinal manifestations in the male offspring of Shank3 mice. Int. J. Mol. Sci. 2025, 26, 5927. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Pernitzsch, S.R.; Bäumler, A.J. Healthy hosts rule within: Ecological forces shaping the gut microbiota. Mucosal Immunol. 2018, 11, 1299–1305. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Bäumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carretero, R.; Lopez-Lomba, M.; Carrasco-Fernandez, B.; Duran-Valle, M.T. Clinical features and outcomes of Fusobacterium species infections in a ten-year follow-up. J. Crit. Care Med. (Targu Mures) 2017, 3, 141–147. [Google Scholar] [CrossRef]
- Perinkulam Sathyanarayanan, S.; Hamid, K.; Hoerschgen, K.; Oliver, T. A rare case of rectal bleeding and Fusobacterium mortiferum sepsis due to solitary fibrous tumour originating from the mesentery. BMJ Case Rep. 2021, 14, e244603. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The genus Alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Fu, J.; Li, G.; Li, X.; Song, S.; Cheng, L.; Rui, B.; Jiang, L. Gut commensal Alistipes as a potential pathogenic factor in colorectal cancer. Discov. Oncol. 2024, 15, 473. [Google Scholar] [CrossRef]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef]
- Rios-Covian, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de Los Reyes-Gavilan, C.G.; Salazar, N. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef]
- Qiu, Z.; Luo, D.; Yin, H.; Chen, Y.; Zhou, Z.; Zhang, J.; Zhang, L.; Xia, J.; Xie, J.; Sun, Q.; et al. Lactiplantibacillus plantarum N-1 improves autism-like behavior and gut microbiota in mouse. Front. Microbiol. 2023, 14, 1134517. [Google Scholar] [CrossRef]
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef]
- Singh, V.; Lee, G.; Son, H.; Koh, H.; Kim, E.S.; Unno, T.; Shin, J.-H. Butyrate producers, “the sentinel of gut”: Their intestinal significance with and beyond butyrate, and prospective use as microbial therapeutics. Front. Microbiol. 2023, 13, 1103836. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Mu, C.L.; Farzi, A.; Zhu, W.Y. Tryptophan metabolism: A link between the gut microbiota and brain. Adv. Nutr. 2020, 11, 709–723. [Google Scholar] [CrossRef]
- Ney, L.M.; Wipplinger, M.; Grossmann, M.; Engert, N.; Wegner, V.D.; Mosig, A.S. Short chain fatty acids: Key regulators of the local and systemic immune response in inflammatory diseases and infections. Open Biol. 2023, 13, 230014. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Martín, A.; Selma, M.V.; Tomás-Barberán, F.A.; González-Sarrías, A.; Espín, J.C. Where to look into the puzzle of polyphenols and health? The postbiotics and gut microbiota associated with human metabotypes. Mol. Nutr. Food Res. 2020, 64, e1900952. [Google Scholar] [CrossRef]
- Brister, D.; Rose, S.; Delhey, L.; Tippett, M.; Jin, Y.; Gu, H.; Frye, R.E. Metabolomic signatures of autism spectrum disorder. J. Pers. Med. 2022, 12, 1727. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- González Fraguela, M.E.; Diaz Hung, M.-L.; Vera, H.; Maragoto, C.; Noris, E.; Blanco, L.; Galvizu, R.; Robinson, M. Oxidative stress markers in children with autism spectrum disorders. Br. J. Med. Med. Res. 2013, 3, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Kuhad, A.; Bhandari, R. Nitric oxide pathway as a plausible therapeutic target in autism spectrum disorders. Expert Opin. Ther. Targets 2022, 26, 659–679. [Google Scholar] [CrossRef]
- Ju, Z.; Li, M.; Xu, J.; Howell, D.C.; Li, Z.; Chen, F.E. Recent development on COX-2 inhibitors as promising anti-inflammatory agents: The past 10 years. Acta Pharm. Sin. B 2022, 12, 2790–2807. [Google Scholar] [CrossRef]
- Li, F.; Ke, H.; Wang, S.; Mao, W.; Fu, C.; Chen, X.; Fu, Q.; Qin, X.; Huang, Y.; Li, B.; et al. Leaky gut plays a critical role in the pathophysiology of autism in mice by activating the lipopolysaccharide-mediated Toll-like receptor 4–myeloid differentiation factor 88–nuclear factor kappa B signaling pathway. Neurosci. Bull. 2023, 39, 911–928. [Google Scholar] [CrossRef] [PubMed]
- Anaclerio, F.; Minelli, M.; Antonucci, I.; Gatta, V.; Stuppia, L. Microbiota and autism: A review on oral and gut microbiome analysis through 16S rRNA sequencing. Biomedicines 2024, 12, 2686. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Zhang, C.; Teng, X.; Cao, Q.; Deng, Y.; Yang, M.; Wang, L.; Rui, D.; Ling, X.; Wei, C.; Chen, Y.; et al. Gut microbiota dysbiosis exacerbates heart failure by the LPS-TLR4/NF-κB signalling axis: Mechanistic insights and therapeutic potential of TLR4 inhibition. J. Transl. Med. 2025, 23, 762. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.J.; Lee, N.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Amphiregulin induces iNOS and COX-2 expression through NF-κB and MAPK signaling in hepatic inflammation. Mediat. Inflamm. 2023, 2023, 2364121. [Google Scholar] [CrossRef]
- Lee, N.; Heo, Y.J.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Anti-inflammatory effects of empagliflozin and gemigliptin on LPS-stimulated macrophage via the IKK/NF-κB, MKK7/JNK, and JAK2/STAT1 signalling pathways. J. Immunol. Res. 2021, 2021, 9944880. [Google Scholar] [CrossRef]
- Guo, M.; Li, R.; Wang, Y.; Ma, S.; Zhang, Y.; Li, S.; Zhang, H.; Liu, Z.; You, C.; Zheng, H. Lactobacillus plantarum ST-III modulates abnormal behavior and gut microbiota in a mouse model of autism spectrum disorder. Physiol. Behav. 2022, 257, 113965. [Google Scholar] [CrossRef]
- Wang, C.; Chen, W.; Jiang, Y.; Xiao, X.; Zou, Q.; Liang, J.; Zhao, Y.; Wang, Q.; Yuan, T.; Guo, R.; et al. A synbiotic formulation of Lactobacillus reuteri and inulin alleviates ASD-like behaviors in a mouse model: The mediating role of the gut–brain axis. Food Funct. 2024, 15, 387–400. [Google Scholar] [CrossRef]
- Song, Y.; Shi, J.; Fu, X.; Fu, D.; Xu, L.; Liu, W.; Cao, J.; Ding, Y.; Huang, S.; Zhou, L.; et al. Effects of Lactobacillus acidophilus-mediated improvement of the intestinal barrier in mice with autism. Probiotics Antimicrob. Proteins 2025, 18, 5000–5011. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, D.; Flores, A.; Uebelhoer, M.; Pasque, V.; Plath, K.; Iruela-Arispe, M.L.; Christofk, H.R.; Lowry, W.E.; Coller, H.A. Mapping metabolism: Monitoring lactate dehydrogenase activity directly in tissue. J. Vis. Exp. 2018, 136, 57760. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of cell viability by the lactate dehydrogenase assay. Cold Spring Harb. Protoc. 2018, 6, pdb.prot095497. [Google Scholar] [CrossRef]
- Klein, R.; Nagy, O.; Tóthová, C.; Chovanová, F. Clinical and diagnostic significance of lactate dehydrogenase and its isoenzymes in animals. Vet. Med. Int. 2020, 2020, 5346483. [Google Scholar] [CrossRef] [PubMed]
- Faddah, L.; Abdel-Hamid, N.; Abul-Naga, Y.; Ibrahim, S.; Mahmoud, A. Lactate dehydrogenase isoenzyme pattern in the liver tissue of chemically-injured rats treated by combinations of diphenyl dimethyl bicarboxylate. J. Appl. Biomed. 2007, 5, 77–80. [Google Scholar] [CrossRef]
- Chaari, A.; Al Ali, D.; Roach, J. Biochemistry course based undergraduate research experience: Purification, characterization, and identification of an unknown lactate dehydrogenase isoenzyme. Biochem. Mol. Biol. Educ. 2020, 48, 369–380. [Google Scholar] [CrossRef]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse microbiota models: Comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front. Physiol. 2018, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Manca, C.; Boubertakh, B.; Leblanc, N.; Deschênes, T.; Lacroix, S.; Martin, C.; Houde, A.; Veilleux, A.; Flamand, N.; Muccioli, G.G.; et al. Germ-free mice exhibit profound gut microbiota-dependent alterations of intestinal endocannabinoidome signaling. J. Lipid Res. 2020, 61, 70–85. [Google Scholar] [CrossRef]
- Aghighi, F.; Salami, M. What we need to know about the germ-free animal models. AIMS Microbiol. 2024, 10, 107–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.J.; Chang, H.C.; Sung, P.C.; Ge, M.C.; Tang, H.Y.; Cheng, M.L.; Cheng, H.T.; Chou, H.H.; Lin, C.Y.; Lin, W.R.; et al. Oral fecal transplantation enriches Lachnospiraceae and butyrate to mitigate acute liver injury. Cell Rep. 2024, 43, 113591. [Google Scholar] [CrossRef]
- Yuan, C.; Fan, J.; Jiang, L.; Ye, W.; Chen, Z.; Wu, W.; Huang, Q.; Qian, L. Integrated analysis of gut microbiome and liver metabolome to evaluate the effects of fecal microbiota transplantation on lipopolysaccharide/D-galactosamine-induced acute liver injury in mice. Nutrients 2023, 15, 1149. [Google Scholar] [CrossRef]
- Sopková, D.; Hertelyová, Z.; Andrejčáková, Z.; Vlčková, R.; Gancarčíková, S.; Petrilla, V.; Ondrašovičová, S.; Krešáková, L. The application of probiotics and flaxseed promotes metabolism of n-3 polyunsaturated fatty acids in pigs. J. Appl. Anim. Res. 2017, 45, 93–98. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.