






Cells 2024, 13(2), 185; https://doi.org/10.3390/cells13020185 - 18 Jan 2024
Cited by 1 | Viewed by 3484
Abstract
►
Show Figures
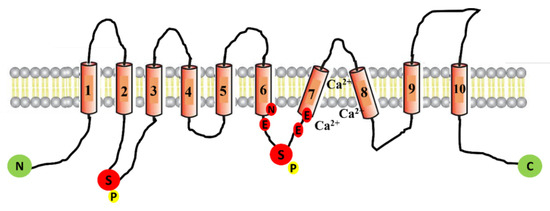
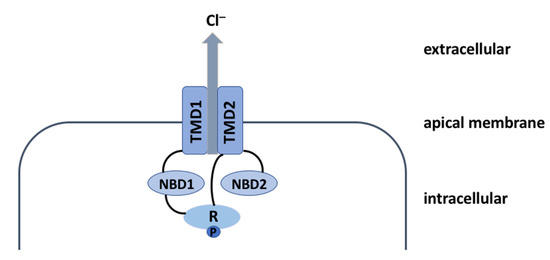
Cystic Fibrosis (CF) is present due to mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, the most frequent variant being p.phe508del. The CFTR protein is a chloride (Cl-) channel which is defective and almost absent of cell membranes when the p.Phe508del
[...] Read more.
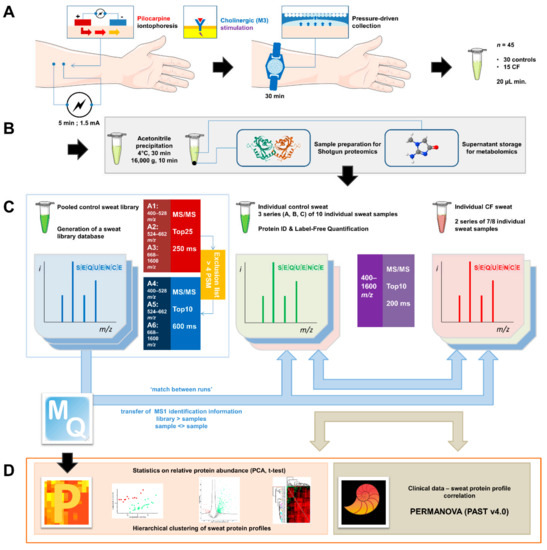
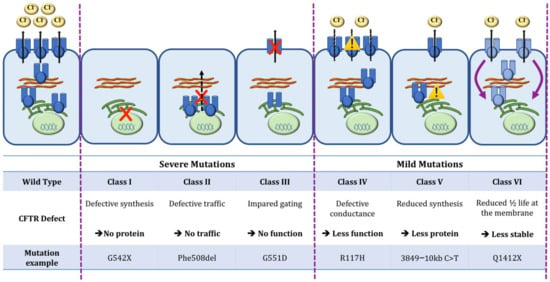
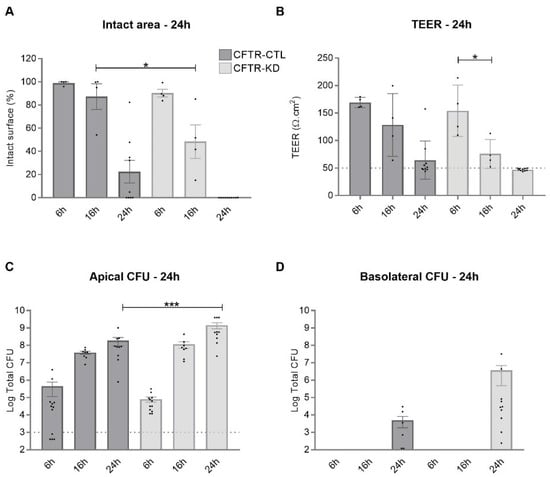
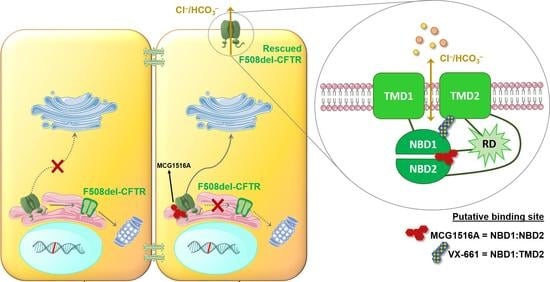
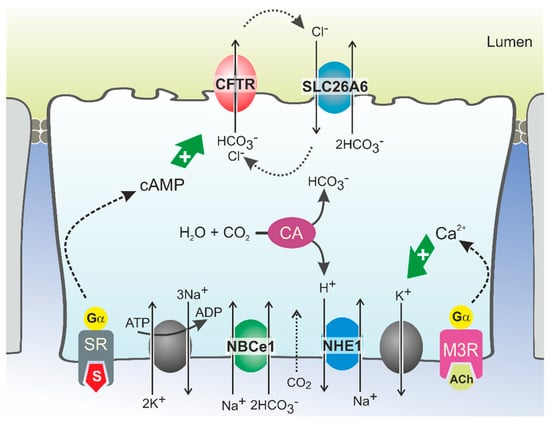
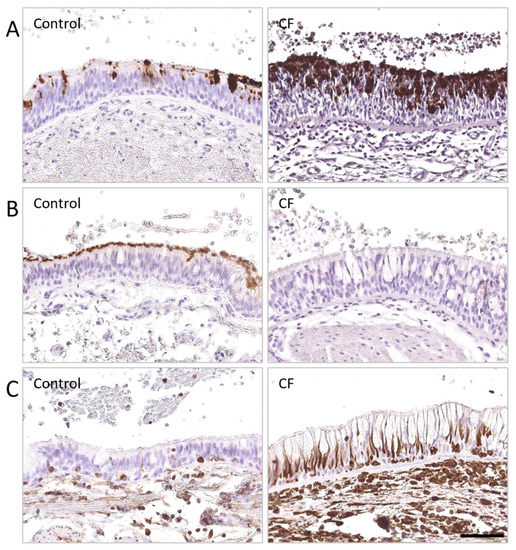
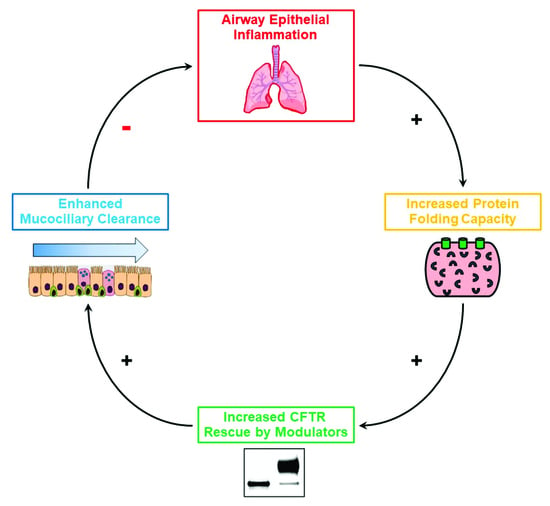
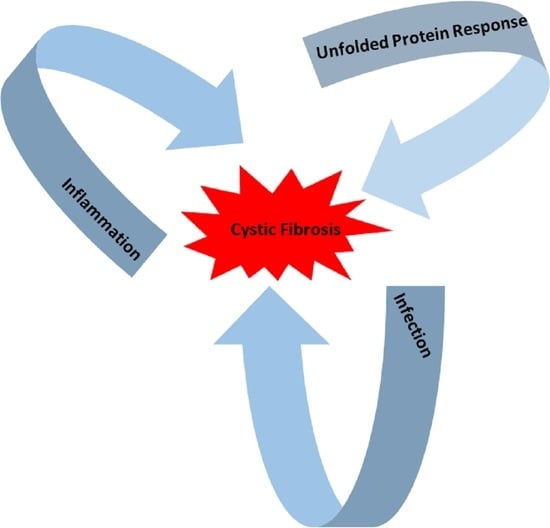
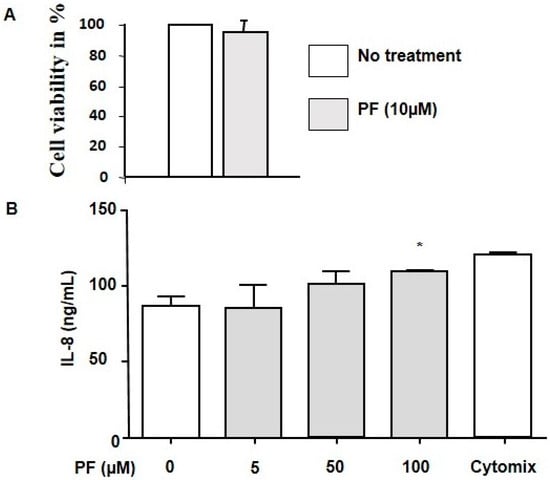
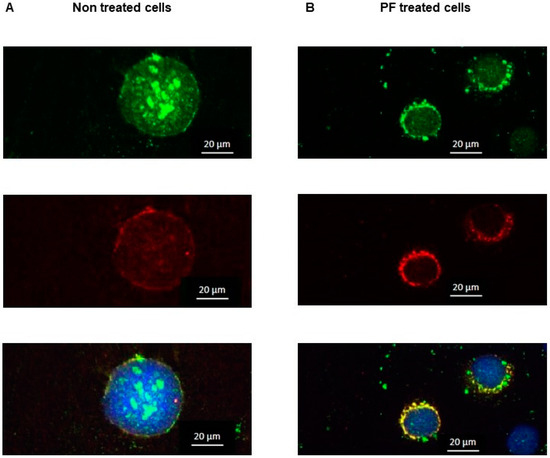
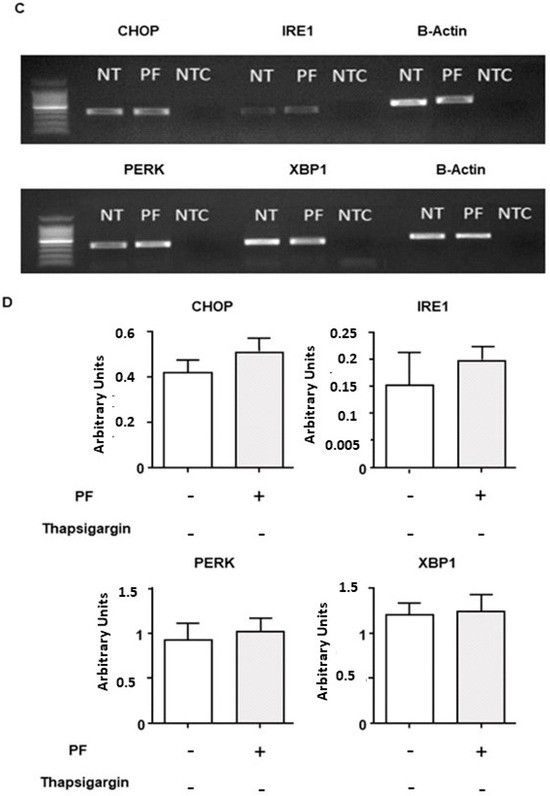
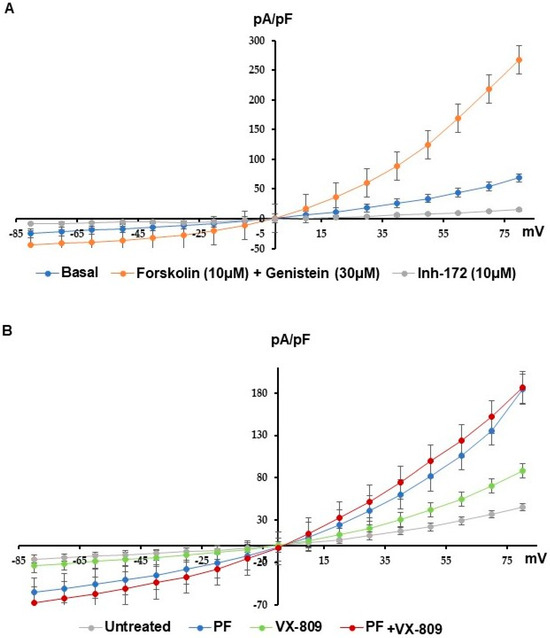
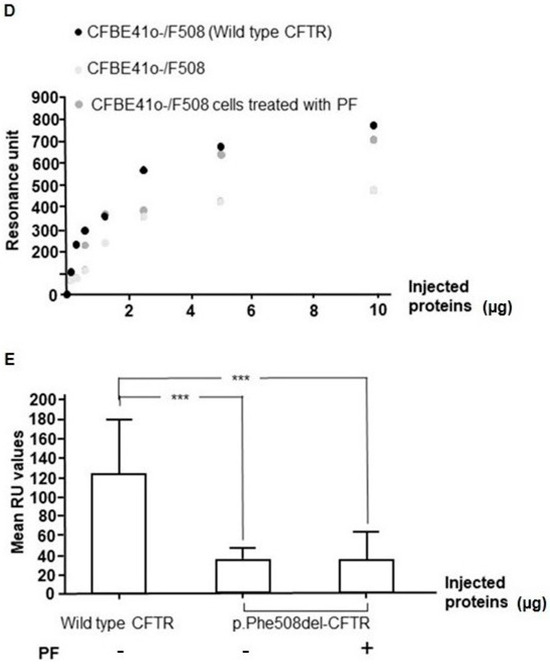
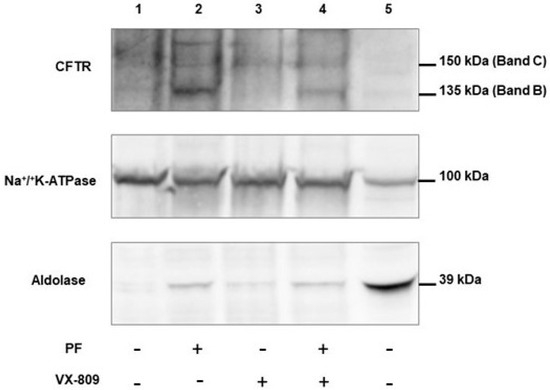
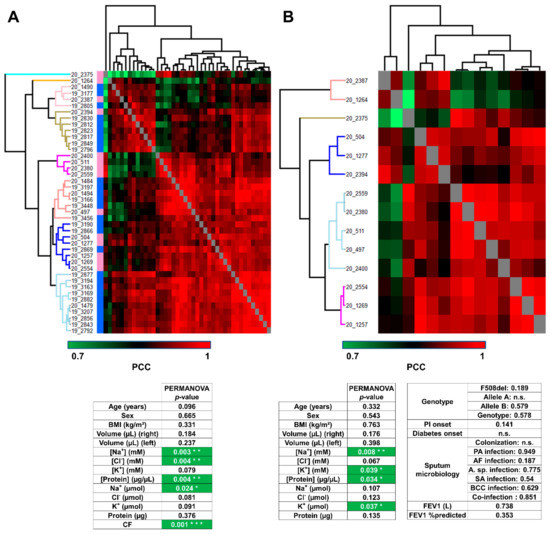
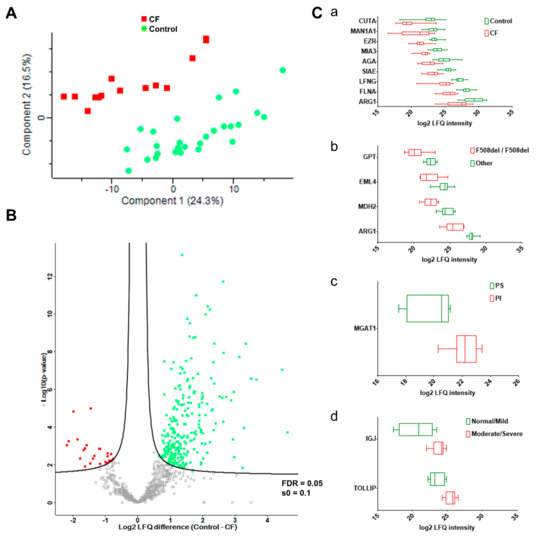
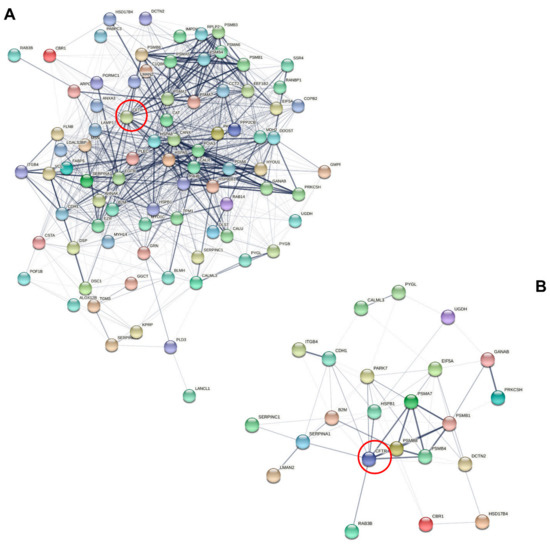
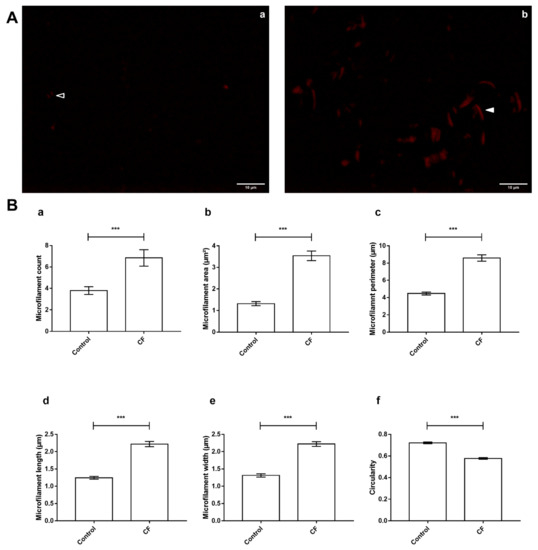
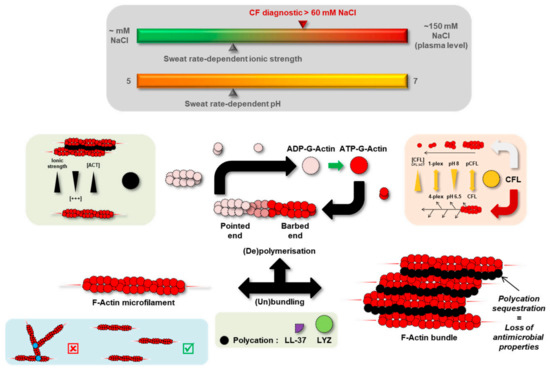
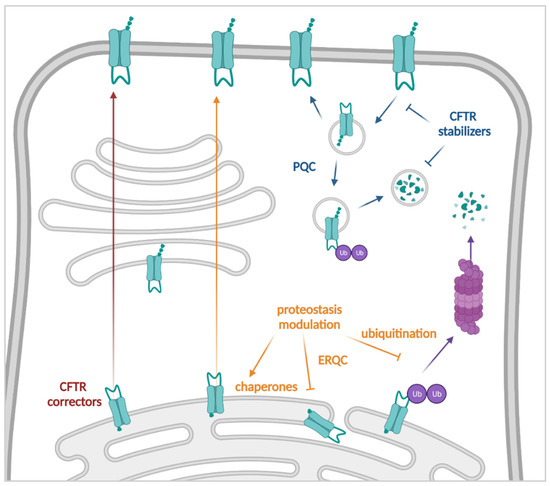
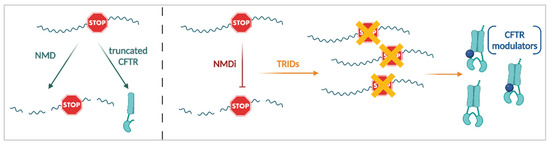
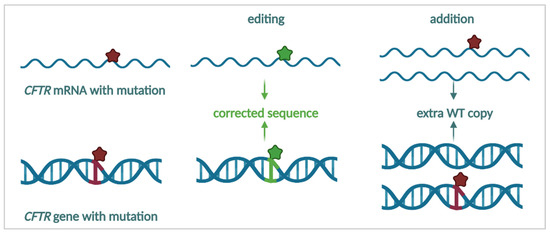
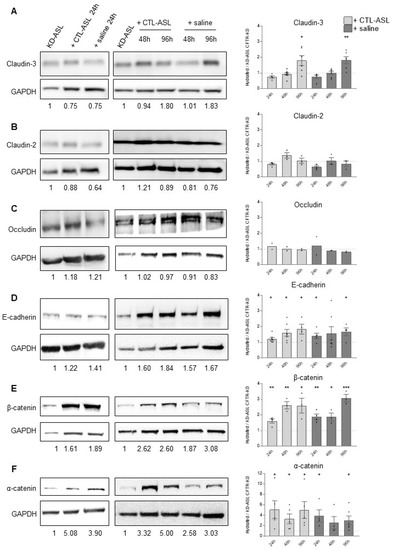
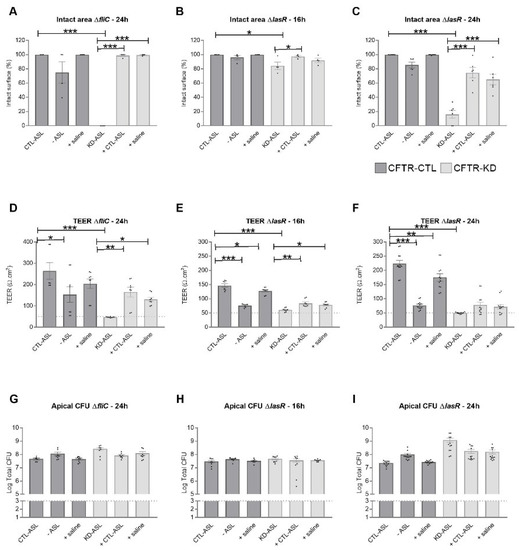
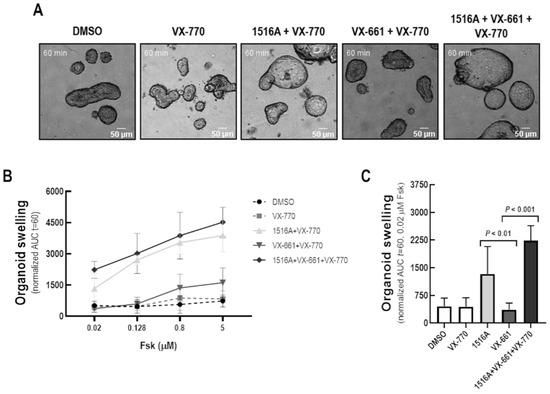
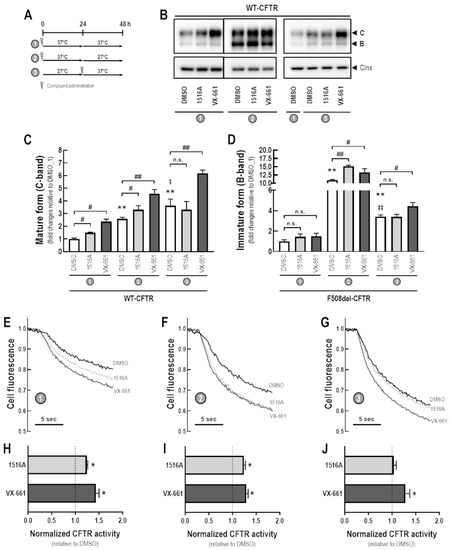
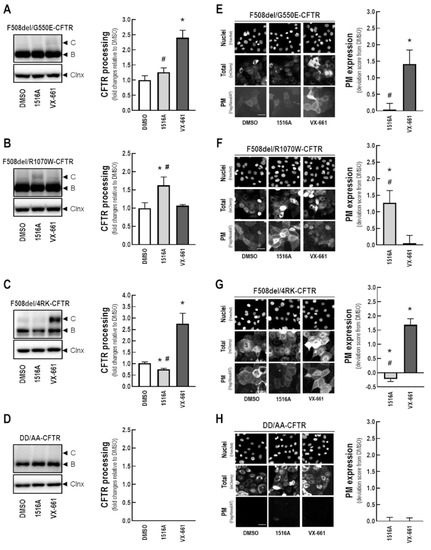
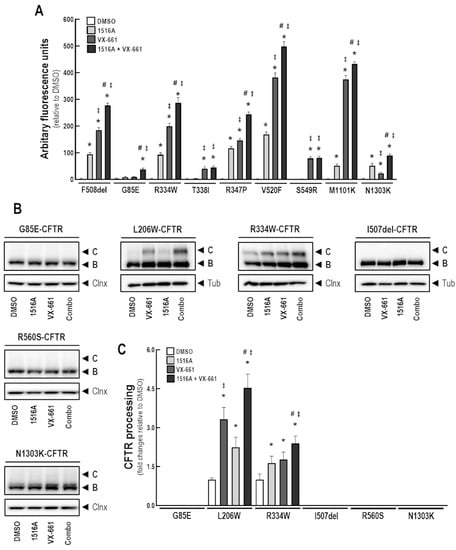
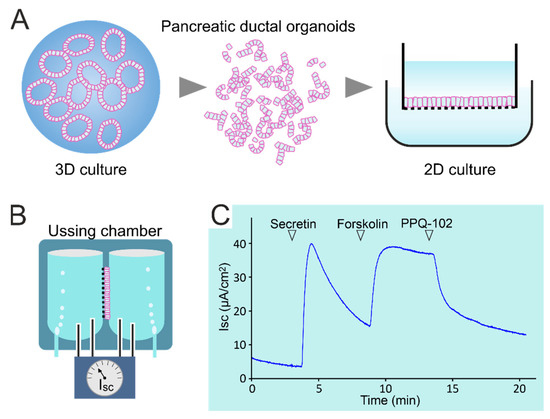
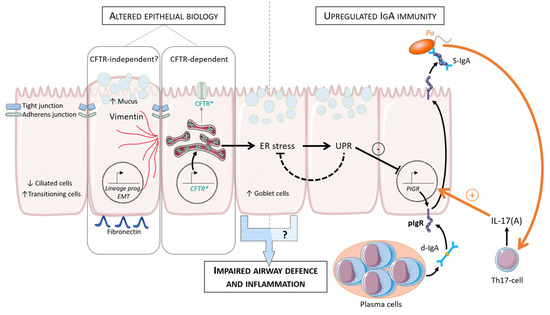
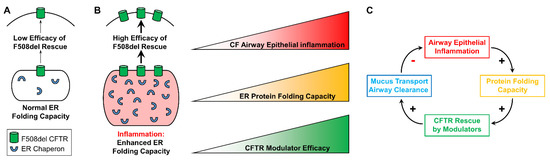
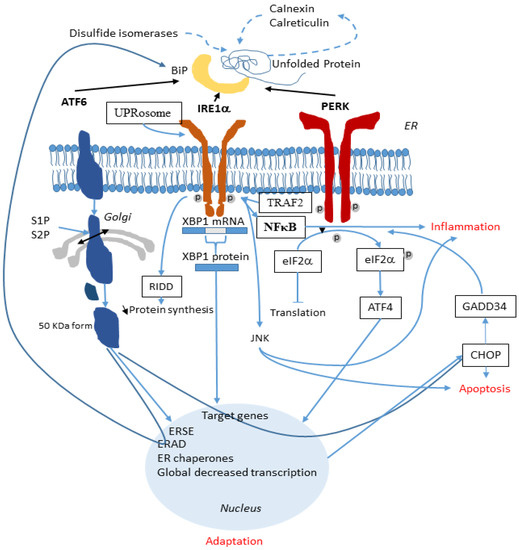
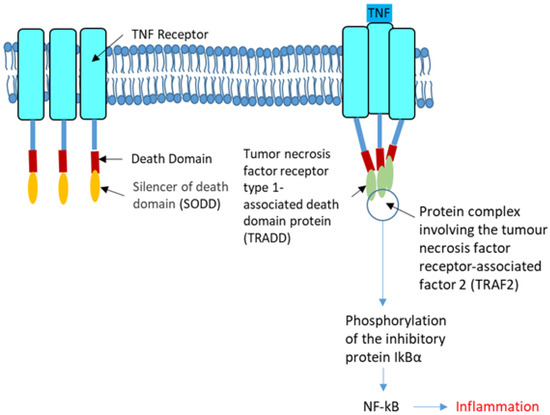
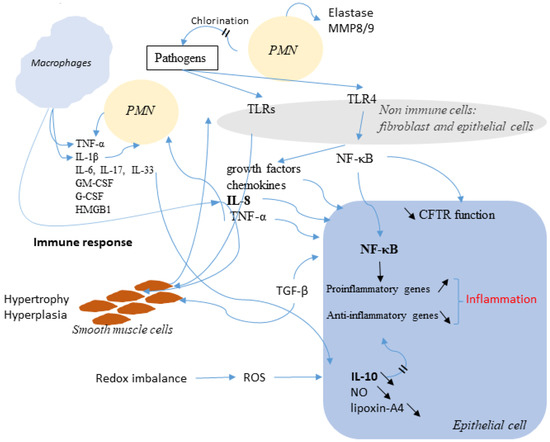
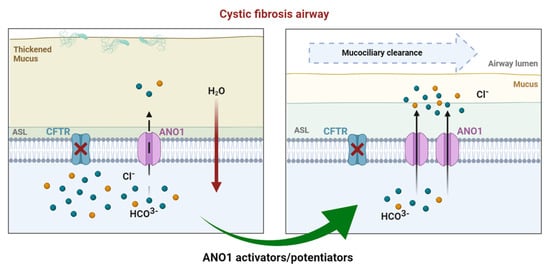
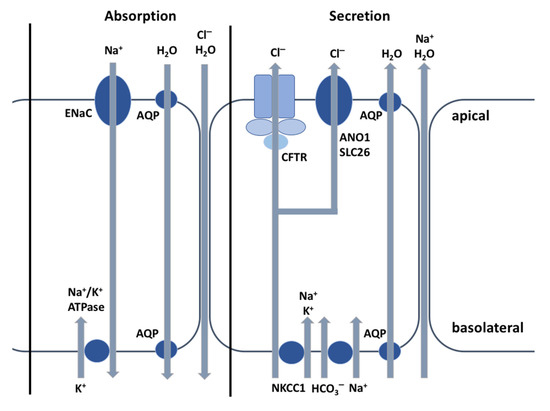
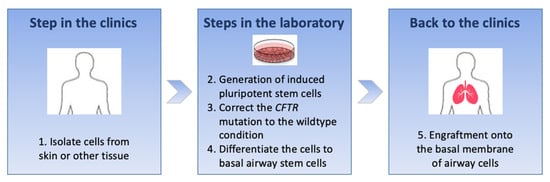
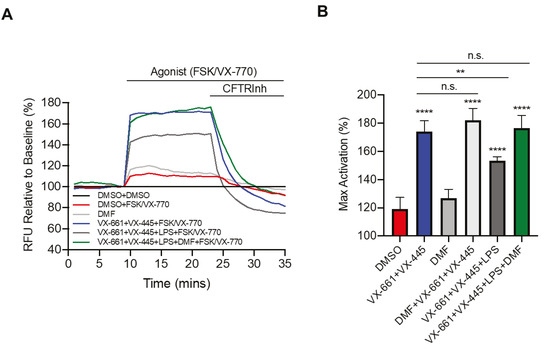
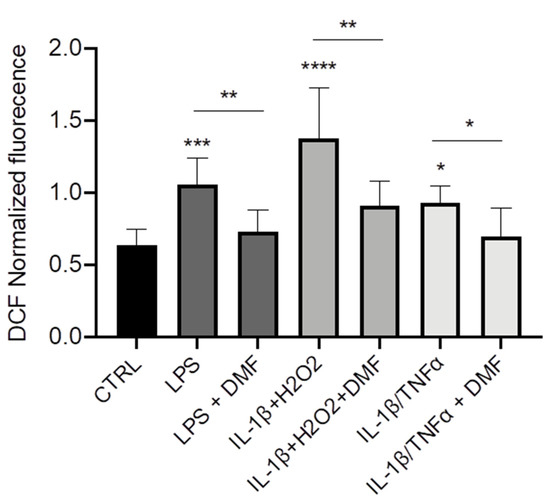
Cystic Fibrosis (CF) is present due to mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, the most frequent variant being p.phe508del. The CFTR protein is a chloride (Cl-) channel which is defective and almost absent of cell membranes when the p.Phe508del mutation is present. The p.Phe508del-CFTR protein is retained in the endoplasmic reticulum (ER) and together with inflammation and infection triggers the Unfolded Protein Response (UPR). During the UPR, the Activating Transcription Factor 6 (ATF6) is activated with cleavage and then decreases the expression of p.Phe508del-CFTR. We have previously shown that the inhibition of the activation of ATF6 alleviates the p.Phe508del-CFTR defects in cells overexpressing the mutated protein. In the present paper, our aim was to inhibit the cleavage of ATF6, and thus its activation in a human bronchial cell line with endogenous p.Phe508del-CFTR expression and in bronchial cells from patients, to be more relevant to CF. This was achieved by inhibiting the protease MBTP1 which is responsible for the cleavage of ATF6. We show here that this inhibition leads to increased mRNA and p.Phe508del-CFTR expression and, consequently, to increased Cl-efflux. We also explain the mechanisms linked to these increases with the modulation of genes when MBTP1 is inhibited. Indeed, RT-qPCR assays show that genes such as HSPA1B, CEBPB, VIMP, PFND2, MAPK8, XBP1, INSIG1, and CALR are modulated. In conclusion, we show that the inhibition of MBTP1 has a beneficial effect in relevant models to CF and that this is due to the modulation of genes involved in the disease.
Full article

Graphical abstract




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}