
Revisiting the Role of Leukocytes in Cystic Fibrosis




Abstract
1. Introduction
2. Leukocytes and Cystic Fibrosis
2.1. Granulocytes
2.2. Monocytes/Macrophages
2.3. Lymphoid Cells
2.4. Platelets
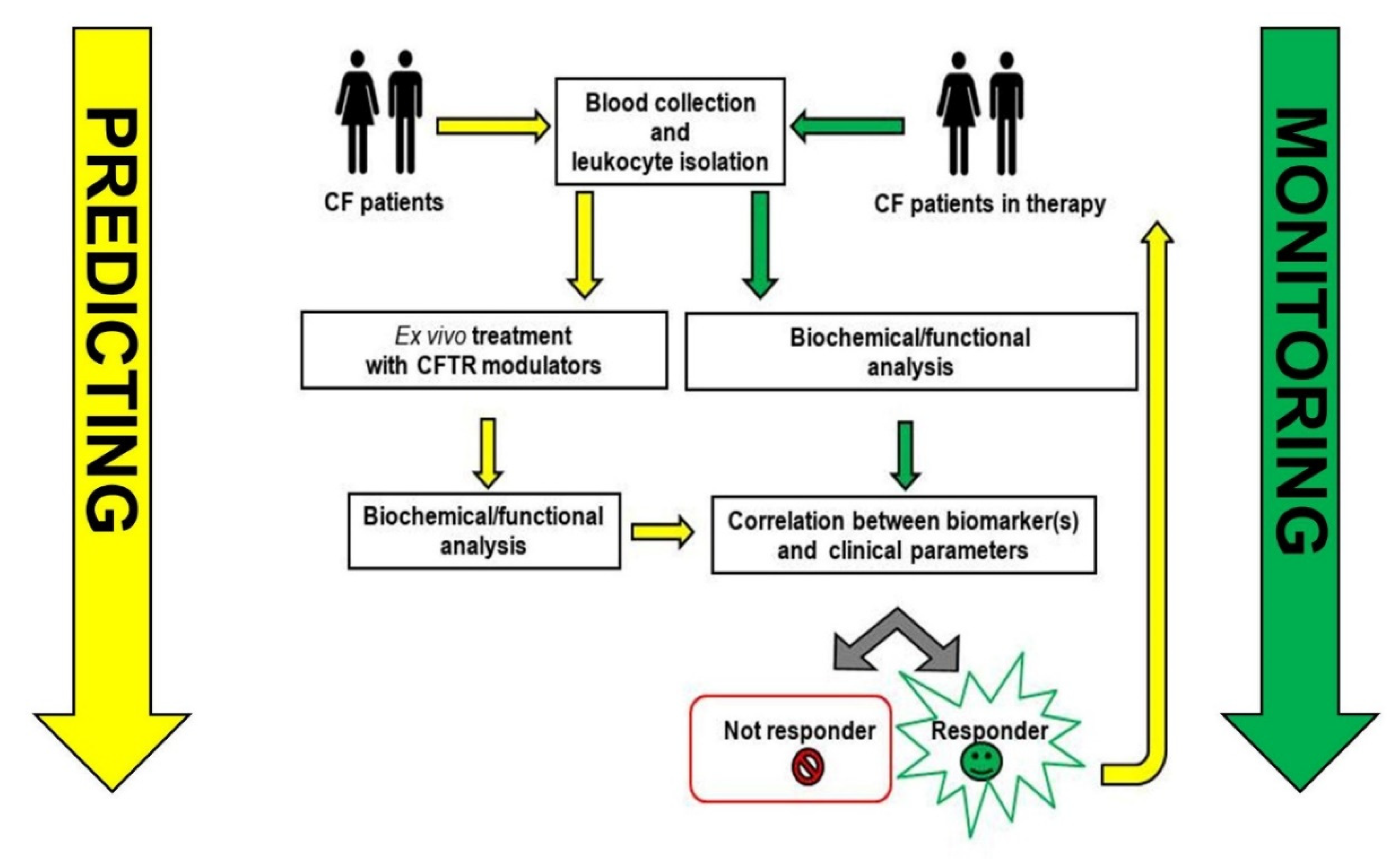
3. Toward Clinical Applications
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef]
- Nichols, D.; Chmiel, J.; Berger, M. Chronic inflammation in the cystic fibrosis lung: Alterations in inter- and intracellular signaling. Clin. Rev. Allergy Immunol. 2008, 34, 146–162. [Google Scholar] [CrossRef]
- Hayes, E.; Pohl, K.; McElvaney, N.G.; Reeves, E.P. The cystic fibrosis neutrophil: A specialized yet potentially defective cell. Arch. Immunol. Ther. Exp. 2011, 59, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, K.; Painter, R.G.; Aiken, M.; Reiser, J.; Stanton, B.A.; Nauseef, W.M.; Wang, G. Cystic fibrosis transmembrane conductance regulator recruitment to phagosomes in neutrophils. J. Innate Immunol. 2013, 5, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef]
- Forrest, O.A.; Ingersoll, S.A.; Preininger, M.K.; Laval, J.; Limoli, D.H.; Brown, M.R.; Lee, F.E.; Bedi, B.; Sadikot, R.T.; Goldberg, J.B.; et al. Frontline Science: Pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J. Leukoc. Biol. 2018, 104, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Tirouvanziam, R.; Gernez, Y.; Conrad, C.K.; Moss, R.B.; Schrijver, I.; Dunn, C.E.; Davies, Z.A.; Herzenberg, L.A.; Herzenberg, L.A. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2008, 105, 4335–4339. [Google Scholar] [CrossRef]
- Makam, M.; Diaz, D.; Laval, J.; Gernez, Y.; Conrad, C.K.; Dunn, C.E.; Davies, Z.A.; Moss, R.B.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Activation of critical, host-induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 5779–5783. [Google Scholar] [CrossRef]
- Corti, A.; Franzini, M.; Cianchetti, S.; Bergamini, G.; Lorenzini, E.; Melotti, P.; Paolicchi, A.; Paggiaro, P.; Pompella, A. Contribution by polymorphonucleate granulocytes to elevated gamma-glutamyltransferase in cystic fibrosis sputum. PLoS ONE 2012, 7, e34772. [Google Scholar] [CrossRef]
- Corti, A.; Pompella, A.; Bergamini, G.; Melotti, P. Glutathione inhalation treatments in cystic fibrosis: The interference of airway gamma-glutamyltransferase. Am. J. Respir. Crit. Care Med. 2014, 189, 233–234. [Google Scholar] [CrossRef]
- Corti, A.; Griese, M.; Hector, A.; Pompella, A. Increasing sputum levels of gamma-glutamyltransferase may identify cystic fibrosis patients who do not benefit from inhaled glutathione. J. Cyst. Fibros. 2017, 16, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, G.; Di Silvestre, D.; Mauri, P.; Cigana, C.; Bragonzi, A.; De Palma, A.; Benazzi, L.; Doring, G.; Assael, B.M.; Melotti, P.; et al. MudPIT analysis of released proteins in Pseudomonas aeruginosa laboratory and clinical strains in relation to pro-inflammatory effects. Integr. Biol. 2012, 4, 270–279. [Google Scholar] [CrossRef]
- Doring, G. Polymorphonuclear leukocyte elastase: Its effects on the pathogenesis of Pseudomonas aeruginosa infection in cystic fibrosis. Antibiot. Chemother. 1989, 42, 169–176. [Google Scholar]
- Hartl, D.; Latzin, P.; Hordijk, P.; Marcos, V.; Rudolph, C.; Woischnik, M.; Krauss-Etschmann, S.; Koller, B.; Reinhardt, D.; Roscher, A.A.; et al. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat. Med. 2007, 13, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S.; Perrett, J.; Forsman-Semb, K.; Marks-Konczalik, J.; Gunawardena, K.; Entwistle, N. Efficacy, safety and effect on biomarkers of AZD9668 in cystic fibrosis. Eur. Respir. J. 2012, 40, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Griese, M.; Latzin, P.; Kappler, M.; Weckerle, K.; Heinzlmaier, T.; Bernhardt, T.; Hartl, D. alpha1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur. Respir. J. 2007, 29, 240–250. [Google Scholar] [CrossRef]
- Yang, C.; Montgomery, M. Dornase alfa for cystic fibrosis. Cochrane Database Syst. Rev. 2018, 9, CD001127. [Google Scholar] [CrossRef]
- Gray, R.D.; Hardisty, G.; Regan, K.H.; Smith, M.; Robb, C.T.; Duffin, R.; Mackellar, A.; Felton, J.M.; Paemka, L.; McCullagh, B.N.; et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 2018, 73, 134–144. [Google Scholar] [CrossRef]
- Bratcher, P.E.; Rowe, S.M.; Reeves, G.; Roberts, T.; Szul, T.; Harris, W.T.; Tirouvanziam, R.; Gaggar, A. Alterations in blood leukocytes of G551D-bearing cystic fibrosis patients undergoing treatment with ivacaftor. J. Cyst. Fibros. 2016, 15, 67–73. [Google Scholar] [CrossRef]
- Hardisty, G.R.; Law, S.M.; Carter, S.; Grogan, B.; Singh, P.K.; McKone, E.F.; Gray, R.D. Ivacaftor modifies cystic fibrosis neutrophil phenotype in subjects with R117H residual function CFTR mutations. Eur. Respir. J. 2021, 57, 2002161. [Google Scholar] [CrossRef]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef]
- Haggie, P.M.; Verkman, A.S. Cystic fibrosis transmembrane conductance regulator-independent phagosomal acidification in macrophages. J. Biol. Chem. 2007, 282, 31422–31428. [Google Scholar] [CrossRef]
- Lamothe, J.; Valvano, M.A. Burkholderia cenocepacia-induced delay of acidification and phagolysosomal fusion in cystic fibrosis transmembrane conductance regulator (CFTR)-defective macrophages. Microbiology 2008, 154, 3825–3834. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A., Jr.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G.; et al. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.G.; Bonvillain, R.W.; Valentine, V.G.; Lombard, G.A.; LaPlace, S.G.; Nauseef, W.M.; Wang, G. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J. Leukoc. Biol. 2008, 83, 1345–1353. [Google Scholar] [CrossRef]
- Zhang, S.; Shrestha, C.L.; Wisniewski, B.L.; Pham, H.; Hou, X.; Li, W.; Dong, Y.; Kopp, B.T. Consequences of CRISPR-Cas9-Mediated CFTR Knockout in Human Macrophages. Front. Immunol. 2020, 11, 1871. [Google Scholar] [CrossRef]
- Wright, A.K.; Rao, S.; Range, S.; Eder, C.; Hofer, T.P.; Frankenberger, M.; Kobzik, L.; Brightling, C.; Grigg, J.; Ziegler-Heitbrock, L. Pivotal Advance: Expansion of small sputum macrophages in CF: Failure to express MARCO and mannose receptors. J. Leukoc. Biol. 2009, 86, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Sorio, C.; Melotti, P. Editorial: The role of macrophages and their scavenger receptors in cystic fibrosis. J. Leukoc. Biol. 2009, 86, 465–468. [Google Scholar] [CrossRef]
- Bruscia, E.M.; Zhang, P.X.; Ferreira, E.; Caputo, C.; Emerson, J.W.; Tuck, D.; Krause, D.S.; Egan, M.E. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 295–304. [Google Scholar] [CrossRef]
- Bonfield, T.L.; Hodges, C.A.; Cotton, C.U.; Drumm, M.L. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J. Leukoc. Biol. 2012, 92, 1111–1122. [Google Scholar] [CrossRef]
- McElvaney, O.J.; Zaslona, Z.; Becker-Flegler, K.; Palsson-McDermott, E.M.; Boland, F.; Gunaratnam, C.; Gulbins, E.; O’Neill, L.A.; Reeves, E.P.; McElvaney, N.G. Specific Inhibition of the NLRP3 Inflammasome as an Antiinflammatory Strategy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1381–1391. [Google Scholar] [CrossRef]
- Rosenow, T.; Mok, L.C.; Turkovic, L.; Berry, L.J.; Sly, P.D.; Ranganathan, S.; Tiddens, H.; Stick, S.M. The cumulative effect of inflammation and infection on structural lung disease in early cystic fibrosis. Eur. Respir. J. 2019, 54, 1801771. [Google Scholar] [CrossRef] [PubMed]
- Scambler, T.; Jarosz-Griffiths, H.H.; Lara-Reyna, S.; Pathak, S.; Wong, C.; Holbrook, J.; Martinon, F.; Savic, S.; Peckham, D.; McDermott, M.F. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. eLife 2019, 8, e49248. [Google Scholar] [CrossRef] [PubMed]
- Kopp, B.T.; Fitch, J.; Jaramillo, L.; Shrestha, C.L.; Robledo-Avila, F.; Zhang, S.; Palacios, S.; Woodley, F.; Hayes, D., Jr.; Partida-Sanchez, S.; et al. Whole-blood transcriptomic responses to lumacaftor/ivacaftor therapy in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 245–254. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Scambler, T.; Wong, C.H.; Lara-Reyna, S.; Holbrook, J.; Martinon, F.; Savic, S.; Whitaker, P.; Etherington, C.; Spoletini, G.; et al. Different CFTR modulator combinations downregulate inflammation differently in cystic fibrosis. eLife 2020, 9, e54556. [Google Scholar] [CrossRef]
- Isopi, E.; Mattoscio, D.; Codagnone, M.; Mari, V.C.; Lamolinara, A.; Patruno, S.; D’Aurora, M.; Cianci, E.; Nespoli, A.; Franchi, S.; et al. Resolvin D1 reduces lung infection and inflammation activating resolution in cystic fibrosis. Front. Immunol. 2020, 11, 581. [Google Scholar] [CrossRef]
- Recchiuti, A.; Patruno, S.; Plebani, R.; Romano, M. The resolution approach to cystic fibrosis inflammation. Front. Pharmacol. 2020, 11, 1129. [Google Scholar] [CrossRef]
- Das, J.; Sharma, A.; Jindal, A.; Aggarwal, V.; Rawat, A. Leukocyte adhesion defect: Where do we stand circa 2019? Genes Dis. 2020, 7, 107–114. [Google Scholar] [CrossRef]
- Sorio, C.; Montresor, A.; Bolomini-Vittori, M.; Caldrer, S.; Rossi, B.; Dusi, S.; Angiari, S.; Johansson, J.E.; Vezzalini, M.; Leal, T.; et al. Mutations of cystic fibrosis transmembrane conductance regulator gene cause a monocyte-selective adhesion deficiency. Am. J. Respir. Crit. Care Med. 2016, 193, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Paemka, L.; McCullagh, B.N.; Abou Alaiwa, M.H.; Stoltz, D.A.; Dong, Q.; Randak, C.O.; Gray, R.D.; McCray, P.B., Jr. Monocyte derived macrophages from CF pigs exhibit increased inflammatory responses at birth. J. Cyst. Fibros. 2017, 16, 471–474. [Google Scholar] [CrossRef]
- Fisher, J.T.; Zhang, Y.; Engelhardt, J.F. Comparative biology of cystic fibrosis animal models. Methods Mol. Biol. 2011, 742, 311–334. [Google Scholar] [CrossRef]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef]
- Sun, X.; Sui, H.; Fisher, J.T.; Yan, Z.; Liu, X.; Cho, H.J.; Joo, N.S.; Zhang, Y.; Zhou, W.; Yi, Y.; et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Investig. 2010, 120, 3149–3160. [Google Scholar] [CrossRef]
- Wine, J.J. The development of lung disease in cystic fibrosis pigs. Sci. Transl. Med. 2010, 2, 29ps20. [Google Scholar] [CrossRef]
- Favia, M.; Gallo, C.; Guerra, L.; De Venuto, D.; Diana, A.; Polizzi, A.M.; Montemurro, P.; Mariggio, M.A.; Leonetti, G.; Manca, A.; et al. Treatment of Cystic Fibrosis Patients Homozygous for F508del with Lumacaftor-Ivacaftor (Orkambi((R))) Restores Defective CFTR Channel Function in Circulating Mononuclear Cells. Int. J. Mol. Sci. 2020, 21, 2398. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Vezzalini, M.; Verze, G.; Caldrer, S.; Bolognin, S.; Buffelli, M.; Bellisola, G.; Tridello, G.; Assael, B.M.; Melotti, P.; et al. Detection of CFTR protein in human leukocytes by flow cytometry. Cytometry A 2014, 85, 611–620. [Google Scholar] [CrossRef]
- Pedrazzi, M.; Vercellone, S.; Barberis, E.; Capraro, M.; De Tullio, R.; Cresta, F.; Casciaro, R.; Castellani, C.; Patrone, M.; Marengo, E.; et al. Identification of Potential Leukocyte Biomarkers Related to Drug Recovery of CFTR: Clinical Applications in Cystic Fibrosis. Int. J. Mol. Sci. 2021, 22, 3928. [Google Scholar] [CrossRef] [PubMed]
- Sorio, C.; Buffelli, M.; Angiari, C.; Ettorre, M.; Johansson, J.; Vezzalini, M.; Viviani, L.; Ricciardi, M.; Verze, G.; Assael, B.M.; et al. Defective CFTR expression and function are detectable in blood monocytes: Development of a new blood test for cystic fibrosis. PLoS ONE 2011, 6, e22212. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.V.; Nghiem, P.T.; Gardner, P.; Martens, C.L. Human lymphocytes transcribe the cystic fibrosis transmembrane conductance regulator gene and exhibit CF-defective cAMP-regulated chloride current. J. Biol. Chem. 1992, 267, 3242–3248. [Google Scholar] [CrossRef]
- Regamey, N.; Tsartsali, L.; Hilliard, T.N.; Fuchs, O.; Tan, H.L.; Zhu, J.; Qiu, Y.S.; Alton, E.W.; Jeffery, P.K.; Bush, A.; et al. Distinct patterns of inflammation in the airway lumen and bronchial mucosa of children with cystic fibrosis. Thorax 2012, 67, 164–170. [Google Scholar] [CrossRef]
- Mueller, C.; Braag, S.A.; Keeler, A.; Hodges, C.; Drumm, M.; Flotte, T.R. Lack of cystic fibrosis transmembrane conductance regulator in CD3+ lymphocytes leads to aberrant cytokine secretion and hyperinflammatory adaptive immune responses. Am. J. Respir. Cell Mol. Biol. 2011, 44, 922–929. [Google Scholar] [CrossRef]
- Tiringer, K.; Treis, A.; Fucik, P.; Gona, M.; Gruber, S.; Renner, S.; Dehlink, E.; Nachbaur, E.; Horak, F.; Jaksch, P.; et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2013, 187, 621–629. [Google Scholar] [CrossRef]
- Iannitti, R.G.; Carvalho, A.; Cunha, C.; De Luca, A.; Giovannini, G.; Casagrande, A.; Zelante, T.; Vacca, C.; Fallarino, F.; Puccetti, P.; et al. Th17/Treg imbalance in murine cystic fibrosis is linked to indoleamine 2,3-dioxygenase deficiency but corrected by kynurenines. Am. J. Respir. Crit. Care Med. 2013, 187, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Hector, A.; Schafer, H.; Poschel, S.; Fischer, A.; Fritzsching, B.; Ralhan, A.; Carevic, M.; Oz, H.; Zundel, S.; Hogardt, M.; et al. Regulatory T-cell impairment in cystic fibrosis patients with chronic pseudomonas infection. Am. J. Respir. Crit. Care Med. 2015, 191, 914–923. [Google Scholar] [CrossRef]
- Mulcahy, E.M.; Hudson, J.B.; Beggs, S.A.; Reid, D.W.; Roddam, L.F.; Cooley, M.A. High peripheral blood th17 percent associated with poor lung function in cystic fibrosis. PLoS ONE 2015, 10, e0120912. [Google Scholar] [CrossRef]
- Siegmann, N.; Worbs, D.; Effinger, F.; Bormann, T.; Gebhardt, M.; Ulrich, M.; Wermeling, F.; Muller-Hermelink, E.; Biedermann, T.; Tighe, M.; et al. Invariant natural killer T (iNKT) cells prevent autoimmunity, but induce pulmonary inflammation in cystic fibrosis. Cell Physiol. Biochem. 2014, 34, 56–70. [Google Scholar] [CrossRef]
- Mulcahy, E.M.; Cooley, M.A.; McGuire, H.; Asad, S.; Fazekas de St Groth, B.; Beggs, S.A.; Roddam, L.F. Widespread alterations in the peripheral blood innate immune cell profile in cystic fibrosis reflect lung pathology. Immunol. Cell Biol. 2019, 97, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Limberis, M.P.; Figueredo, J.; Calcedo, R.; Wilson, J.M. Activation of CFTR-specific T Cells in cystic fibrosis mice following gene transfer. Mol. Ther. 2007, 15, 1694–1700. [Google Scholar] [CrossRef]
- Calcedo, R.; Griesenbach, U.; Dorgan, D.J.; Soussi, S.; Boyd, A.C.; Davies, J.C.; Higgins, T.E.; Hyde, S.C.; Gill, D.R.; Innes, J.A.; et al. Self-reactive CFTR T cells in humans: Implications for gene therapy. Hum. Gene Ther. Clin. Dev. 2013, 24, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Lu, B.; Quintero, J.R.; Vargas, S.O.; Patel, A.S.; Owen, C.A.; Gerard, N.P.; Gerard, C.; Cernadas, M. CFTR regulates B cell activation and lymphoid follicle development. Respir. Res. 2019, 20, 133. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Weber, C. Platelets as immune cells: Bridging inflammation and cardiovascular disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.P.; Linden, M.D.; Frelinger, A.L., III; Barnard, M.R.; Spencer-Manzon, M.; Morris, J.E.; Salem, R.O.; Laposata, M.; Michelson, A.D. Platelet activation in cystic fibrosis. Blood 2005, 105, 4635–4641. [Google Scholar] [CrossRef] [PubMed]
- Mattoscio, D.; Evangelista, V.; De Cristofaro, R.; Recchiuti, A.; Pandolfi, A.; Di Silvestre, S.; Manarini, S.; Martelli, N.; Rocca, B.; Petrucci, G.; et al. Cystic fibrosis transmembrane conductance regulator (CFTR) expression in human platelets: Impact on mediators and mechanisms of the inflammatory response. FASEB J. 2010, 24, 3970–3980. [Google Scholar] [CrossRef]
- O’Sullivan, B.P.; Michelson, A.D. The inflammatory role of platelets in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2006, 173, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Munoz, G.; Yu, M.A.; Lefrancais, E.; Mallavia, B.; Valet, C.; Tian, J.J.; Ranucci, S.; Wang, K.M.; Liu, Z.; Kwaan, N.; et al. Cystic fibrosis transmembrane conductance regulator dysfunction in platelets drives lung hyperinflammation. J. Clin. Investig. 2020, 130, 2041–2053. [Google Scholar] [CrossRef]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Ahmadi, S.; Bozoky, Z.; Di Paola, M.; Xia, S.; Li, C.; Wong, A.P.; Wellhauser, L.; Molinski, S.V.; Ip, W.; Ouyang, H.; et al. Phenotypic profiling of CFTR modulators in patient-derived respiratory epithelia. NPJ Genom. Med. 2017, 2, 12. [Google Scholar] [CrossRef]
- Hubert, D.; Chiron, R.; Camara, B.; Grenet, D.; Prevotat, A.; Bassinet, L.; Dominique, S.; Rault, G.; Macey, J.; Honore, I.; et al. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J. Cyst. Fibros. 2017, 16, 388–391. [Google Scholar] [CrossRef]
- Jennings, M.T.; Dezube, R.; Paranjape, S.; West, N.E.; Hong, G.; Braun, A.; Grant, J.; Merlo, C.A.; Lechtzin, N. An observational study of outcomes and tolerances in patients with cystic fibrosis initiated on lumacaftor/ivacaftor. Ann. Am. Thorac. Soc. 2017, 14, 1662–1666. [Google Scholar] [CrossRef]
- Masson, A.; Schneider-Futschik, E.K.; Baatallah, N.; Nguyen-Khoa, T.; Girodon, E.; Hatton, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; et al. Predictive factors for lumacaftor/ivacaftor clinical response. J. Cyst. Fibros. 2019, 18, 368–374. [Google Scholar] [CrossRef]
- Adam, R.J.; Hisert, K.B.; Dodd, J.D.; Grogan, B.; Launspach, J.L.; Barnes, J.K.; Gallagher, C.G.; Sieren, J.P.; Gross, T.J.; Fischer, A.J.; et al. Acute administration of ivacaftor to people with cystic fibrosis and a G551D-CFTR mutation reveals smooth muscle abnormalities. JCI Insight 2016, 1, e86183. [Google Scholar] [CrossRef]
- Guerra, L.; D’Oria, S.; Favia, M.; Castellani, S.; Santostasi, T.; Polizzi, A.M.; Mariggio, M.A.; Gallo, C.; Casavola, V.; Montemurro, P.; et al. CFTR-dependent chloride efflux in cystic fibrosis mononuclear cells is increased by ivacaftor therapy. Pediatr. Pulmonol. 2017, 52, 900–908. [Google Scholar] [CrossRef]
- Hisert, K.B.; Birkland, T.P.; Schoenfelt, K.Q.; Long, M.E.; Grogan, B.; Carter, S.; Liles, W.C.; McKone, E.F.; Becker, L.; Manicone, A.M. Ivacaftor decreases monocyte sensitivity to interferon-gamma in people with cystic fibrosis. ERJ Open Res. 2020, 6, 00318–2019. [Google Scholar] [CrossRef]
- Hisert, K.B.; Schoenfelt, K.Q.; Cooke, G.; Grogan, B.; Launspach, J.L.; Gallagher, C.G.; Donnelly, S.C.; Welsh, M.J.; Singh, P.K.; McKone, E.F.; et al. Ivacaftor-induced proteomic changes suggest monocyte defects may contribute to the pathogenesis of cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 54, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Sun, Z.; Jiang, Y.; Ferguson, A.A.; Pilewski, J.M.; Kolls, J.K.; Chen, W.; Chen, K. Transcriptomic responses to ivacaftor and prediction of ivacaftor clinical responsiveness. Am. J. Respir. Cell Mol. Biol. 2019, 61, 643–652. [Google Scholar] [CrossRef]
- Ettorre, M.; Verze, G.; Caldrer, S.; Johansson, J.; Calcaterra, E.; Assael, B.M.; Melotti, P.; Sorio, C.; Buffelli, M. Electrophysiological evaluation of cystic fibrosis conductance transmembrane regulator (CFTR) expression in human monocytes. Biochim. Biophys. Acta 2014, 1840, 3088–3095. [Google Scholar] [CrossRef] [PubMed]
- Caldrer, S.; Bergamini, G.; Sandri, A.; Vercellone, S.; Rodella, L.; Cerofolini, A.; Tomba, F.; Catalano, F.; Frulloni, L.; Buffelli, M.; et al. Cystic fibrosis transmembrane conductance regulator functional evaluations in a G542X+/− IVS8Tn:T7/9 patient with acute recurrent pancreatitis. World J. Clin. Cases 2019, 7, 3757–3764. [Google Scholar] [CrossRef] [PubMed]
- Caldrer, S.; Verze, G.; Johansson, J.; Sorio, C.; Angiari, C.; Buffelli, M.; Assael, B.M.; Melotti, P. Challenging the diagnosis of cystic fibrosis in a patient carrying the 186-8T/C allelic variant in the CF transmembrane conductance regulator gene. BMC Pulm. Med. 2014, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Sorio, C.; Angiari, C.; Johansson, J.; Verze, G.; Ettorre, M.; Buffelli, M.; Castellani, C.; Assael, B.M.; Melotti, P. Impaired CFTR function in mild cystic fibrosis associated with the S977F/T5TG12complex allele in trans with F508del mutation. J. Cyst. Fibros. 2013, 12, 821–825. [Google Scholar] [CrossRef][Green Version]
- Ng, H.P.; Valentine, V.G.; Wang, G. CFTR targeting during activation of human neutrophils. J. Leukoc. Biol. 2016, 100, 1413–1424. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| CFTR Function/Currents | CFTR Function/Others | Protein | mRNA | References | |
|---|---|---|---|---|---|
| Mono/mac | Yes | Yes | FC/WB/IF | Yes | [21,46,48,77] |
| PMN | ND | ND | FC/WB/IF | Yes | [4,24,81] |
| B cells | Yes | ND | FC | Yes | [49] |
| T cells | ND | ND | FC | Yes | [49] |
| platelets | ND | Yes | ICH/WB/EM | undetectable | [63,65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Averna, M.; Melotti, P.; Sorio, C. Revisiting the Role of Leukocytes in Cystic Fibrosis. Cells 2021, 10, 3380. https://doi.org/10.3390/cells10123380
Averna M, Melotti P, Sorio C. Revisiting the Role of Leukocytes in Cystic Fibrosis. Cells. 2021; 10(12):3380. https://doi.org/10.3390/cells10123380
Chicago/Turabian StyleAverna, Monica, Paola Melotti, and Claudio Sorio. 2021. "Revisiting the Role of Leukocytes in Cystic Fibrosis" Cells 10, no. 12: 3380. https://doi.org/10.3390/cells10123380
APA StyleAverna, M., Melotti, P., & Sorio, C. (2021). Revisiting the Role of Leukocytes in Cystic Fibrosis. Cells, 10(12), 3380. https://doi.org/10.3390/cells10123380