
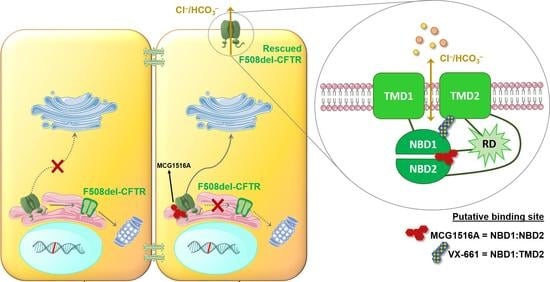
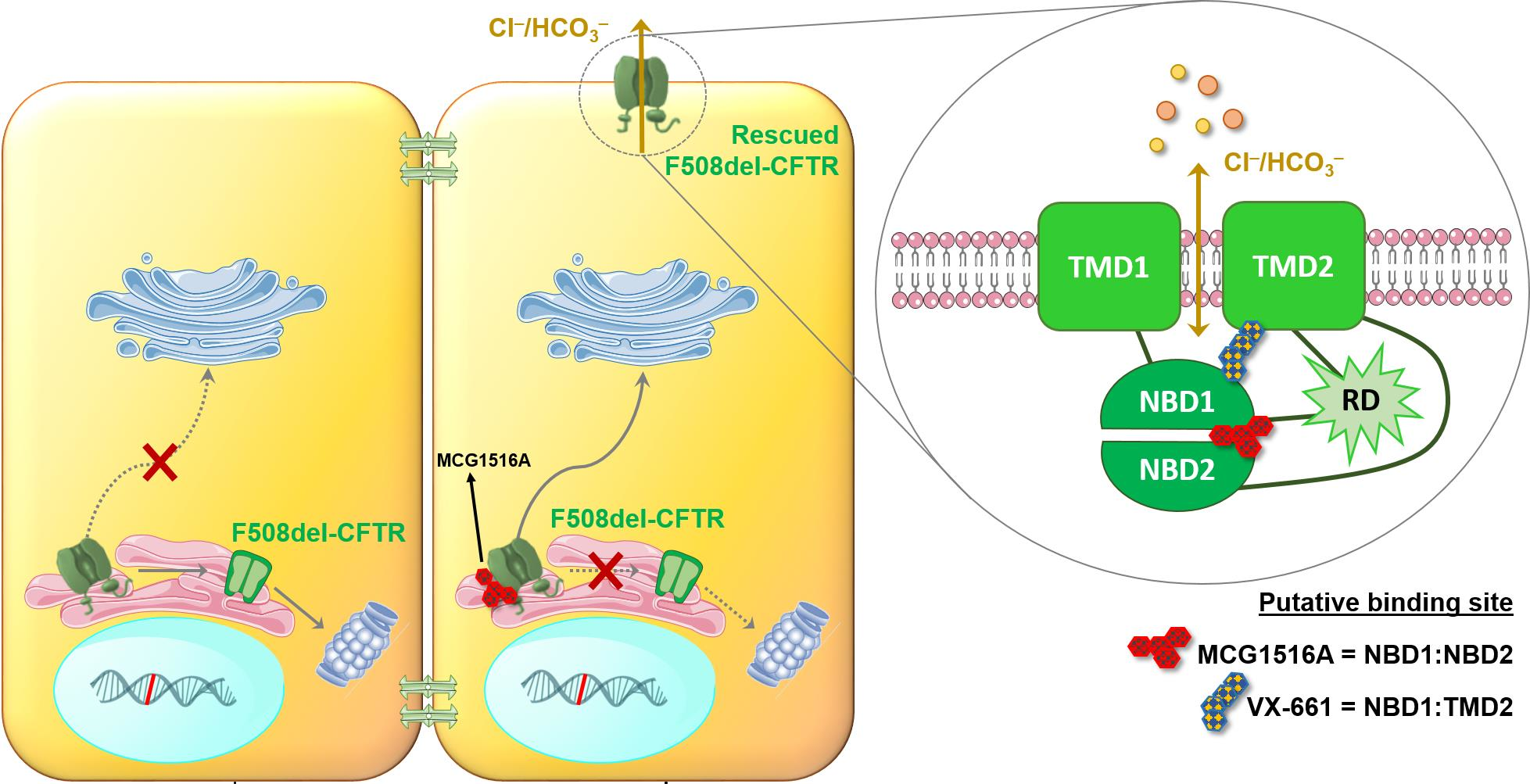
Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A

 ,
,  , , , ,
, , , ,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Compounds
2.2. Cell Culture
2.3. Generation of Novel Cell Lines
2.4. Immunostaining and Fluorescence Microscopy
2.5. HS-YFP Assay on the Plate Reader
2.6. Western Blot
2.7. FLIPR Membrane Potential (FMP) Assay
2.8. Fsk-Induced Swelling (FIS) Assay of Intestinal Organoids
2.9. Statistical Analyses
3. Results
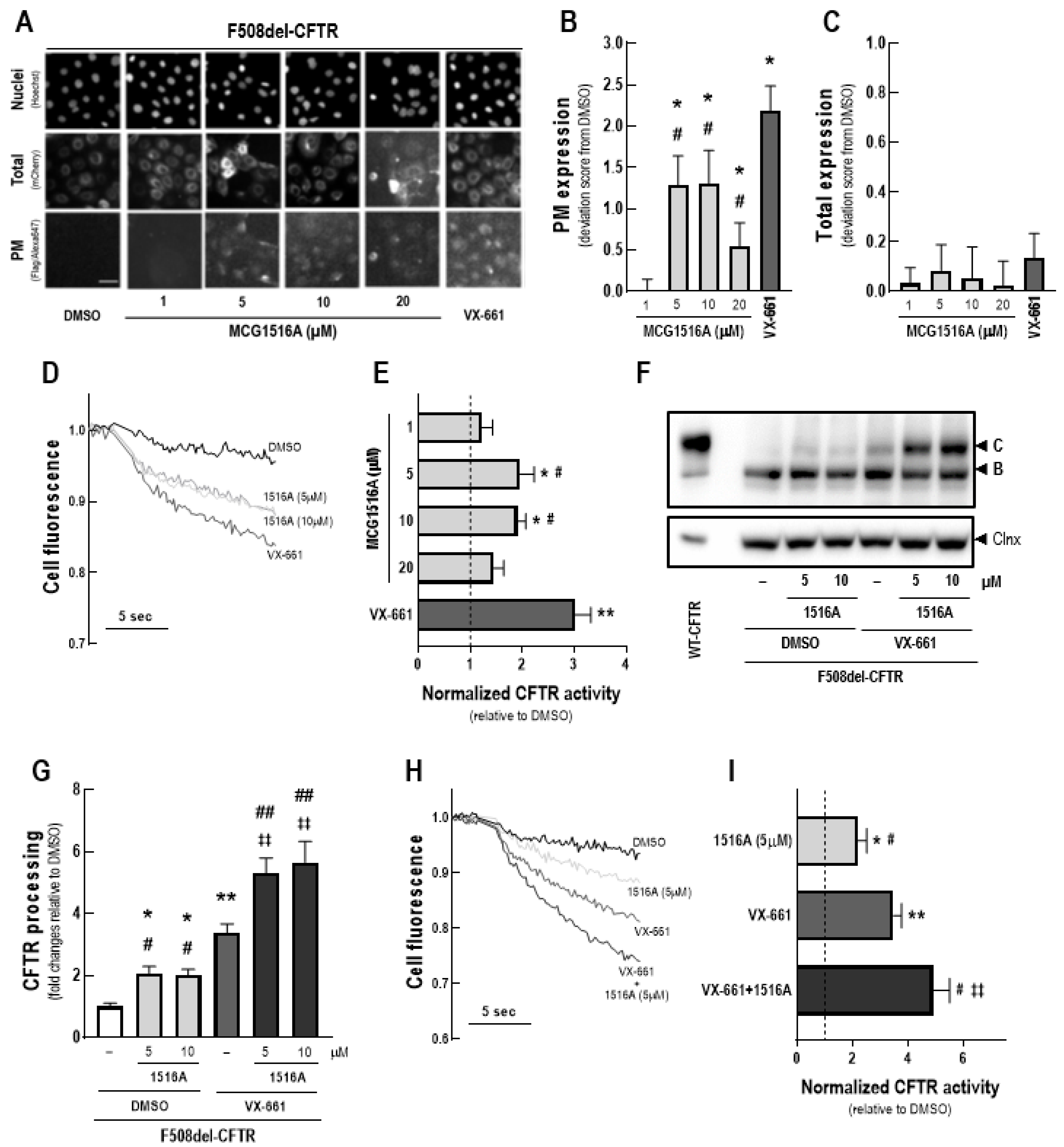
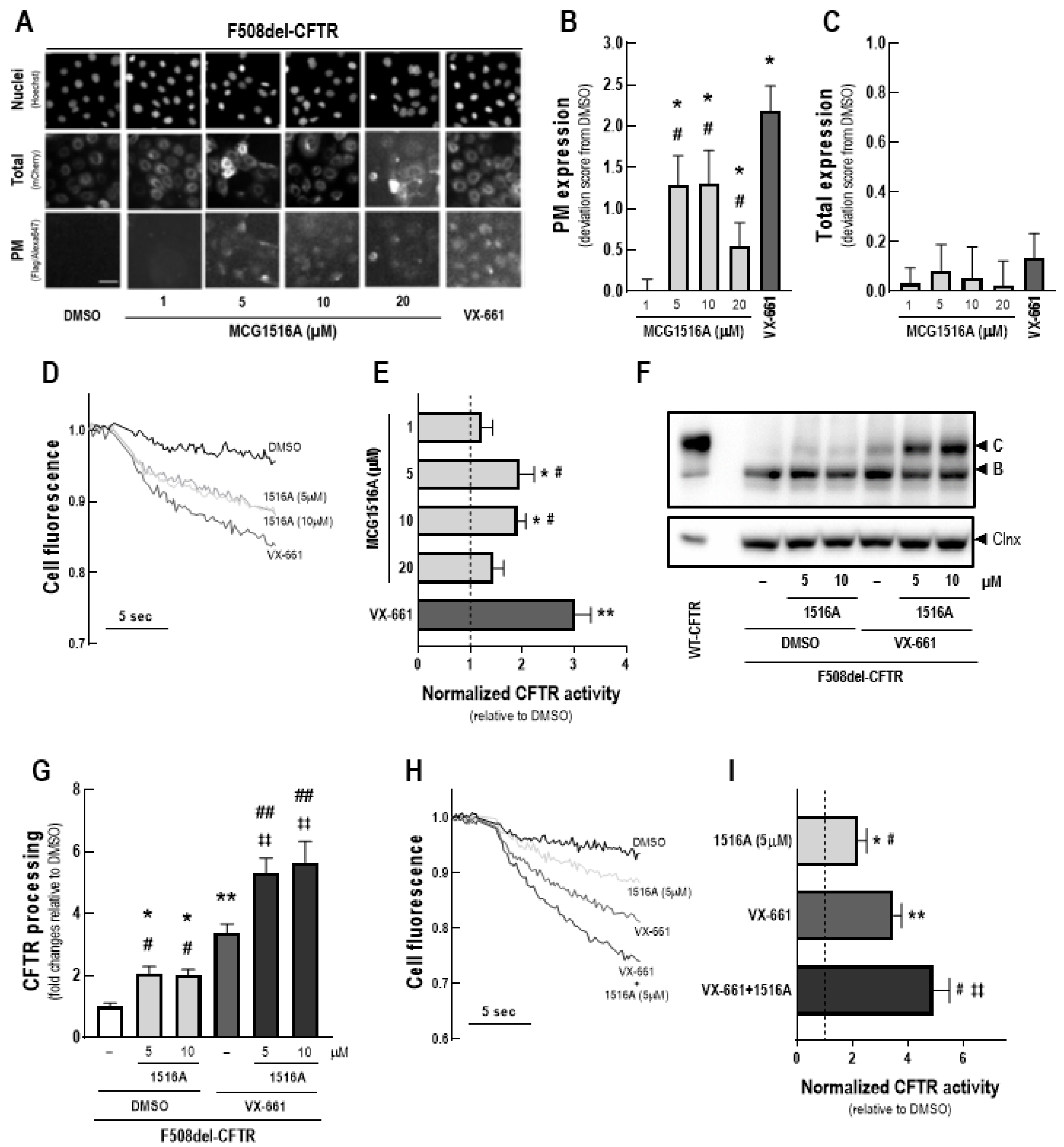
3.1. Assessment of MCG1516A Effects on F508del-CFTR Processing, PM Expression and Channel Activity and Its Additive Effects to Those of the FDA-Approved Corrector VX-661
3.2. Assessment of Rescue of CFTR-Dependent Swelling in F508del/F508del Intestinal Organoids by MCG1516A
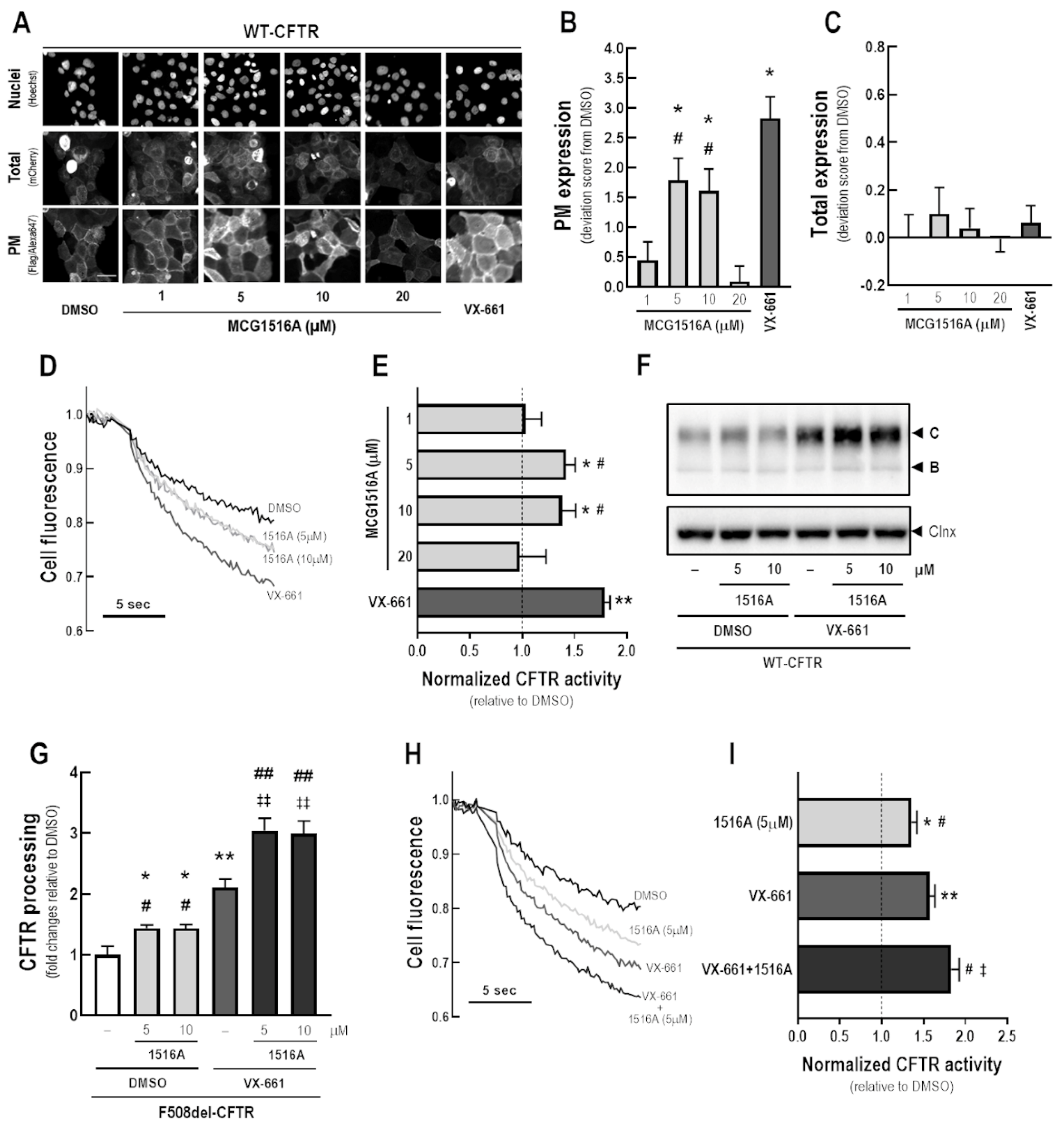
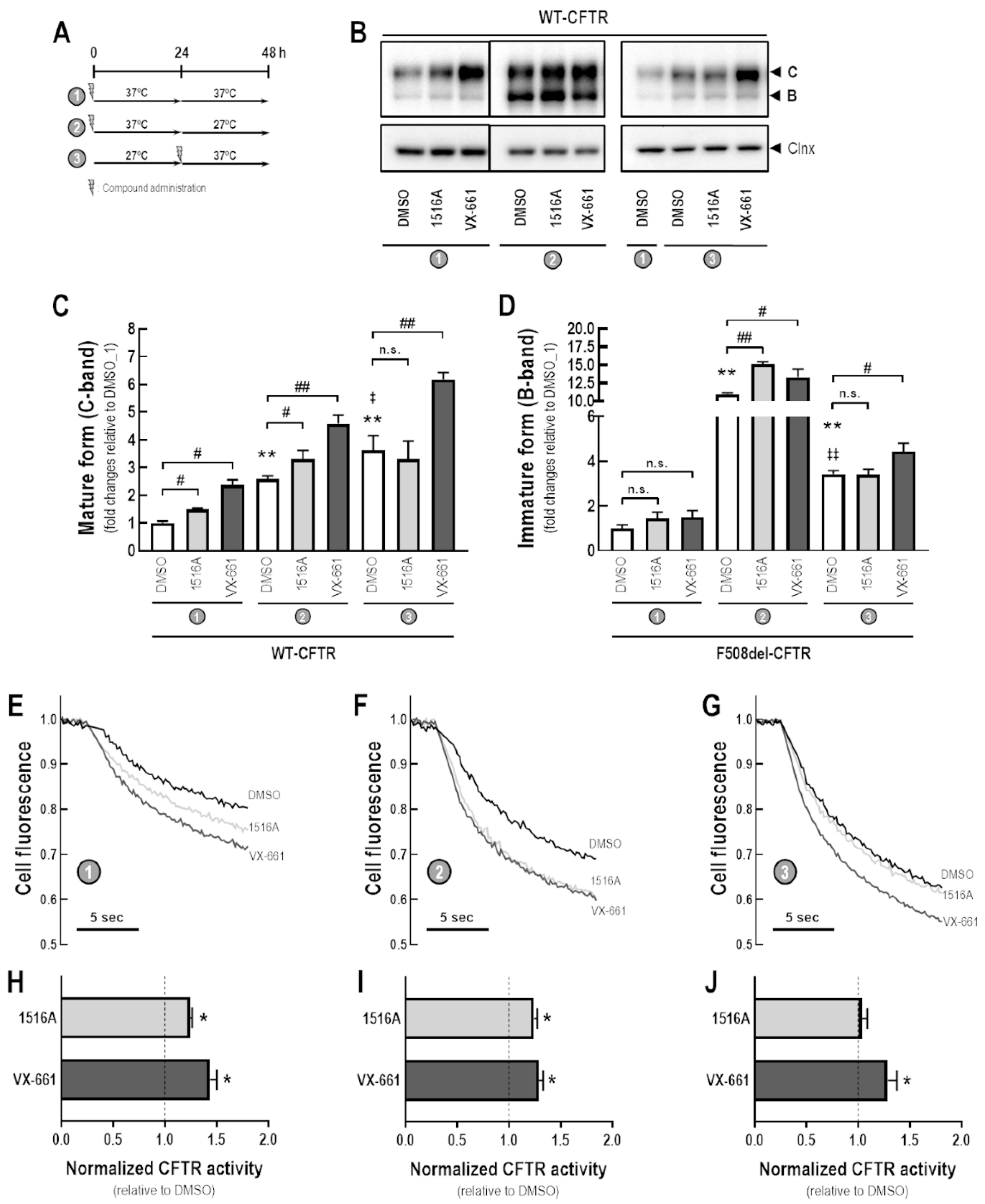
3.3. Assessment of MCG1516A Effects on WT-CFTR Processing, PM Expression and Channel Activity and Its Additive Effects with the FDA-Approved Corrector VX-661
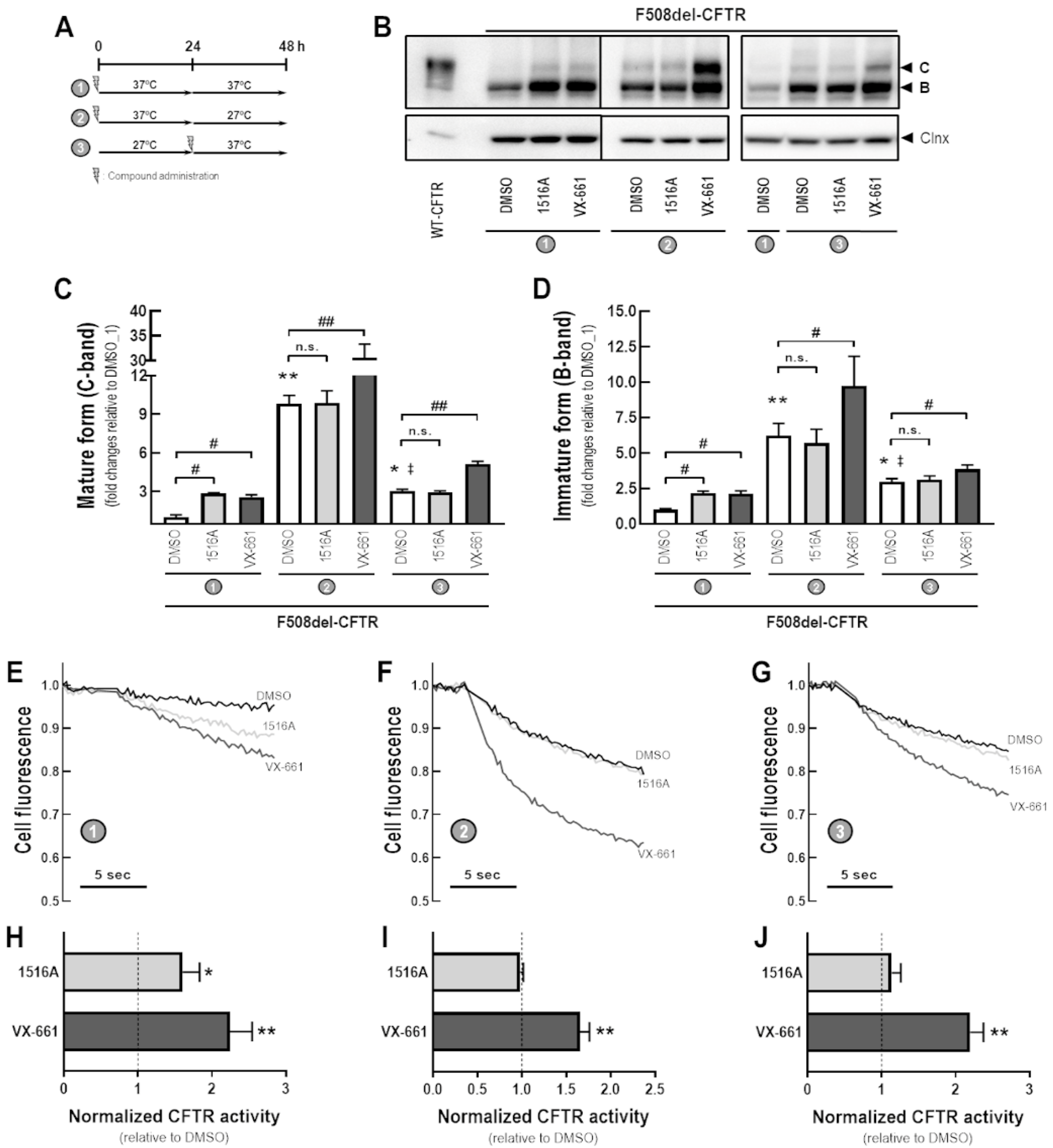
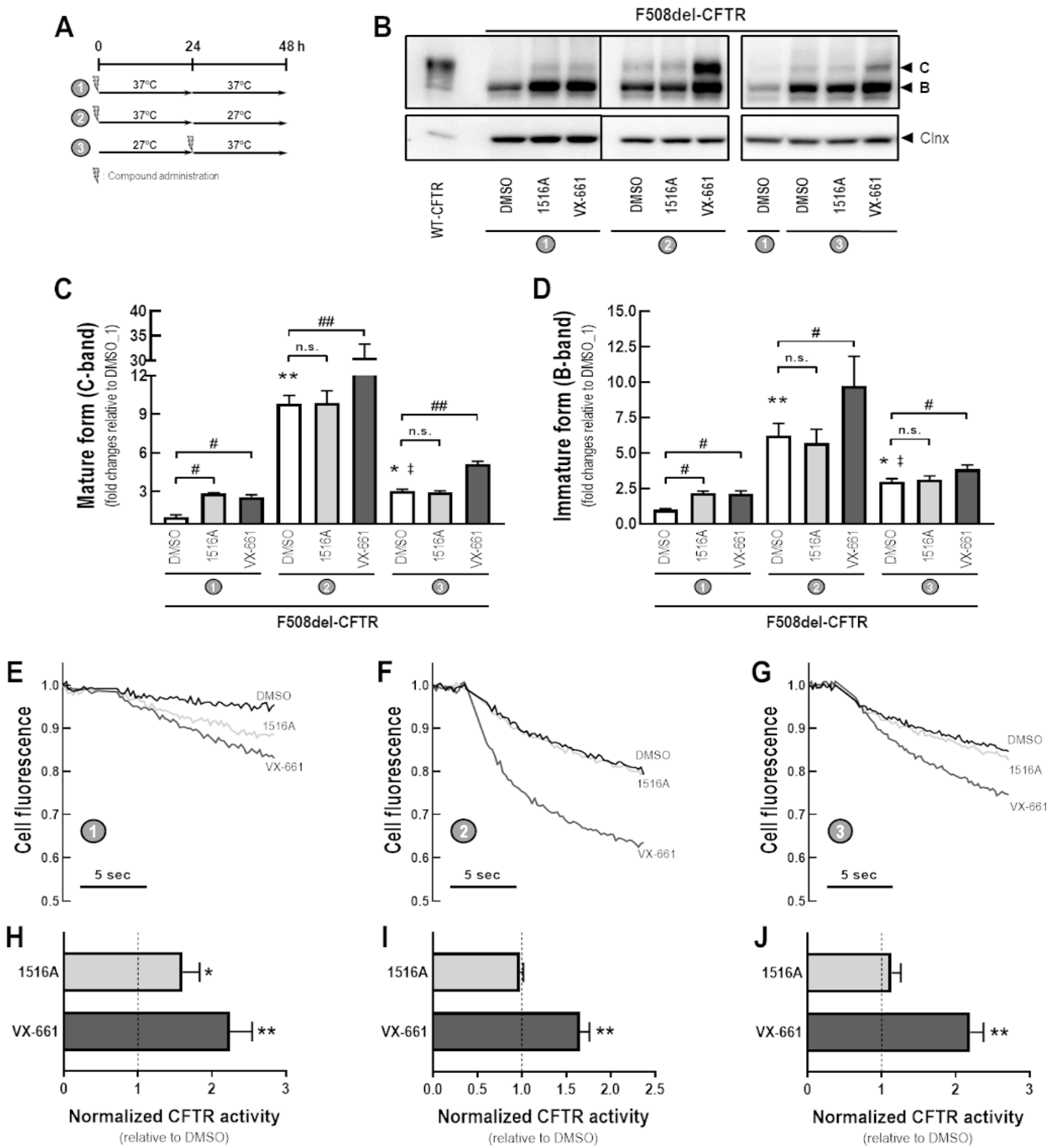
3.4. Assessment of Additive Effects of MCG1516A to Low Temperature in Rescuing F508del-CFTR Processing and Activity
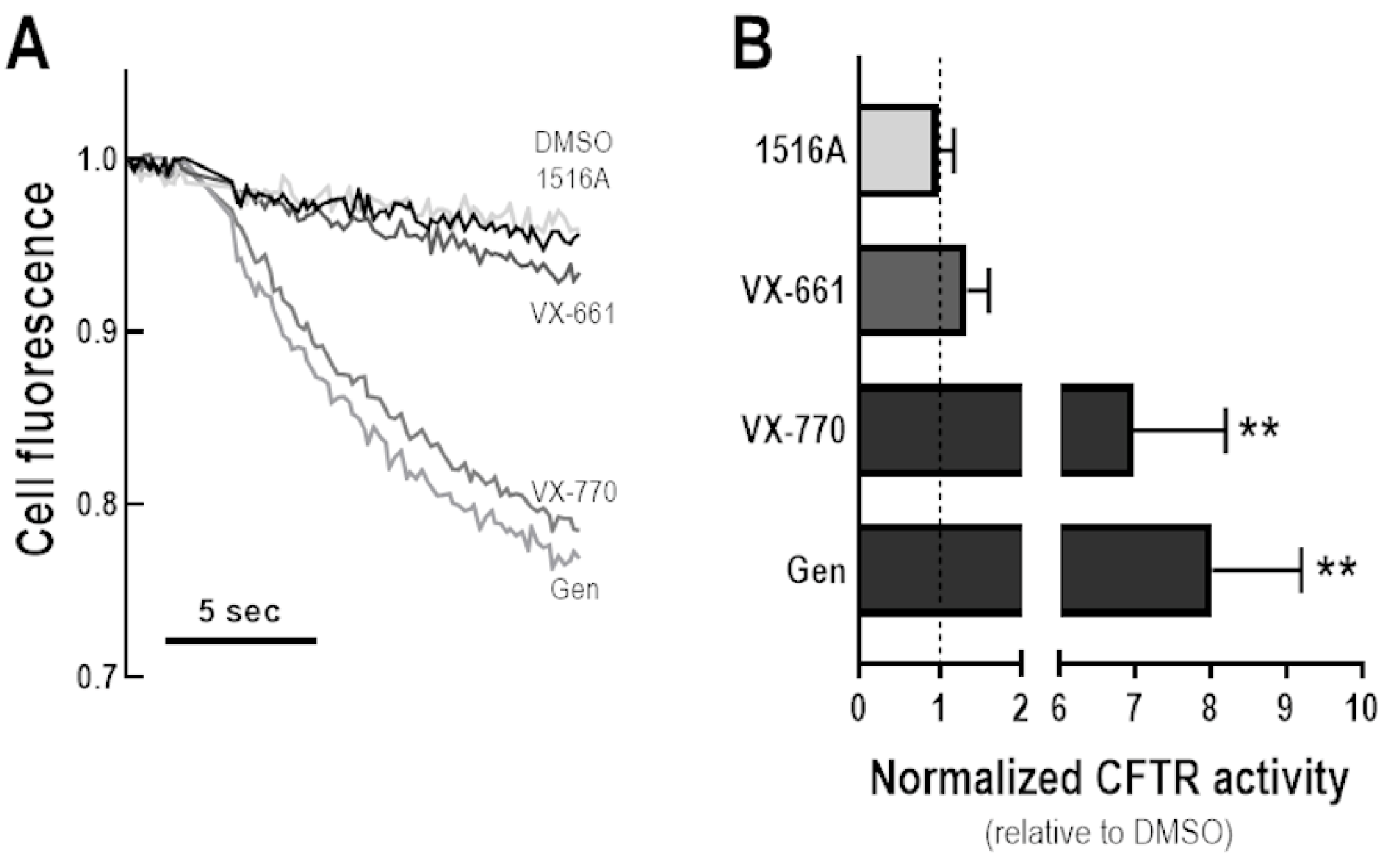
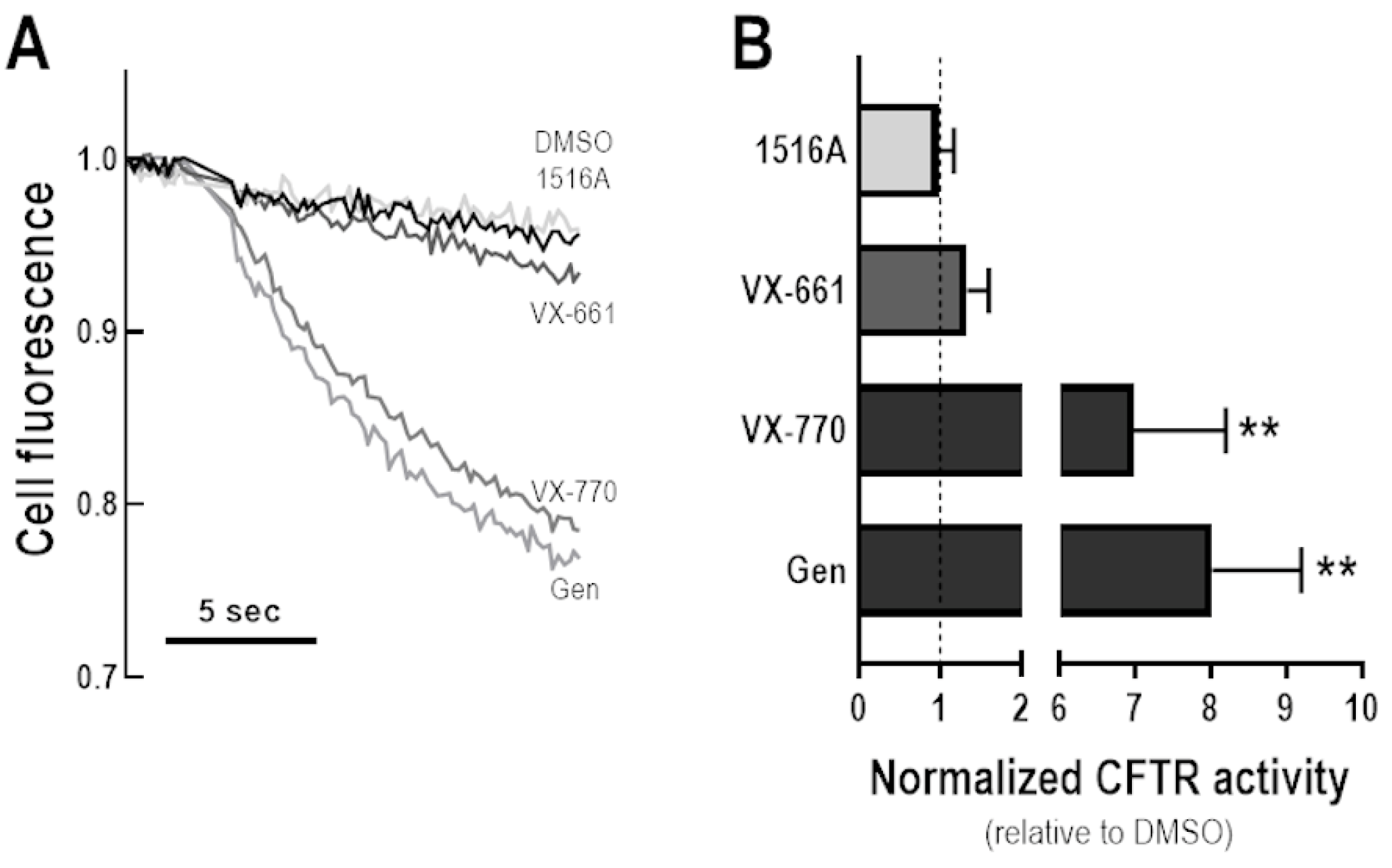
3.5. Assessment of Potentiator Activity of MCG1516A on the Rescued F508del-CFTR
3.6. Assessment of Additive Effects of MCG1516A to a Low Temperature in Enhancing WT-CFTR Processing and Activity
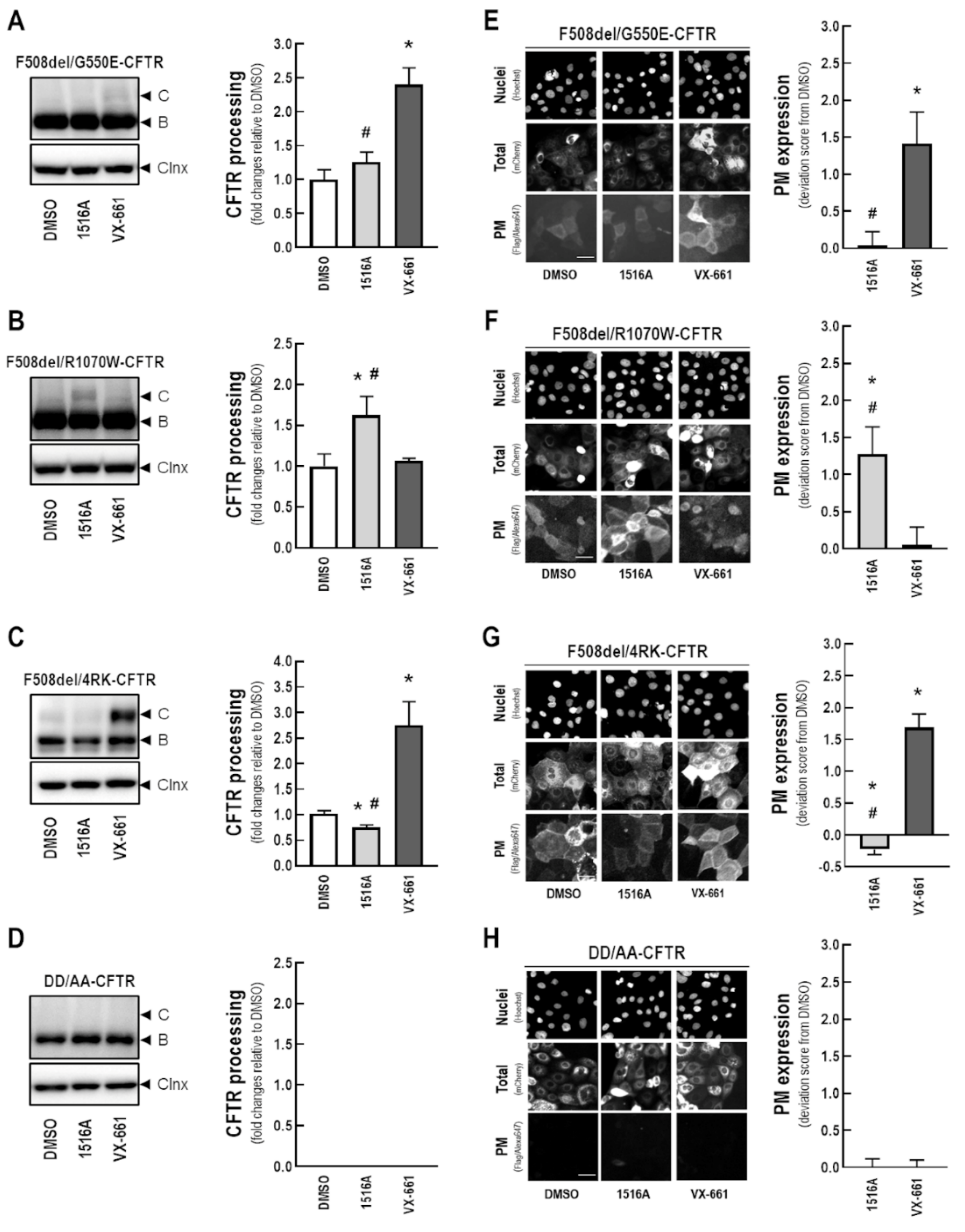
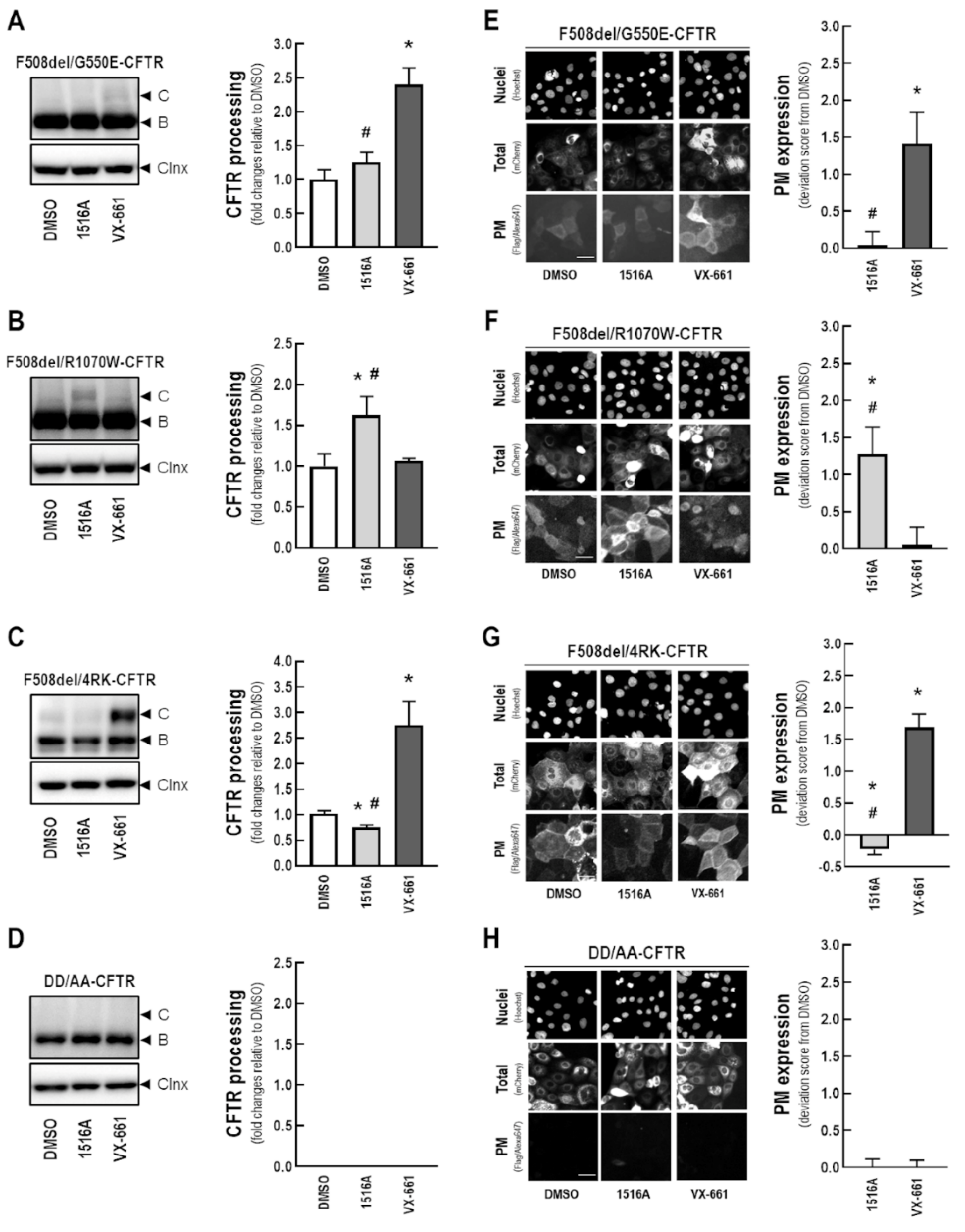
3.7. Assessment of the Effects of MCG1516A on Processing and PM Expression of F508del-CFTR Genetic Revertants and the Traffic-Null Variant DD/AA
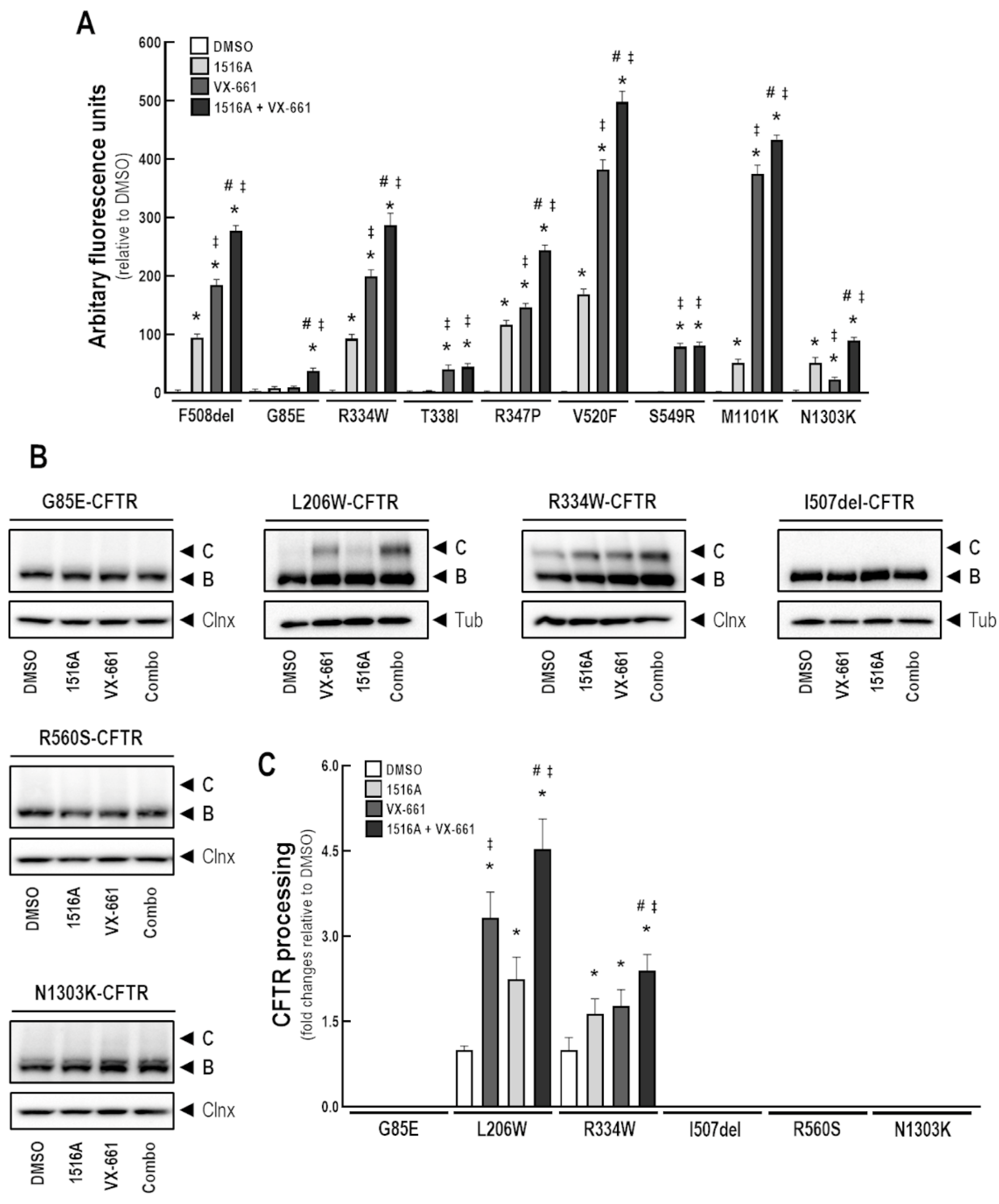
3.8. Assessment of MCG1516A Effects on Other CF-Causing Mutations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Skach, W.R. Mechanisms of CFTR Folding at the Endoplasmic Reticulum. Front. Pharmacol. 2012, 3, 201. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Jensen, T.J.; Loo, M.A.; Pind, S.; Williams, D.B.; Goldberg, A.L.; Riordan, J.R. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995, 83, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Varga, K.; Goldstein, R.F.; Jurkuvenaite, A.; Chen, L.; Matalon, S.; Sorscher, E.J.; Bebok, Z.; Collawn, J.F. Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochem. J. 2008, 410, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Ciciriello, F.; Anjos, S.M.; Carissimo, A.; Liao, J.; Carlile, G.W.; Balghi, H.; Robert, R.; Luini, A.; Hanrahan, J.W.; et al. Ouabain mimics low temperature rescue of F508del-CFTR in cystic fibrosis epithelial cells. Front. Pharmacol. 2012, 3, 176. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Combination of Correctors Rescue ΔF508-CFTR by Reducing Its Association with Hsp40 and Hsp27. J. Biol. Chem. 2015, 290, 25636–25645. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Cui, L.; Hou, Y.; Jensen, T.J.; Aleksandrov, A.A.; Mengos, A.; Riordan, J.R. Removal of multiple arginine-framed trafficking signals overcomes misprocessing of ΔF508 CFTR present in most patients with cystic fibrosis. Mol. Cell 1999, 4, 137–142. [Google Scholar] [CrossRef]
- Roxo-Rosa, M.; Xu, Z.; Schmidt, A.; Neto, M.; Cai, Z.; Soares, C.M.; Sheppard, D.H.; Amaral, M.D. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc. Natl. Acad. Sci. USA 2006, 103, 17891–17896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Pacheco, M.; Silva, I.A.L.; Turner, M.J.; Carlile, G.W.; Sondo, E.; Thomas, D.Y.; Pedemonte, N.; Hanrahan, J.W.; Amaral, M.D. Characterization of the mechanism of action of RDR01752, a novel corrector of F508del-CFTR. Biochem. Pharmacol. 2020, 180, 114133. [Google Scholar] [CrossRef]
- Thibodeau, P.H.; Richardson, J.M., 3rd; Wang, W.; Millen, L.; Watson, J.; Mendoza, J.L.; Du, K.; Fischman, S.; Senderowitz, H.; Lukacs, G.L.; et al. The Cystic Fibrosis-causing Mutation deltaF508 Affects Multiple Steps in Cystic Fibrosis Transmembrane Conductance Regulator Biogenesis. J. Biol. Chem. 2010, 285, 35825–35835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Bertozzi, S.M.; et al. Partial rescue of f508del-cftr stability and trafficking defects by double corrector treatment. Int. J. Mol. Sci. 2021, 22, 5262. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awatade, N.T.; Ramalho, S.; Silva, I.A.L.; Felício, V.; Botelho, H.M.; de Poel, E.; Vonk, A.; Beekman, J.M.; Farinha, C.M.; Amaral, M.D. R560S: A class II CFTR mutation that is not rescued by current modulators. J. Cyst. Fibros. 2019, 18, 182–189. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Sabirzhanova, I.; Rapino, D.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Correctors Rescue CFTR Mutations in Nucleotide-Binding Domain 1 (NBD1) by Modulating Proteostasis. ChemBioChem 2016, 17, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Rapino, D.; Cebotaru, L. Combination of Correctors Rescues CFTR Transmembrane-Domain Mutants by Mitigating their Interactions with Proteostasis. Cell. Physiol. Biochem. 2017, 41, 2194–2210. [Google Scholar] [CrossRef] [Green Version]
- Carlile, G.W.; Yang, Q.; Matthes, E.; Liao, J.; Radinovic, S.; Miyamoto, C.; Robert, R.; Hanrahan, J.W.; Thomas, D.Y. A novel triple combination of pharmacological chaperones improves F508del-CFTR correction. Sci. Rep. 2018, 8, 11404. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Matteson, J.; An, Y.; Moyer, B.; Yoo, J.; Bannykh, S.; Wilson, I.A.; Riordan, J.R.; Balch, W.E.; Wang, X.; et al. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J. Cell Biol. 2004, 167, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Galietta, L.J. Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol.-Cell Physiol. 2010, 298, C866–C874. [Google Scholar] [CrossRef] [Green Version]
- Sondo, E.; Tomati, V.; Caci, E.; Esposito, A.I.; Pfeffer, U.; Pedemonte, N.; Galietta, L.J. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol.-Cell Physiol. 2011, 301, C872–C885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botelho, H.M.; Uliyakina, I.; Awatade, N.T.; Proença, M.C.; Tischer, C.; Sirianant, L.; Kunzelmann, K.; Pepperkok, R.; Amaral, M.D. Protein Traffic Disorders: An Effective High-Throughput Fluorescence Microscopy Pipeline for Drug Discovery. Sci. Rep. 2015, 12, 9038. [Google Scholar] [CrossRef] [Green Version]
- Farinha, C.M.; Sousa, M.; Canato, S.; Schmidt, A.; Uliyakina, I.; Amaral, M.D. Increased efficacy of VX-809 in different cellular systems results from an early stabilization effect of F508del-CFTR. Pharmacol. Res. Perspect. 2015, 3, e00152. [Google Scholar] [CrossRef]
- Ward, C.L.; Kopito, R.R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994, 269, 25710–25718. [Google Scholar] [CrossRef]
- Varga, K.; Jurkuvenaite, A.; Wakefield, J.; Hong, J.S.; Guimbellot, J.S.; Venglarik, C.J.; Niraj, A.; Mazur, M.; Sorscher, E.J.; Collawn, J.F.; et al. Efficient intracellular processing of the endogenous cystic fibrosis transmembrane conductance regulator in epithelial cell lines. J. Biol. Chem. 2004, 279, 22578–22584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Kota, P.; Aleksandrov, A.A.; Cui, L.; Jensen, T.; Dokholyan, N.V.; Riordan, J.R. Correctors of F508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013, 27, 536–545. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Benharouga, M.; Hu, W.; Lukacs, G.L. Conformational and Temperature-sensitive Stability Defects of the ΔF508 Cystic Fibrosis Transmembrane Conductance Regulator in Post-endoplasmic Reticulum Compartments. J. Biol. Chem. 2001, 276, 8942–8950. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Vergani, P.; Lockless, S.W.; Nairn, A.C.; Gadsby, D.C. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 2005, 433, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Rapino, D.; Sabirzhanova, I.; Lopes-Pacheco, M.; Grover, R.; Guggino, W.B.; Cebotaru, L. Rescue of NBD2 Mutants N1303K and S1235R of CFTR by Small-Molecule Correctors and Transcomplementation. PLoS ONE 2015, 10, e0119796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor+tezacaftor+ ivacaftor mediated in part by the dual activities of elexacaftor as both corrector and potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes-Pacheco, M.; Bacalhau, M.; Ramalho, S.S.; Silva, I.A.L.; Ferreira, F.C.; Carlile, G.W.; Thomas, D.Y.; Farinha, C.M.; Hanrahan, J.W.; Amaral, M.D. Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A. Cells 2022, 11, 136. https://doi.org/10.3390/cells11010136
Lopes-Pacheco M, Bacalhau M, Ramalho SS, Silva IAL, Ferreira FC, Carlile GW, Thomas DY, Farinha CM, Hanrahan JW, Amaral MD. Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A. Cells. 2022; 11(1):136. https://doi.org/10.3390/cells11010136
Chicago/Turabian StyleLopes-Pacheco, Miquéias, Mafalda Bacalhau, Sofia S. Ramalho, Iris A. L. Silva, Filipa C. Ferreira, Graeme W. Carlile, David Y. Thomas, Carlos M. Farinha, John W. Hanrahan, and Margarida D. Amaral. 2022. "Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A" Cells 11, no. 1: 136. https://doi.org/10.3390/cells11010136
APA StyleLopes-Pacheco, M., Bacalhau, M., Ramalho, S. S., Silva, I. A. L., Ferreira, F. C., Carlile, G. W., Thomas, D. Y., Farinha, C. M., Hanrahan, J. W., & Amaral, M. D. (2022). Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A. Cells, 11(1), 136. https://doi.org/10.3390/cells11010136