Abstract
The initial formation of corrosion products in pure humid air on magnesium alloys AZ91 and AZ31 was studied using infrared reflection absorption spectroscopy (IRRAS), infrared spectroscopic imaging, and SEM-EDS. The kinetics of corrosion product formation were monitored in situ with IRRAS during exposure to humid air (95% relative humidity) under two different CO2 concentrations: low (≤1 ppm) and ambient (400 ppm). For low CO2 concentrations, the primary corrosion product detected on both alloys was magnesium hydroxide (Mg(OH)2). In contrast, under ambient CO2 conditions (400 ppm), magnesium hydroxy carbonate was the dominant product. After 16 h of exposure, the amount of magnesium converted into corrosion products was approximately 8–10 times higher under low-CO2 conditions compared to ambient levels. The smaller formation of corrosion products but increased magnesium carbonate formation on AZ91D is attributed to its higher aluminium content compared to AZ31. Corrosion attack and product formation were largely localised to the centre of the α-phase in AZ91D, with the β-phase likely serving as sites for cathodic reactions.
1. Introduction
Magnesium (Mg) alloys are some of the lightest metallic structural alloys and are of interest for a wide range of uses because of their mechanical properties, for example, high strength-to-weight ratio and good machinability. However, their range of applications is restricted due to poor corrosion behaviour compared to other light materials, such as aluminium alloys. Magnesium alloys are frequently used where resistance to aggressive environments is not a critical property. For example, in automotive interior components such as instrumental panels, cross bar beams, seat frames, roof frames, and more []. In the electronics industry, magnesium alloy plays an important role in the shells and components of 3C devices like mobile phones, notebook computers, and optical equipment. For these applications, high thermal and electrical conductivity, as well as excellent electromagnetic shielding properties are highly desirable []. In both of these typical applications, the magnesium alloy is seldom exposed to harsh and highly corrosive conditions, such as high chloride loads, but often to intermittent high humidity and moisture.
Numerous studies have been devoted to the corrosion behaviour of magnesium alloys during the last decades. Many studies are performed in aqueous NaCl electrolyte solution where the electrochemical and corrosion properties variations in composition, microstructure, and surface conditions have been studied extensively. These results have been summarised in several reviews [,,,,,]. Fewer investigations have studied the corrosion behaviour of magnesium alloys under atmospheric conditions [,,]. Many of these studies have been focusing on exposures to aggressive conditions with high chloride deposition on the surfaces or in polluted industrial atmospheres []. Fewer investigations have been published on the atmospheric corrosion of magnesium alloys in high humidity but milder atmospheric conditions without chloride and corrosive gases [,,,]. Arrabal et al. [] studied the atmospheric corrosion in humid conditions for Mg/Al alloys, including AZ91D and AZ31, and found that both aluminium content and alloy microstructure influenced corrosion. M. Shahabi-Navid et al. [] studied the initial atmospheric corrosion of Mg/Al alloys in humid air, with low and ambient CO2 content in the air and concluded that the surface film formed was thinner after exposure in the presence of CO2 compared to when CO2 was absent. It was also observed that alloy AZ91 exhibited significantly less localised corrosion in humid air than alloy AM50 and pure magnesium. In another study, R. Lindström et al. showed that the mass gain of pure magnesium exposed in humid air was higher when exposed to an environment with lower CO2 content []. Several studies have shown that the atmospheric corrosion rate decreases with higher aluminium content in the alloy. The effect of aluminium has been attributed to several factors. For example, Al content influences the formation of the oxide film, which could slow down corrosion in the initial stages. Additionally, micro-galvanic coupling of β-phase can initially cause accelerated attack on the a-Mg, but as corrosion proceeds, the fraction of the aluminium rich β-phase increase at the surface exposed to the corrosive medium. This leads to an overall decrease in the corrosion rate with the β-phase acting as a physical barrier against corrosion attack and hinder corrosion propagation in the matrix [,]. For studies of the initial stages of atmospheric corrosion under milder conditions, the latter is probably less relevant because of the low severity of the corrosion attack. In contrast, aluminium content and how it influences the stability of the initial surface oxide/surface film effects can severely affect even the early stages of corrosion. It has been suggested that the effects of Al content are associated with an Al-enriched layer which has been detected in the thin surface film on Mg/Al alloys [,,,,,]. It was found that Al can diffuse and be incorporated into Mg-oxide/hydroxide during atmospheric corrosion [].
Studies investigating corrosion of metals under atmospheric conditions are difficult to perform with conventional electrochemical techniques, especially when soluble salts are absent on the surface and the water layer on the surface is thin. Infrared spectroscopy is a powerful method for identifying corrosion and reaction products formed during atmospheric corrosion of metals under in situ conditions. It can also provide information about the kinetics of corrosion product formation upon exposure in humid air [,]. FTIR-microscopy can be used for chemical imaging of corrosion products related to the microstructure of a material, which can provide a deeper understanding of corrosion mechanisms []. The recent development of nanoscale infrared microscopy has overcome the diffraction-limited restriction in lateral resolution for conventional optical IR-microscopy and can provide information about corrosion product formation with submicron and nanoscale resolution []. Although the atmospheric corrosion of magnesium alloys with different aluminium content has been investigated in several studies upon exposure to humid conditions, the initial kinetics of formation of different corrosion products has not been determined under in situ conditions.
In the present work, the initial atmospheric corrosion on AZ91 and AZ31 during 19 h exposure in humid air was studied using in situ infrared reflection absorption spectroscopy (IRRAS), FTIR-ATR FPA chemical imaging, and AFM-IR. Highly sensitive in situ IRRAS measurements were used to provide unique insights into the initial formation of various corrosion products and their kinetics of formation. Complementary techniques such as FTIR-FPA imaging and AFM-IR provide additional information, particularly regarding the spatial localisation of corrosion product formation. Additionally, SEM-EDS measurements were employed to determine the localisation of the corrosion attacks and the morphology of corrosion products. The main objective was to provide more comprehensive view of the initial corrosion product formation in humid air on magnesium alloys with different aluminium content and the effect of CO2 on corrosion product formation.
2. Materials and Methods
2.1. Experimental
Materials
The materials studied were two commercial alloys (KG Fridman AB, Karlstad, Sweden) with different aluminium content, e.g., AZ31 and AZ91, with nominal compositions given in Table 1. The microstructures of the materials studied here are shown in Figure 1 and Figure 2. AZ91 has a dendritic solidification microstructure with very clear dendrites and secondary phases in the inter-dendritic spacings. Al and Zn are strongly segregated, and the main secondary phase consists of Al12Mg17. AZ31 has a more heterogeneous grain structure with greater variability in grain size. AlMn intermetallic particles and some particles contains Zn (and Al) can be found mainly at grain boundaries. The samples used for in situ IRRAS measurements were sequentially ground, with the last step using # 4000 grinding paper, while samples used for FTIR-FPA imaging and AFM-IR were diamond-polished to 1 µm.

Table 1.
Nominal composition in wt% of alloys tested as specified by the manufacturer.

Figure 1.
SEM BSE image (a) and SEM-EDS maps (b) of AZ91 prior to exposure.

Figure 2.
SEM BSE image of AZ31 prior to exposure.
2.2. Exposure Conditions
The exposures of the samples were performed with pure humid air containing a low content of CO2, (≤1 ppm) as produced by an FTIR purge gas generator (adsorption dryer). Ambient CO2 levels were 400 ± 20 ppm. Humid air with 95% ± 2% relative humidity (RH) was produced by mixing dry air and humidified air at 98–99% RH in an apparatus described previously []. The maximum exposure time was 19 h, with in situ IRRAS measurements being performed during the exposure period in accordance with Section 2.3. Additionally, shorter exposures of 5 h were performed to perform FTIR-ATR FPA imaging and AFM-IR of surfaces with less coverage of corrosion products. SEM-EDS was performed on samples after 19 h of exposure.
2.3. In Situ Infrared Reflection Absorption Spectroscopy
In situ infrared reflection absorption spectroscopy (IRRAS) during exposure to humid air was performed using an experimental set-up which allows measurements at grazing angle (78°) on the metal surface using p-polarised light []. The in situ measurements were performed in a stainless steel cell with ZnSe windows. Infrared spectra were first recorded after 5 min and then every 30 min after the introduction of the humid air into the cell. In situ spectra of the corrosion products formed on the surface were obtained by removing bands related to gas-phase water and CO2 in the cell. This was performed by subtracting the first spectra, collected after five minutes, from each subsequent spectrum collected. The air flow through the cell was 1 L/min. Optical simulations of IRRAS spectra were conducted with the transfer matrix method using Matlab (R2023b). Transmission spectra used for the calculations were made by mixing the compounds with KBr and recording transmission spectra of a pressed tablet in accordance with previous studies [,].
2.4. FTIR-ATR FPA Imaging
FTIR imaging was performed using a Bruker Vertex 70 spectrometer equipped with a Hyperion 3000 microscope (Bruker Optics, Ettlingen, Germany) with a 64 × 64 focal plane array (FPA) detector. FTIR-FPA imaging was performed in the attenuated total reflectance (ATR) mode with a 20 × ATR objective equipped with a 100 µm diameter Ge internal reflection element. The field of view was ≈33 × 33 µm2 using the Ge-ATR objective (Bruker Optics, Ettlingen, Germany), corresponding to a pixel resolution of ≈0.5 µm. Infrared spectra were acquired in the region 880 to 3860 cm−1 by adding 500 scans at 8 cm−1 resolution.
2.5. AFM-IR
Atomic Force Microscopy–InfraRed (AFM-IR) measurements were performed in contact mode using resonance-enhanced AFM-IR with a Bruker NanoIR 3 (Bruker. The light source was an MIRCAT POINT spectra QCL Laser (four chip) (DRS Daylight Solutions, San Diego, CA, USA)with a spectral range of 800–1800 cm−1. IR-images were recorded by measuring at a fixed wavelength using a phase-locked loop. A gold-coated contact NIR-probe (Bruker) with a resonance frequency of 13 ± 4 kHz was used for AFM-IR measurements. AFM-IR spectra were measured with 2 cm−1 spectral resolution. Laser power during spectral acquisition and IR imaging was ≈15% of the maximum laser power of approximately 800 mW. Step discontinuities which occurred when the QCL laser shifted between the four chips, which results in sharp steps in the broadband spectra, were corrected by post-processing the spectra using the Analysis Studio software (version 3.17). During AFM-IR measurements, dry nitrogen was flowed through the AFM-IR instrument to minimise contributions from water vapour and CO2 in the spectra.
2.6. Scanning Kelvin Probe Force Microscopy (SKPFM)
The variation in Volta potential along an unexposed polished (0.25 mm diamond) AZ91 surface was measured using a Bruker NanoIR3 system with an SKPFM attachment. The probes had resonant frequencies of 47–76 kHz, spring constants in the range of 1.2–6.4 N/m, and a 25 ± 5 nm thick conductive Pt/Ir coating. Areas of interest, containing both beta and alpha phase, were identified using the attached optical microscope. The Volta potential maps are presented as potential with respect to the sample, and the values were offset to be centred around 0 V.
2.7. SEM and EDS
Field Emission Gun—Scanning Electron Microscope (FEG-SEM) imaging was performed using an LEO 1530 (Carl Zeiss Microscopy, Jena, Germany) equipped with a Gemini column, upgraded to a Zeiss Supra 55 (Carl Zeiss Microscopy, Jena, Germany). The system included a 50 mm2 X-Max Silicon Drift Detector (SDD) from Oxford Instruments for Energy Dispersive Spectroscopy (EDS) (Oxford Instruments, Abingdon, UK). SEM imaging was conducted at an accelerating voltage of 5 kV in Backscattered Electron (BSE) mode.
3. Results
3.1. In Situ Infrared Reflection Absorption Spectroscopy
IRRAS absorption spectra of the AZ91 during exposure to humid air with low and ambient CO2 content are shown in Figure 3. The spectra obtained in low CO2 atmosphere show a sharp peak at 3703 cm−1, which is due to hydroxyl stretching of Mg(OH)2 [,], and a broad asymmetric band with a maximum at 600 cm−1, mainly attributed to Mg-O vibrations in Mg(OH)2. A weaker band around 1420–1490 cm−1 is due to asymmetric stretching vibrations of carbonate ions (CO32−) in the corrosion products. A band at around 1615 cm−1 can be explained by the presence of water molecules in the corrosion products or adsorbed on the corrosion products that covered the surface. Contributions from carboxylate ions originating from volatile organic acids contaminants cannot be excluded [,]. For the AZ91 sample exposed in humid air with ambient (≈400 ppm) CO2 concentration (Figure 3), Mg(OH)2 was not detected. Instead, a stronger band from carbonate around 1470 cm−1 was present in the spectra, and in the hydroxyl stretching region, bands at 3654 cm−1 (sharp peak) and 3380 cm−1 (wider and less distinct band) appeared. These bands could originate from a poorly crystalline magnesium hydroxy carbonate, such as hydromagnesite, Mg5(CO3)4(OH)2·5H2O [,]. The experimental spectra obtained for AZ91 can be compared with calculated IRRAS spectra of Mg(OH)2 in Figure 4.
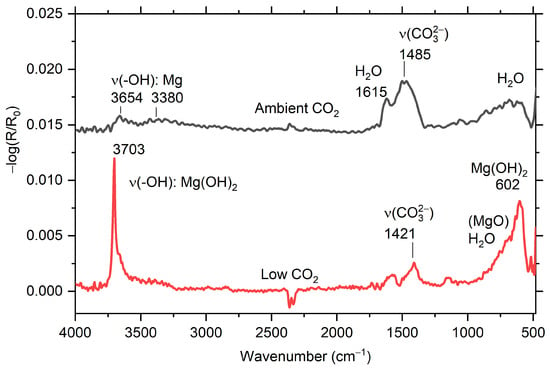
Figure 3.
In situ infrared reflection absorption subtraction spectra for AZ91 after 5 h of exposure in humid air (94% RH) with low CO2 (≤1 ppm) and ambient CO2 (≈400 ppm).
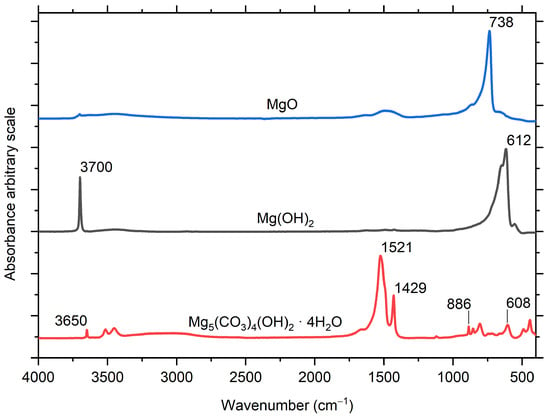
Figure 4.
Calculated IRRAS spectra of MgO, Mg(OH)2, and Mg5(CO3)4(OH)2·4H2O on a magnesium substrate.
It cannot be ruled out that there is a minor contribution from MgO to the asymmetric band with the maximum at 602 cm−1 for samples exposed at low CO2 in Figure 3 and Figure 5, due to the LO mode of MgO, which has been reported to be located at 718 cm−1 [] and 737 cm−1 []. This vibrational mode should be visible in IRRAS spectra recorded at grazing angles []. Calculated IRRAS spectra of MgO on a Mg surface indeed have a strong band at 738 cm−1. However, the similarity between calculated IRRAS spectra of Mg(OH)2 in Figure 4 and the spectra obtained during exposures with low CO2, with an asymmetric band with a maximum at 612 cm−1 and 602 cm−1, respectively, indicates that the spectra for the Mg alloys exposed at low CO2 level is mainly explained by Mg(OH)2, and that the contribution from MgO is probably small. Note that the MgO standard sample used for calculations contained small amounts of impurities in the form of additional hydroxyl groups (small band around 3700 cm−1) and carbonates (wide band around 1450 cm−1).
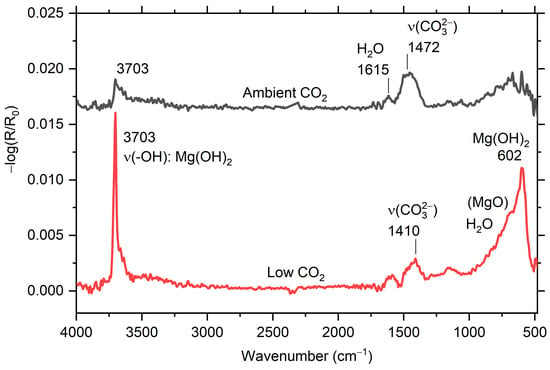
Figure 5.
In situ infrared reflection absorption subtraction spectra for AZ31 after 5 h exposure in humid air (94% RH) with low CO2 (≤1 ppm) and ambient CO2 (≈400 ppm).
For the AZ31 alloy, the spectra are similar for low CO2 content in humid air, with the formation of Mg(OH)2 and weak bands from carbonate, see Figure 5. The spectra obtained at ambient CO2 level also have a weak peak at 3703 cm−1 due to Mg(OH)2, and hydroxyl and carbonate bands associated with formation of magnesium hydroxy carbonate.
Formation of magnesium hydroxy carbonate, hydromagnesite (Mg5(CO3)4(OH)2·4H2O was detected after 28 days of exposure to humid air by Arrabal et al. []. Magnesium hydroxy carbonates such as hydromagnesite [] and dypingite (Mg5(CO3)4(OH)2·4H2O [,] have been reported in previous exposures to humid air.
A detailed comparison of the spectra in the hydroxyl stretching region is shown in Figure 6, together with reference spectra of brucite (Mg(OH)2) and hydromagnesite, (Mg5(CO3)4(OH)2·4H2O). The sharp hydroxyl band associated with Mg(OH)2 is clearly present for AZ91 and AZ31 after exposure to humid air with low CO2 content, but also for AZ31 exposed at ambient CO2. However, for AZ91 exposed at ambient CO2, a peak around 3650 cm−1, which has a similar position as that of hydromagnesite (Mg5(CO3)4(OH)2·4H2O, may be observed. However, the other characteristic bands at 3520 and 3450 cm−1 for Mg5(CO3)4(OH)2·4H2O are not clearly discerned in the IRRAS spectra of the surface, which could be due to the formation of a more disordered magnesium hydroxy carbonate at this stage of exposure.
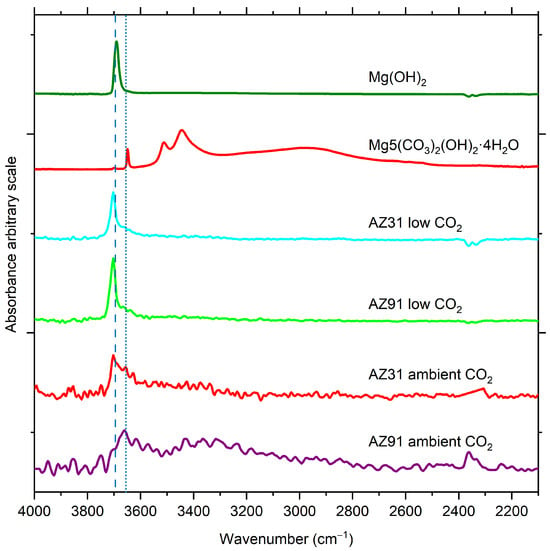
Figure 6.
Comparison of spectra (hydroxyl stretch region 4000–2200 cm−1) for AZ31 and AZ91 after exposure and after 5 h exposure reference spectra of Mg(OH)2 and Mg5(CO3)4(OH)2·4H2O. The dashed and dotted lines illustrate the difference in position of the -OH stretching bands.
By plotting the intensities of different IR bands in the spectra, information about the kinetics of corrosion product formation may be obtained. Figure 7 shows the intensities of the hydroxyl stretching band for Mg(OH)2 at 3704 cm−1 and the carbonate band in the region 1400–1500 cm−1 vs. exposure time for the two different alloys and exposure conditions. The kinetics of corrosion show a rapid formation of corrosion products during the first 8 h of exposure, with a decreasing rate of formation after 8–10 h exposure.
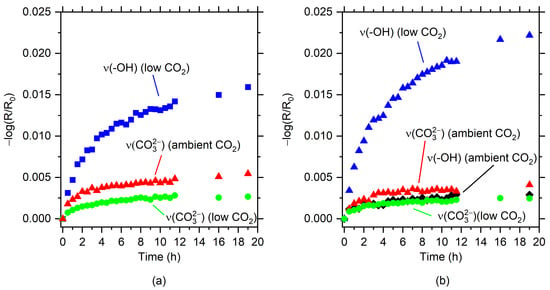
Figure 7.
Intensities (−log(R/R0) of hydroxyl at 3704 cm−1 (Mg(OH)2) and Mg carbonate (1400–1500 cm−1) vs. exposure time for AZ91 (a) and AZ31 (b).
The intensities of Mg(OH)2 are clearly higher for AZ31, which has a lower aluminium content, while the carbonate bands are higher for AZ91 compared to AZ31 in humid air with ambient CO2 content.
The IR intensities vs. time for the corrosion products formed were used to roughly estimate the amount of metallic magnesium converted into corrosion products, assuming that they mainly consist of Mg(OH)2 and Mg5(CO3)4(OH)2·4H2O. Estimation of the average thickness of the corrosion product layers was made using a simplified version of Francis and Ellison’s equation []. The formula approximates the intensity of a band in reflection–absorption spectra for a certain material thickness and assumes that the underlying material is a good reflector for IR wavelengths:
where d is the average thickness of surface film, n1 is the refractive index of the surface film, n2 is the refractive index of the surface film, and α is the absorption coefficient of the actual IR band of the surface film. The absorption coefficients for the hydroxyl stretching peak of Mg(OH)2 and the carbonate asymmetric stretching band of Mg5(CO3)4(OH)2·4H2O were estimated from transmission spectra of these compounds. The average thickness of the corrosion products was estimated from the peak intensities in the experimental spectra and Equation (1). The amount of magnesium in the corrosion products Mg(OH)2 and Mg5(CO3)4(OH)2·4H2O, assuming that this is the magnesium hydroxy carbonate formed, is determined using the density of these compounds. The estimated amount of Mg formed in the corrosion products is considerably higher during exposure in humid air with low CO2 content, as seen in Figure 8. The corrosion products formation is also higher on AZ31 compared to AZ91, which is in accordance with the results by Arrabal et al. [], who showed that alloys with higher aluminium content (AZ80 and AZ91D) presented lower mass gain values compared to AZ31 and pure Mg. Similar results were obtained by Feliu Jr et al. [], who found two times lower mass gain for AZ91 compared to AZ31 after exposure in air with 98% RH. At the same time, higher Al content resulted in increased carbonate formation and less MgO/Mg(OH)2. Studies of the effect of low and ambient levels of CO2 for pure magnesium showed that the mass gain was approximately four times higher after four weeks of exposure in humid air with low CO2 content compared to air with 330 ppm CO2, which is in line with the present results []. Thinner surface films were formed on Mg/Al alloys in the presence of higher CO2 content in the humid air during short-term exposures []. Thus, the results obtained in this work are consistent with previous studies of magnesium and magnesium alloys in humid conditions.
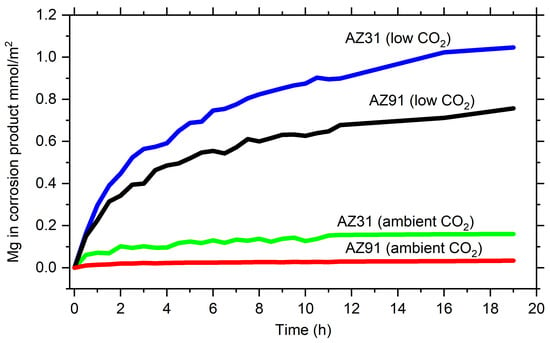
Figure 8.
Magnesium formed in corrosion products (assumed to consist of Mg(OH)2 + Mg5(CO3)4(OH)2·4H2O) during exposure to humid air in low and ambient CO2 of AZ31 and AZ91 estimated from in situ IRRAS measurements.
3.2. SEM and EDS
After exposure, the surfaces were examined by SEM. Figure 9 shows SEM images of AZ91 exposed in humid air with ambient and low CO2 content in the air showing corrosion attack in the α-phase. The severity of the attacks, with numerous initiation points, seems to be located largely in the interior of α-grains, while a zone in the α-phase close to the β-phase is much less affected by corrosion. For the AZ31 alloy, the corrosion also seems to be initiated in the α-Mg phase but not directly in the vicinity of the bright intermetallic particles. Preferred attacks toward the middle of the α-phase have been observed for AZ91 after humid exposures [] and for AM50 after short exposure in NaCl electrolyte []. The preferential attacks in the centre of the grains were attributed to the Al content in the α-phase, which decreased from the vicinity of the β-phase towards the grain centres. This gradient in the Al content is due to coring during non-equilibrium solidification of the alloy. Detailed studies of the AM50 alloy by Schwarz et al. [] using atom probe tomography revealed that a thin Al-rich layer was present beneath an amorphous MgO and Mg(OH)2, at the alloy/oxide interfaces under the atmospheric corrosion of a magnesium alloy. However, they could not find evidence of the formation of an aluminium oxide layer.

Figure 9.
SEM BSD images of AZ91 exposed for 19 h in humid air (95% RH) with ambient CO2 content (a) and low CO2 content (b), and AZ31 exposed for 19 h in humid air (95% RH) at ambient CO2 content (c).
SEM-EDS analysis of AZ91 exposed for 19 h in humid air (95% RH) with ambient CO2 content in Figure 10 indicate a lower aluminium content in the area with preferential corrosion attacks compared to areas closer to the β-phase, which has been observed in a previous study of the same alloy []. In the corroded areas, both carbon and oxygen contents are enhanced, which is consistent with the formation of magnesium hydroxide carbonate as detected by IRRAS.

Figure 10.
SEM image of AZ91 exposed for 19 h in humid air (95% RH) with ambient CO2. Location of where EDS analysis (Table 2) was performed are marked with the corresponding number and analyzed area (white square).
Table 2.
EDS elemental analysis of the areas in Figure 10.
Table 2.
EDS elemental analysis of the areas in Figure 10.
| Area | Mg at% | Al at% | Zn at% | O at% | C at% | Mn at% |
|---|---|---|---|---|---|---|
| a | 71.3 | 2.7 | 19.4 | 6.7 | ||
| b | 79.7 | 2.0 | 14.1 | 4.3 | ||
| c | 0.7 | 52.1 | 2.7 | 0.8 | 42.5 | |
| d | 59.7 | 35.6 | 1.9 | 1.8 | 1.2 | |
| e | 61.0 | 35.0 | 1.7 | 1.4 | 0.9 | |
| f | 91.8 | 5.9 | 1.0 | 1.9 | ||
| g | 91.4 | 6.5 | 1.2 | 0.9 |
For AZ31, the corrosion attacks were also largely localised sone distance from the intermetallic particles, but the numerous initiation points observed for AZ91 seems to be absent, and the corrosion attacks seems to be more uniform compared to AZ91. Some corrosion effects are also observed in the vicinity of the intermetallic particles. A more uniform corrosion of AZ31 compared to AZ91 was also observed by Arrabal et al. after longer exposure in humid air [].
When the surfaces at corrosion attacks are examined in detail, a striking difference may be observed (see Figure 11). The corrosion products formed under ambient CO2 content, which mainly consist of magnesium hydroxy carbonate, seem to form a compact layer on the surface at corrosion initiation sites. On the other hand, the Mg(OH)2 corrosion products formed with low CO2 content have a different morphology with more porous, sponge-like structure growing outward from the surface. Similar corrosion product morphology was also observed for corrosion of pure Mg exposed to humid air with low CO2 content by Lindstrom et al. []. The morphology of Mg(OH)2 indicates that this product is less protective and porous compared to the magnesium hydroxy carbonate, which can lead to a higher corrosion rate under low-CO2 conditions.

Figure 11.
SEM images with high magnification in areas where corrosion products are located on AZ91 exposed for 19 h in humid air with (a) ambient CO2 and (b) low CO2, and (c) AZ31 exposed for 19 h in humid air with ambient CO2.
3.3. Infrared Spectroscopy Chemical Imaging
The exposed sample surfaces were also studied with FTIR-ATR FPA chemical imaging, which can provide information about the distribution of the corrosion products on the alloy surface. Chemical images of the carbonate band at 1470 cm−1 for AZ91 after 5 and 19 h of exposure show that the carbonate-containing corrosion product forms on the α-Mg, with nothing or very small amounts on the β-phase, as seen in Figure 12.
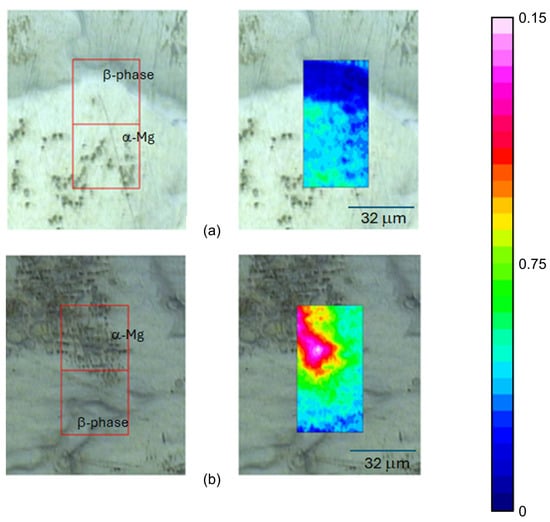
Figure 12.
FTIR-ATR FPA chemical imaging of AZ91 after (a) 5 h and (b) 19 h of exposure in humid air (95%) with ambient CO2 content. Optical images of the surface (to the left) and false colour images of the intensities of the carbonate band at 1470 cm−1 overlaid the optical images.
The chemical and optical images also indicated how the amount of corrosion products increases with exposure time. Similar results are also observed for low-CO2 conditions, where the distribution of Mg(OH)2 on the surface is shown in Figure 13.
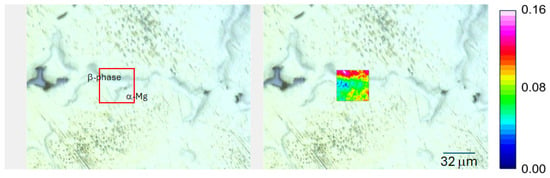
Figure 13.
FTIR-ATR FPA chemical imaging of AZ91 exposed for 19 h in humid air (95%) with low CO2 content. Optical images of the surface (to the left) and false colour images of the intensity of the hydroxyl stretching band of Mg(OH)2 (centred around 3700 cm−1) are overlaid on the optical image.
The surface of AZ91 exposed to humid air at ambient CO2 content was also investigated with AFM-IR in an area where the corrosion product/corrosion attack was observed. The topographical image and the IR image of the carbonate band at 1420 cm−1 are shown in Figure 14. Note that the scale bar is in mV, which is connected to the AFM cantilever deflection. Although it is related to the infrared absorbance of the materials, the absolute values are not directly comparable to, e.g., FITR-ATR or IRRAS spectra. A corrosion product cluster is seen, together with numerous submicron/nanosized corrosion initiation sites. These are located mainly at the scratches on the surfaces created by the polishing process. The AFM-IR results show that there are numerous corrosion initiation sites within the active area, and that the corrosion is initiated at many sites in the attacked area, rather than a few single pit-like attacks. Initiations could be related to sites at the scratches with enhanced dissolution at defect sites and dislocation loops produced during the grinding process []. The presence of the scratches might also promote water clustering [] and the formation of microdroplets of water, which initiate corrosion attack.
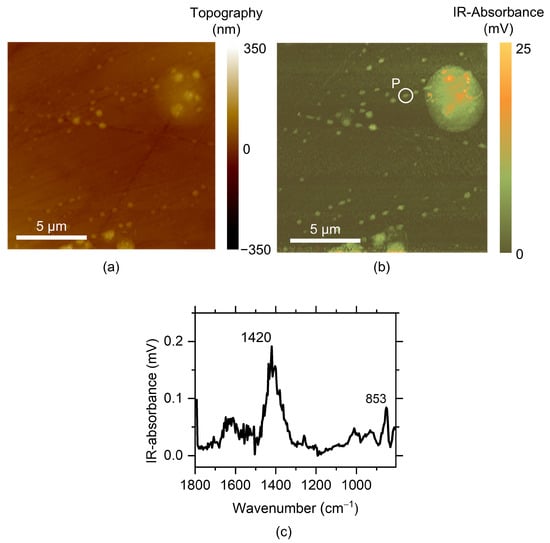
Figure 14.
AFM-IR measurements showing a topographical map (a), an intensity map of the 1420 cm−1 band (b), and a typical spectrum of the corrosion products (c) from a corroded area of the AZ91 sample, after 5 h of exposure in humid air with ambient CO2 content. The spectrum was taken from position “P”.
3.4. Scanning Kelvin Probe Force Microscopy Measurements
SKPFM was employed to study the Volta potential variations in AZ91, focusing on different microstructural features on the surface of unexposed samples. These measurements can provide insights into the likelihood of forming micro-galvanic couples between more noble constituents and components in the microstructure with lower Volta potential. Figure 15 presents Volta potential maps and line profiles showing the potential differences (ΔV) between α-Mg and the β-phase, as well as for AlMn intermetallic particles. The Volta potential difference between the β-phase and the central regions of the α-Mg grains were in the range from 100 to 150 mV, whereas the difference between the β-phase and the α-Mg grains in its vicinity was much smaller.
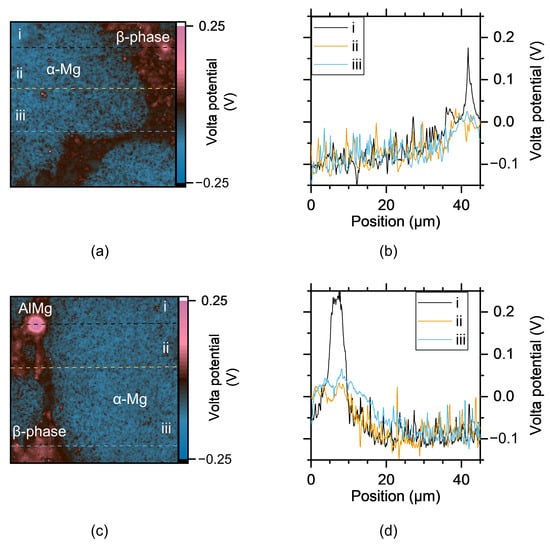
Figure 15.
Volta potential maps of unexposed AZ91 surfaces (a,c), and three Volta potential line profiles (i, ii and iii) for each area (b,d). Line profiles show the Volta potential along the dashed line with the corresponding index (i, ii, and iii) shown in the potential maps.
The Volta potential line profiles indicate a potential gradient within the α-Mg phase near the more noble phases. This gradient is likely linked to the observation that corrosion tends to initiate in the central regions of the α-Mg grains. For AlMn particles, the ΔV between them and the α-Mg phase is higher, consistent with previous findings [,]. The Volta potential differences between the various phases are slightly lower compared to earlier studies []. Arrabal et al. [] reported only minor differences between α-Mg and the β-phase, but their SKPFM measurements were limited to 5–7 μm into the α-Mg from the interface between the two phases. This is consistent with the results obtained here, showing low Volta potential differences close to the β-phase. However, they did observe a gradient in Al content within the α-Mg near the β-phase, which is also supported by the SEM-EDS measurements in this study. The gradient in the aluminium content is due to coring which occurs during solidification of the alloy.
This gradient in Al content could explain the small Volta potential differences observed near the β-phase. In contrast, the central regions of the α-Mg grains exhibit a higher ΔV, sufficient to form micro-galvanic couples. It should be noted that there is no corrosion attack localised in the proximity of the AlMn particles despite the large potential difference between AlMn and the a-Mg close to these particles.
4. Discussion
The results of the surface analysis of corroded magnesium alloys using IRRAS, SEM-EDS, and IR-spectroscopy imaging show that corrosion is initiated on the α-Mg phase, and in the central parts of the grains for AZ91:
Mg → Mg2+ (aq) + 2e−
The main sites for the cathodic reaction in AZ91 are probably the β-phase, on which oxygen reduction can take place according to the following:
O2 + 4e− + 2H2O → 4OH− (aq)
Water reduction may also contribute to the cathodic process according to the following:
H2O +2e− → H2(g) + 2OH− (aq)
The formation of hydroxyl ions will cause alkalinisation of the surface electrolyte, which affects the type of corrosion products that can form and may also dissolve aluminium-containing surface oxides on the β-phase [], which can facilitate the cathodic reactions on the β-phase and contribute to higher corrosion in low CO2 content conditions.
In exposures in humid air with ambient CO2 content, absorption of CO2 in the thin adsorbed water layer occurs. The following reactions describe the equilibria for CO2 in water:
H2CO3 (aq) + OH− → HCO3− (aq) + H2O
HCO3− (aq) + OH− → CO32− (aq)+ H2O
Formation of corrosion products occurs by ion pairing and precipitation in the surface electrolyte layer by the following dominating reaction in low CO2 content humid air:
Mg2+ (aq) + 2OH− (aq) → Mg(OH)2
Both cathodic reaction routes in Equations (3) and (4) give rise to alkalinisation of the surface electrolyte, which promotes the formation of Mg(OH)2 at low CO2 content in the air. In humid air with ambient CO2 content, the formation of magnesium hydroxy carbonate prevails according to the following equation:
5Mg2+ (aq) + 6OH− (aq) + 4HCO32− (aq) → Mg5(CO3)4(OH)2·4H2O
The effect of the CO2 content on the corrosion behaviour is significant and can be related to the protective properties of the corrosion products formed. As seen in Figure 11, the corrosion products formed on AZ91 at low CO2 content seems to have a porous, sponge-like structure, which is probably less protective and promotes further corrosion. Furthermore, the solubility of Mg(OH)2 is higher (log Ks = −17.1) [] compared to, for instance, hydromagnesite Mg5(CO3)4(OH)2·4H2O (log Ks = −37.08) [], which can lead to a higher ionic content in the thin water layer, higher ionic conductivity, and lower water activity. The latter promotes further water uptake in the surface electrolyte and leads to the formation of a thicker electrolyte layer, stimulating the corrosion process.
As seen from the IRRAS results, a small amount of Mg(OH)2 was also formed on AZ31 during exposure in humid air with ambient CO2 content, while Mg(OH)2 was not detected on AZ91. Furthermore, larger amounts of magnesium hydroxy carbonate were found on AZ91 compared to AZ31 at ambient CO2 concentrations. This indicates that the Al content in the alloys affects the composition of the corrosion products formed. This is in line with the study by Feliu et al., which found that increasing Al content in the alloys leads to more magnesium carbonate formation and less MgO and Mg(OH)2 []. The suppression of Mg(OH)2 formation could be related to the dissolution of Al3+ ions from the surface film into the surface electrolyte layer. This, in turn, could affect the pH and promote the formation of magnesium carbonates. It is clear from the IRRAS measurements that the rate of formation of Mg(OH)2 is higher for AZ31 compared to AZ91. This could probably be linked to a lower protective ability of the surface oxide/surface film on AZ31, caused by its lower Al content in the surface film. The formation of less protective Mg(OH)2 promotes a higher corrosion rate on AZ31. The corrosion attacks are more localised on AZ91 and mainly initiated in areas with lower aluminium content, while the corrosion of AZ31 seems to be more uniform. This should be linked to the generally lower Al content in the latter alloy.
Although water reduction is frequently regarded as the predominant cathodic reaction under aqueous conditions, it has been found that oxygen reduction is an important contributor to the cathodic process under atmospheric conditions [,], as well as in electrolyte solutions [,]. For AZ91, the β-phase is probably the cathodic areas, and as indicated by the results of SEM-EDS and IR-chemical imaging. The formation of corrosion products is low on the β-phase, and the cathodic processes can be sustained during prolonged exposures. The Volta potential difference between the β-phase and the centre of the α-phase was 100–150 mV, which could be enough for the formation of micro-galvanic couples. However, as the SKPFM results indicate, the Volta potential differences are reached after approximately 10 μm into the α-phase, which could explain why corrosion is initiated in the central parts and not close to the β-phase. The Volta potential differences are larger between the AlMn intermetallics and the α-phase. Although these are located near the β-phase in many cases, corrosion attacks are mainly initiated in the interior of the α-phase during the initial stages of corrosion. This suggests that aluminium content has a crucial role in stabilising the surface film on the α-phase, particularly near the secondary phases and intermetallics. However, additional research is required to gain a more comprehensive understanding of this.
We believe that oxygen reduction is the dominating cathode reaction for AZ91 under these conditions, with very thin water adlayers on the surface. For AZ31, Strebl et al. [] showed that oxygen reduction contributed less to the cathodic process compared to AZ91, and water reduction could be more important. For AZ31, the intermetallic AlMn particles are probably the main cathodic sites. A schematic description of the initial atmospheric corrosion of AZ91, with micro-galvanic couples formed between the β-phase and the central parts of the α-phase, is shown in Figure 16. However, it is also likely that AlMn intermetallics will act as cathodic sites on AZ91 as well, but corrosion initiation and propagation probably occur, at least during the initial stages, in locations of the α-phase with lower aluminium content. The intermetallics are probably the main cathodic sites for AZ31, but due to its generally lower aluminium content, the corrosion seems to be less localised.
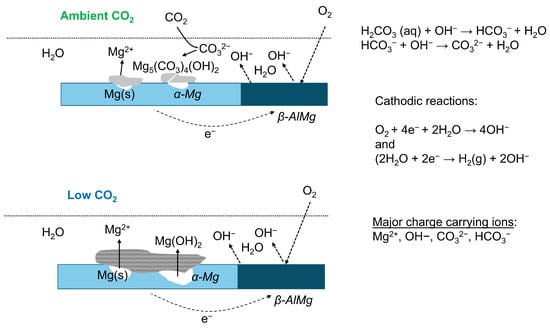
Figure 16.
Schematic description of atmospheric corrosion processes on AZ91 in humid air with ambient and low CO2 content. Formation of micro-galvanic couples with the β-phase and α-Mg.
5. Conclusions
The initial atmospheric corrosion of magnesium alloys AZ91 and AZ31 was investigated in pure humid air with 95% RH under the following two conditions: low CO2 concentration (≤1 ppm) and ambient CO2 levels (≈400 ppm). The study employed in situ infrared reflection absorption spectroscopy (IRRAS), FTIR-ATR FPA imaging, Scanning Electron Microscopy with Energy Dispersive Spectroscopy (SEM-EDS), Scanning Kelvin Probe Force Microscopy, and Atomic Force Microscopy with Infrared Spectroscopy (AFM-IR) to study the initial atmospheric corrosion during 19 h of exposure.
The in situ IRRAS measurements provided new insights into the composition and kinetics of corrosion product formation in humid air. Under low-CO2 conditions, Mg(OH)2 was the predominant corrosion product on AZ91. In contrast, at ambient CO2 levels, magnesium hydroxy carbonate was the dominant product, with a notably higher carbonate content observed on AZ91. For AZ31, a small amount of Mg(OH)2 was also detected with ambient CO2. Quantitative analysis of corrosion rates based on IRRAS data indicated that corrosion was significantly more severe, by an order of magnitude, in humid air with low CO2 for both alloys. Additionally, AZ31 exhibited a higher corrosion rate than AZ91 under both low and ambient CO2 conditions.
Initial corrosion of AZ91 was localised, primarily occurring in the central regions of the α-Mg phase, where corrosion product formation was observed, and significant Volta potential differences were measured relative to the β-phase. No significant corrosion product formation was detected on the β-phase. These findings suggest that the corrosion behaviour is strongly influenced by the microstructure of AZ91, with the β-phase (AlMg) acting as the primary site for cathodic reactions and the central parts of the α-phase serving as the site for anodic processes, forming micro-galvanic cells. The lower corrosion rate and more localised corrosion attacks observed for AZ91 is likely related to its higher aluminium content compared to AZ31.
The reduced corrosion rate in ambient CO2 content is likely attributed to the formation of a physically blocking and more protective magnesium carbonate layer, such as hydromagnesite (Mg5(CO3)4(OH)2·4H2O). In contrast, Mg(OH)2 forms a more porous and less protective layer, resulting in higher corrosion rates. Additionally, the lower solubility of magnesium carbonate may contribute to a less conductive surface electrolyte and a thinner water layer, further reducing corrosion compared to the more soluble Mg(OH)2.
Author Contributions
Conceptualisation, D.P., N.L., and D.T.; methodology, D.P.; software, D.P.; validation, D.P.; formal analysis, D.P.; investigation, D.P. and A.W.; resources, D.P. and D.T.; data curation, D.P. and A.W.; writing—original draft preparation, D.P. and A.W.; writing—review and editing, D.P., A.W., DT., and N.L.; visualisation, D.P. and A.W.; funding acquisition, D.T. and N.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We acknowledge Oskar Karlsson, Swerim, for performing SEM-EDS.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Zhan, L.; Kong, C.; Zhao, C.; Cui, X.; Zhang, L.; Song, Y.; Lu, Y.; Xia, L.; Ma, K.; Yang, H.; et al. Recent advances on magnesium alloys for automotive cabin components: Materials, applications, and challenges. J. Mater. Res. Technol. 2025, 36, 9924–9961. [Google Scholar] [CrossRef]
- Ashraf, M.N.; Guo, Z.; Wu, R.; Jhiao, W.; Chun, M.X.; Khan Gorar, A.A. Historical Progress in Electromagnetic Interference Shielding Effectiveness of Conventional Mg Alloys Leading to Mg-Li-Based Alloys: A Review. Adv. Eng. Mater. 2023, 25, 2300732. [Google Scholar] [CrossRef]
- Song, G.-L. Corrosion electrochemistry of magnesium (Mg) and its alloys. In Corrosion of Magnesium Alloys; Song, G.L., Ed.; Woodhead: Cambridge, UK, 2011; pp. 3–65. [Google Scholar]
- Ghali, E. Activity and passivity of magnesium (Mg) and its alloys. In Corrosion of Magnesium Alloys; Song, G.L., Ed.; Woodhead: Cambridge, UK, 2011; pp. 66–109. [Google Scholar]
- Atrens, A.; Liu, M.; Zainal Abidin, N.I.; Song, G.-L. Corrosion of magnesium alloys and metallurgical influence alloys. In Corrosion of Magnesium Alloys; Song, G.L., Ed.; Woodhead: Cambridge, UK, 2011; pp. 117–161. [Google Scholar]
- Huang, J.; Song, G.-L.; Atrens, A.; Dargusch, M. What activates the Mg surface—A comparison of Mg dissolution mechanisms. J. Mat. Sci. Technol. 2020, 57, 204–220. [Google Scholar] [CrossRef]
- Song, G.-L.; Atrens, A. Recently deepened insights regarding Mg corrosion and advanced engineering applications of Mg alloys. J. Magnes. Alloys 2023, 11, 3948–3991. [Google Scholar] [CrossRef]
- Esmaily, M.; Svensson, J.E.; Fajardo, S.; Birbilis, N.; Frankel, G.S.; Virtanen, S.; Arrabal, R.; Thomas, S.; Johansson, L.G. Fundamentals and advances in magnesium alloy corrosion. Prog. Mater. Sci. 2017, 89, 92–193. [Google Scholar] [CrossRef]
- Liu, H.; Cao, F.; Song, G.-L.; Zheng, D.; Shi, Z.; Dargush, M.S.; Atrens, A. Review of the atmospheric corrosion of magnesium alloys. J. Mat. Sci. Technol. 2019, 35, 2003–2016. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, L.; Wang, H.; Liu, T.; Li, H.; Tan, B.; Pan, Y.; Liu, A.; Cheng, J. Research progress on the corrosion behavior of magnesium alloys in natural environments. Mater. Today Commun. 2025, 48, 113433. [Google Scholar] [CrossRef]
- Wang, M.; Yang, L.; Liu, H.; Wang, X.; Li, Y.; Huang, Y. Recent Progress on Atmospheric Corrosion of Field-Exposed Magnesium Alloys. Metals 2024, 14, 1000. [Google Scholar] [CrossRef]
- Arrabal, R.; Pardo, A.; Merino, M.C.; Merino, S.; Mohedano, M.; Casajús, P. Corrosion behaviour of Mg/Al alloys in high humidity atmospheres. Mater. Corros. 2011, 62, 326–334. [Google Scholar] [CrossRef]
- Shahabi-Navid, M.; Cao, Y.; Svensson, J.E.; Allanore, A.; Birbilis, N.; Johansson, L.G.; Esmaily, M. On the early stages of localised atmospheric corrosion of magnesium–aluminium alloys. Sci. Rep. 2020, 10, 20972. [Google Scholar] [CrossRef] [PubMed]
- Feliu, S., Jr.; Pardo, A.; Merino, M.C.; Coy, A.E.; Viejo, F.; Arrabal, R. Correlation between the surface chemistry and the atmospheric corrosion of AZ31, AZ80 and AZ91D magnesium alloys. Appl. Surf. Sci. 2009, 255, 4102–4108. [Google Scholar] [CrossRef]
- Lindström, R.; Johansson, L.-G.; Thompson, G.E.; Skeldon, P.; Svensson, J.-E. Corrosion of magnesium in humid air. Corros. Sci. 2004, 46, 1141–1158. [Google Scholar] [CrossRef]
- Zhao, M.C.; Liu, M.; Song, G.; Atrens, A. Influence of the β-phase morphology on the corrosion of the Mg alloy AZ91. Corros. Sci. 2008, 50, 1939–1953. [Google Scholar] [CrossRef]
- Jönsson, M.; Persson, D. The influence of the microstructure on the atmospheric corrosion behaviour of magnesium alloys AZ91D and AM50. Corros. Sci. 2010, 52, 1077–1085. [Google Scholar] [CrossRef]
- Esmaily, M.; Blücher, D.B.; Svensson, J.E.; Halvarsson, M.; Johansson, L.G. New insights into the corrosion of magnesium alloys —The role of aluminum. Scr. Mater. 2016, 115, 91–95. [Google Scholar] [CrossRef]
- Danaie, M.; Asmussen, R.M.; Jakupi, P.; Shoesmith, D.W.; Botton, G.A. The role of aluminum distribution on the local corrosion resistance of the microstructure in a sand-cast AM50 alloy. Corros. Sci. 2013, 77, 151–163. [Google Scholar] [CrossRef]
- Schwarz, T.M.; Birbilis, N.; Gault, B.; McCarroll, I. Understanding the Al diffusion pathway during atmospheric corrosion of a Mg-Al alloy using atom probe tomography. Corros. Sci. 2025, 252, 112951. [Google Scholar] [CrossRef]
- Feliu, S., Jr.; Merino, M.C.; Arrabal, R.; Coy, A.E.; Matykina, E. XPS study of the effect of aluminium on the atmospheric corrosion of the AZ31magnesium alloy. Surf. Interface Anal. 2009, 41, 143–150. [Google Scholar] [CrossRef]
- Feliu, S., Jr.; Maffiotte, C.; Galván, J.C.; Barranco, V. Atmospheric corrosion of magnesium alloys AZ31 and AZ61 under continuous condensation conditions. Corros. Sci. 2011, 53, 1865–1872. [Google Scholar] [CrossRef]
- Persson, D.; Leygraf, C. In Situ Infrared Reflection Absorption Spectroscopy for Studies of Atmospheric Corrosion. J. Electrochem. Soc. 1993, 140, 1256. [Google Scholar] [CrossRef]
- Persson, D.; Thierry, D.; LeBozec, N.; Prosek, T. In situ infrared reflection spectroscopy studies of the initial atmospheric corrosion of Zn–Al–Mg coated steel. Corros. Sci. 2013, 72, 54–63. [Google Scholar]
- Persson, D.; Thierry, D.; LeBozec, N. The Effect of Microstructure on Local Corrosion Product Formation during Initial SO2-Induced Atmospheric Corrosion of ZnAlMg Coating Studied by FTIR-ATR FPA Chemical Imaging. Corros. Mater. Degrad. 2023, 4, 503–515. [Google Scholar] [CrossRef]
- Mathurin, J.; Deniset-Besseau, A.; Bazin, D.; Dartois, E.; Wagner, M.; Dazzi, A. Photothermal AFM-IR spectroscopy and imaging: Status, challenges, and trends. J. Appl. Phys. 2022, 131, 010901. [Google Scholar] [CrossRef]
- Farmer, V.C. (Ed.) The Infrared Spectra of Minerals; Mineralogical Society: London, UK, 1974. [Google Scholar]
- Jönsson, M.; Persson, D.; Thierry, D. Corrosion product formation during NaCl induced atmospheric corrosion of magnesium alloy AZ1D. Corr. Sci. 2007, 49, 1540–1558. [Google Scholar] [CrossRef]
- Fotea, C.; Callaway, J.; Alexander, M.R. Characterisation of the surface chemistry of magnesium exposed to the ambient atmosphere. Surf. Interface Anal. 2006, 38, 1363–1371. [Google Scholar] [CrossRef]
- Harms, L.; Brand, I. Application of PM IRRAS to study structural changes of the magnesium surface in corrosive environments. Vib. Spectrosc. 2018, 97, 106–113. [Google Scholar] [CrossRef]
- Hofmeister, A.M.; Keppel, E.; Speck, A.K. Absorption and reflection infrared spectra of MgO and other diatomic compounds. Mon. Not. R. Astron. Soc. 2003, 345, 16–38. [Google Scholar] [CrossRef]
- Harbecke, B.; Heinz, B.; Grosse, P. Optical Properties of Thin Films and the Berreman Effect. Appl. Phys. A 1985, 38, 263–267. [Google Scholar] [CrossRef]
- Francis, S.A.; Ellison, A.H. Infrared Spectra of Monolayers on Metal Mirrors. J. Opt. Soc. Am. 1959, 49, 131–138. [Google Scholar] [CrossRef]
- He, H.; Cao, J.; Duan, N. Defects and their behaviors in mineral dissolution under water environment: A review. Sci. Total Environ. 2019, 651, 2208–2217. [Google Scholar] [CrossRef]
- Wang, K.; Paulus, B. Cluster Formation Effect of Water on Pristine and Defective MoS2, Monolayers. Nanomaterials 2023, 13, 229. [Google Scholar] [CrossRef]
- Jönsson, M.; Thierry, D.; Lebozec, N. The influence of microstructure on the corrosion behaviour of AZ91D studied by scanning Kelvin probe force microscopy and scanning Kelvin probe. Corr. Sci. 2006, 48, 1193–1208. [Google Scholar] [CrossRef]
- Jönsson, M.; Persson, D.; Gubner, R. The Initial Steps of Atmospheric Corrosion on Magnesium Alloy AZ91D. J. Electrochem. Soc. 2007, 154, C684. [Google Scholar] [CrossRef]
- Liu, M.; Zanna, S.; Ardelean, H.; Frateur, I.; Schmutz, P.; Song, G.; Atrens, A.; Marcus, P. A first quantitative XPS study of the surface films formed, by exposure to water, on Mg and on the Mg–Al intermetallics: Al3Mg2 and Mg17Al12. Corros. Sci. 2009, 51, 1115–1127. [Google Scholar] [CrossRef]
- Altmaier, M.; Metz, V.; Neck, V.; Müller, R.; Fanghänel, T. Solid-liquid equilibria of Mg(OH)2(cr) and Mg2(OH)3Cl·4H2O(cr) in the system Mg-Na-H-OH-Cl-H2O at 25 °C. Geochim. Cosmochim. Acta 2003, 67, 3595–3601. [Google Scholar] [CrossRef]
- Gautier, Q.; Benezeth, P.; Mavromatis, V.; Schott, J. Hydromagnesite solubility product and growth kinetics in aqueous solution from 25 to 75 °C. Geochim. Cosmochim. Acta 2014, 138, 1–20. [Google Scholar] [CrossRef]
- Strebl, M.; Virtanen, S. Real-Time Monitoring of Atmospheric Magnesium Alloy Corrosion. J. Electrochem. Soc. 2019, 166, C3001–C3009. [Google Scholar] [CrossRef]
- Strebl, M.; Bruns, M.; Virtanen, S. Editors’ Choice—Respirometric in Situ Methods for Real-Time Monitoring of Corrosion Rates: Part I. Atmospheric Corrosion. J. Electrochem. Soc. 2020, 167, 021510. [Google Scholar] [CrossRef]
- Silva, E.; Lamaka, S.V.; Mei, D.; Zheludkevich, M. The Reduction of Dissolved Oxygen During Magnesium Corrosion. ChemistryOpen 2018, 7, 664–668. [Google Scholar] [CrossRef]
- Wang, C.; Qian, K.; Wu, Y.; Mei, D.; Chu, C.; Xue, F.; Bai, J.; Zheludkevich, M.L.; Lamaka, S.V. Consistent high rate oxygen reduction reaction during corrosion of Mg-Ag Alloy. Corros. Sci. 2024, 229, 111893. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).