Abstract
This study used density functional theory (DFT)-based first-principles calculations to investigate the desolvation effect of single-walled carbon nanotubes (SWCNTs) modified with hydroxyl (-OH), carbonyl (-C=O), and carboxyl (-COOH) groups. SWCNTs have great potential as supercapacitor electrode materials due to their unique structural and electronic properties, but their practical application is limited by poor solvation-induced dispersibility and low ion transport efficiency. To solve this, this study constructed functionalized SWCNT models, simulated their interaction with lithium ion (Li+) complexes in acetonitrile (AN) solvent, and analyzed Li+ desolvation behavior, relative capacitance, and post-desolvation density of states (DOSs). The key research results are as follows: [Li(AN)]+ complete desolvation sizes differed: 5.91 Å (pristine SWCNTs), 6.26 Å (hydroxylated SWCNTs, HCNT), 6.11 Å (carbonylated SWCNTs, CNCNT; carboxylated SWCNTs, CXCNT). HCNT showed the largest relative capacitance enhancement (max 1.4× pristine), while CNCNT and CXCNT both had a max 1.3× improvement. Post-desolvation DOS analysis revealed distinct electronic property changes: HCNT-Li+ enhanced metallicity and conductivity; CNCNT-Li+ increased metallicity but reduced conductivity; and CXCNT-Li+ decreased metallicity with nearly unchanged conductivity. This study provides an atomic-scale theoretical basis for optimizing the properties of SWCNT solutions, supporting their application in high-performance supercapacitors, particularly in enhancing device energy density and cycle stability.
1. Introduction
Carbon nanotubes (CNTs), leveraging their high specific surface area, excellent electronic conductivity, and mechanical stability derived from their one-dimensional tubular structure, demonstrate core application potential in the field of supercapacitor electrode materials [1,2,3,4]. Their unique structure can effectively construct ion transport channels and enhance the charge storage efficiency between electrodes and electrolytes, making them one of the key materials for breaking through the energy density bottleneck of traditional supercapacitors [5]. However, pristine CNTs exhibit strong surface inertness, poor dispersibility, and are susceptible to the strong adsorption of solvent molecules in electrolyte environments (such as aqueous or organic electrolytes) [6,7]. This not only hinders effective contact between electrolyte ions and the electrode surface but also reduces the charge transfer rate, significantly limiting the practical application efficiency of supercapacitors [8,9].
To improve the research system for the capacitance of carbon materials and enhance the capacitance and electrical conductivity of supercapacitors, single-walled carbon nanotubes (SWCNTs) are modified accordingly. The functionalization of CNTs through the grafting of functional groups such as hydroxyl, carbonyl, and carboxyl can simultaneously regulate their surface electronic structure and lyophilicity, as well as optimize the electrode–electrolyte interface compatibility [10]. The desolvation effect accompanying this process (i.e., the process of solvent molecules desorbing from the surface of functionalized CNTs) directly determines the ion desorption–adsorption kinetics, interface charge storage stability, and cycle life during the charge–discharge process of supercapacitors [11]. Currently, there is still a lack of atomic-level understanding regarding the microscopic mechanisms of the desolvation effect, such as the desorption energy barrier of electrolyte solvent molecules, the structure–activity relationship between the type of functional groups and the degree of desolvation, and the evolution law of the electronic structure on the CNTs surface during the desorption process. Experimental methods are limited by the accuracy of characterization and are thus difficult to conduct precise analyses. Nevertheless, first-principles calculations, which can simulate atomic-level electronic behavior and energy changes based on quantum mechanics principles, provide an ideal tool for revealing the essence of this effect.
In this study, the first-principles calculation method was employed to systematically investigate the regulatory effect of hydroxylation and carboxylation modifications on the surface electronic structure of CNTs. Emphasis was placed on analyzing the adsorption–desorption energy barrier of solvent molecules in acetonitrile (a commonly used electrolyte in supercapacitors) on the surface of functionalized CNTs, and clarifying the thermodynamic and kinetic mechanisms of the desolvation process. The research results are expected to provide theoretical support for optimizing the preparation process of functionalized CNT electrodes for supercapacitors, thereby improving the energy density and cycle stability of the devices. At the same time, they are expected to offer new ideas for regulating the interface of low-dimensional nanomaterials in the field of electrochemical energy storage. In a previous related work, the group of authors used two different forms of bilayer graphene to simulate flat micropores and investigate their desolvation behavior towards [Li(H2O)]+ [12]. Currently, there are few reported studies on the desolvation behavior of [Li(AN)]+ in functionalized cylindrical micropores [3].
By constructing cylindrical pores composed of oxygen-containing functionalized carbon nanotubes (CNTs), the reaction energies of Li+, acetonitrile solvent molecules, and Li+ complexes in oxygen-containing functionalized CNTs with different pore sizes were calculated. The desolvation behavior of Li+ was investigated, as stable structures not only enabled the acquisition of relative capacitance values from the data but also allowed for the determination of the size of corresponding desolvation pores in the solution and the influence of oxygen-containing functionalized CNTs on capacitance values. Furthermore, the complete desolvation pore size of cylindrical pores and the mechanism of relative capacitance enhancement were obtained. Through density of states (DOSs) analysis of the oxygen-containing functionalized CNT-Li+ system, the relationship regarding the conductivity strength of the CNT-Li+ system after desolvation was derived. The research results indicate that the study of desolvation in the micropores of carbon materials is a research field with broad prospects.
2. Calculation Method
This study conducts computational optimization using the CASTEP [13] software (2019 Version) module based on density functional theory (DFT) [14,15,16]. The Perdew–Burke–Ernzerhof (PBE) functional [17,18] is used under the generalized gradient approximation (GGA) [19,20,21,22], and the Grimme method with van der Waals correction is utilized. Ultra-soft pseudopotentials [23,24] are used to describe the interactions between valence electrons and atoms. Considering the large number of atoms in the system structure, a 1 × 1 × 2 k-point grid [25,26,27,28] in the Brillouin zone is selected, and its convergence is verified. During the geometric structure optimization, the convergence criteria are set as follows: the interatomic force convergence standard is 0.03 eV/Å, the internal stress convergence standard is 5 × 10−2 GPa, the single-atom energy convergence standard is 1 × 10−5 eV, and the maximum ionic displacement is within 0.01 Å.
In this study, the different types of single-walled carbon nanotubes (SWCNTs) studied are composed of periodic hexagonal supercell structures. Taking the (5, 5) single-walled carbon nanotube structure as an example, three types of oxygen-containing functional groups are placed in the middle position inside the carbon nanotube, as shown in Figure 1 (in Figure 1, the gray atoms on the pore base are carbon atoms, the white atoms are hydrogen atoms, and the red atoms are oxygen atoms). As listed in Table 1, typical single-walled carbon nanotubes with diameters ranging from 5 to 10 Å are considered within the scope of this study.
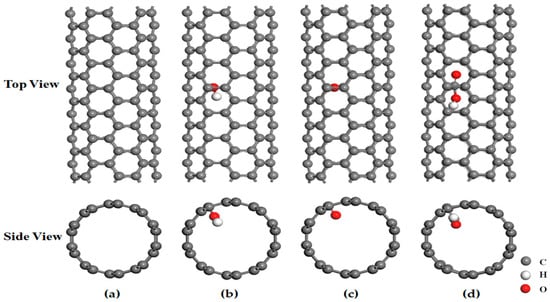
Figure 1.
Stable structure of SWCNTs. (a) Stable Structure of (5, 5) SWCNTs. (b) Stable Structure of Hydroxylated (5, 5) SWCNTs. (c) Stable Structure of Carbonylated (5, 5) SWCNTs. (d) Stable structure of Carboxylated (5, 5) SWCNTs.

Table 1.
Structure forms and diameters of single-walled carbon nanotubes.
3. Results and Discussion
3.1. Reaction Principle
During the intercalation of Li ions into single-walled carbon nanotubes (SWCNTs) or oxygen-containing functionalized single-walled carbon nanotubes (OSWCNTs), the following three reactions may occur. The first is that Li ions can stably exist in the pores, while AN solvent molecules escape from the pores. The second is that AN solvent molecules can be intercalated into the pores, while Li ions remain outside the pores. The third is that Li ion complexes are intercalated into the pores. These three reactions can be represented by (1) to (3), respectively:
Li (AN) + SWCNTs/OSWCNTs → Li (SWCNTs/OSWCNTs) + AN
Li (AN) + SWCNTs/OSWCNTs → AN (SWCNTs/OSWCNTs) + Li
Li (AN) + SWCNTs/OSWCNTs → Li (AN) SWCNTs/OSWCNTs
Here, Li (AN) represents the Li ion complex; SWCNTs stands for single-walled carbon nanotubes; OSWCNTs denotes oxygen-containing functionalized single-walled carbon nanotubes, which include hydroxyl carbon nanotubes (CNT-OH), carbonyl carbon nanotubes (CNT-O), and carboxyl carbon nanotubes (CNT-COOH); Li (SWCNTs/OSWCNTs) refers to the intercalation compound of Li ions in single-walled carbon nanotubes or oxygen-containing functionalized single-walled carbon nanotubes; AN (SWCNTs/OSWCNTs) represents the intercalation compound of AN molecules in single-walled carbon nanotubes or oxygen-containing functionalized single-walled carbon nanotubes; Li (AN) SWCNTs/OSWCNTs indicates the intercalation compound of Li ion complexes in single-walled carbon nanotubes or oxygen-containing functionalized single-walled carbon nanotubes. To evaluate the possibility of intercalation of Li ions, solvated Li ions, and solvents into carbon nanotubes with different diameter sizes, the reaction energies for the three reaction scenarios were calculated and defined as Eint1, Eint2, Eint3, Eint4, and Eint5, respectively, which can be expressed by Equations (4)–(8):
Eint1 = ELi(OSWCNTs) + EAN − ELi(AN) − EOSWCNTs
Eint2 = EAN(OSWCNTs) + ELi − ELi(AN) − EOSWCNTs
Eint3 = ELi(AN)OSWCNTs − E Li(AN) − EOSWCNTs
Eint4 = ELi(SWCNTs) + EAN − ELi(AN) − ESWCNTs
Eint5 = ELi(AN)SWCNTs − ELi(AN) − ESWCNTs
Herein, ELi(AN) represents the energy of the Li ion complex; EOSWCNTs denotes the energy of oxygen-containing functionalized single-walled carbon nanotubes; ESWCNTs stands for the energy of single-walled carbon nanotubes; ELi(OSWCNTs) indicates the energy of the intercalation compound of Li ions in oxygen-containing functionalized single-walled carbon nanotubes; ELi(SWCNTs) represents the energy of the intercalation compound of Li ions in single-walled carbon nanotubes; EAN is the energy of acetonitrile solvent molecules; EAN(OSWCNTs) denotes the energy of the intercalation compound of acetonitrile solvent molecules in oxygen-containing functionalized single-walled carbon nanotubes; ELi(AN)SWCNTs represents the energy of the intercalation compound of Li ion complexes in single-walled carbon nanotubes; ELi is the energy of Li ions; ELi(AN)OSWCNTs indicates the energy of the intercalation compound of Li ion complexes in oxygen-containing functionalized single-walled carbon nanotubes; and ELi(AN)SWCNTs represents the energy of the intercalation compound of Li ion complexes in single-walled carbon nanotubes. The smaller the values of Eint1, Eint2, Eint3, Eint4, and Eint5, the greater the possibility of the reaction occurring.
3.2. Desolvation of Li+ Complexes
Figure 2 shows the desolvation reaction energy curves of [Li(AN)]+ with hydroxylated single-walled carbon nanotubes and single-walled carbon nanotube models. To obtain the critical size of the desolvation pore diameter for [Li(AN)]+, the reaction energies of Li+, AN, [Li(AN)]+ in hydroxylated single-walled carbon nanotubes, and Li+ and [Li(AN)]+ in single-walled carbon nanotubes were calculated, and are denoted as Eint1, Eint2, Eint3, Eint4, and Eint5, respectively. When the diameter of the hydroxylated carbon nanotube d < 6.26 Å, Eint2 < 0, indicating that AN molecules can exist alone in the hydroxylated single-walled carbon nanotube. When the diameter of the hydroxylated carbon nanotube d = 6.26 Å, the energy value of Eint1 (−4.09 eV) is close to that of Eint3 (−4.04 eV), meaning 6.26 Å is the critical point for the desolvation of [Li(AN)]+ in the hydroxylated carbon nanotube. When the diameter of the hydroxylated carbon nanotube is less than 6.26 Å and Eint1 < Eint3 < 0, Li+ exists more stably in the pore than [Li(AN)]+. Therefore, when the pore diameter is less than 6.26 Å, [Li(AN)]+ can undergo complete desolvation in the hydroxylated carbon nanotube. When the diameter of the hydroxylated carbon nanotube is greater than 6.26 Å, Eint3 drops rapidly to the minimum value, which indicates that no desolvation occurs in the system, and [Li(AN)]+ exists more stably in the pore. When the diameter of the carbon nanotube (d) is 5.91 Å, the energy value of Eint4 (−2.98 eV) is close to that of Eint5 (−3.06 eV). Therefore, 5.91 Å is the critical point for the desolvation of [Li(AN)]+ in the carbon nanotube. When the diameter of the carbon nanotube is less than 5.91 Å and Eint4 < Eint5 < 0, Li+ exists more stably in the pore than [Li(AN)]+. Thus, when the pore diameter is less than 5.91 Å, [Li(AN)]+ can undergo complete desolvation in the carbon nanotube. The results show that, compared with carbon nanotubes, Li+ and [Li(AN)]+ require less energy to enter hydroxylated carbon nanotubes and are more easily incorporated. Hydroxyl groups improve the adsorption capacity of carbon nanotubes for Li+ and enhance the ability of carbon nanotubes to store Li+.
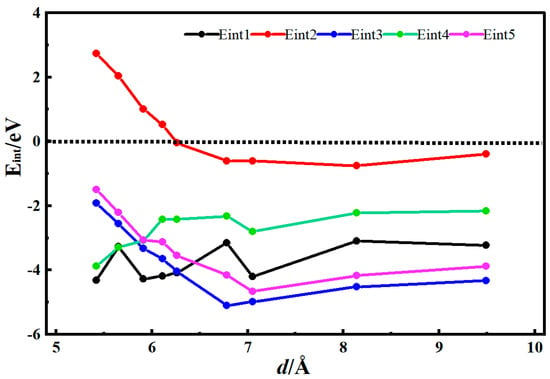
Figure 2.
Reaction energy curves of Li+, AN, [Li(AN)]+ in hydroxylated functional carbon nanotubes and Li+ and [Li(AN)]+ in carbon nanotubes.
Figure 3 shows the desolvation reaction energy curves of [Li(AN)]+ with carbonylated single-walled carbon nanotubes and single-walled carbon nanotube models. To obtain the critical size of the desolvation pore diameter for [Li(AN)]+, the reaction energies of Li+, AN, [Li(AN)]+ in carbonylated single-walled carbon nanotubes, and Li+ and [Li(AN)]+ in single-walled carbon nanotubes were calculated, denoted as Eint1, Eint2, Eint3, Eint4, and Eint5, respectively. When the diameter of the carbonylated carbon nanotube d < 6.78 Å, Eint2 < 0, indicating that AN molecules can exist alone in the carbonylated single-walled carbon nanotube. When the diameter of the carbonylated carbon nanotube d = 6.11 Å, the energy value of Eint1 (−3.52 eV) is close to that of Eint3 (−3.49 eV), meaning 6.11 Å is the critical point for the desolvation of [Li(AN)]+ in the carbonylated carbon nanotube. When the diameter of the carbonylated carbon nanotube is less than 6.11 Å and Eint1 < Eint3 < 0, Li+ exists more stably in the pore than [Li(AN)]+. Therefore, when the pore diameter is less than 6.11 Å, [Li(AN)]+ can undergo complete desolvation in the carbonylated carbon nanotube. When the diameter of the hydroxylated carbon nanotube is greater than 6.11 Å, Eint3 drops rapidly to the minimum value, which indicates that no desolvation occurs in the system, and [Li(AN)]+ exists more stably in the pore. When the diameter of the carbon nanotube d = 5.91 Å, the energy value of Eint4 (−2.98 eV) is close to that of Eint5 (−3.06 eV), so 5.91 Å is the critical point for the desolvation of [Li(AN)]+ in the carbon nanotube. When the diameter of the carbon nanotube is less than 5.91 Å and Eint4 < Eint5 < 0, Li+ exists more stably in the pore than [Li(AN)]+. Thus, when the pore diameter is less than 5.91 Å, [Li(AN)]+ can undergo complete desolvation in the carbon nanotube. The results show that, compared with carbon nanotubes, Li+ and [Li(AN)]+ require less energy to enter carbonylated carbon nanotubes and are more easily incorporated. Carbonyl groups also improve the adsorption capacity of carbon nanotubes for Li+ and enhance the ability of carbon nanotubes to store Li+.
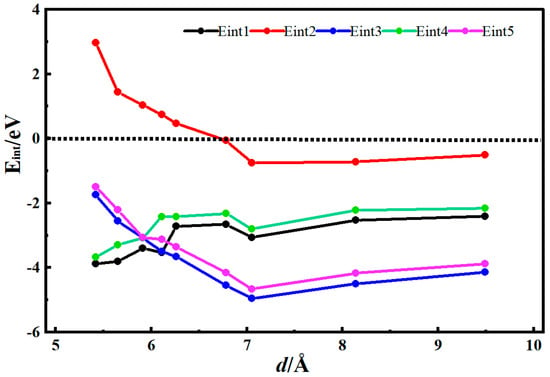
Figure 3.
Reaction energy curves of Li+, AN, [Li(AN)]+ in carbonylated functional carbon nanotubes and Li+ and [Li(AN)]+ in carbon nanotubes.
Figure 4 shows the desolvation reaction energy curves of [Li(AN)]+ with carboxylated single-walled carbon nanotubes and single-walled carbon nanotube models. To obtain the critical size of the desolvation pore diameter for [Li(AN)]+, the reaction energies of Li+, AN, [Li(AN)]+ in carboxylated single-walled carbon nanotubes, and Li+ and [Li(AN)]+ in single-walled carbon nanotubes were calculated, denoted as Eint1, Eint2, Eint3, Eint4, and Eint5, respectively. When the diameter of the carboxylated carbon nanotube d < 6.26 Å, Eint2 < 0, indicating that AN molecules can exist alone in the carboxylated single-walled carbon nanotube. When the diameter of the carboxylated carbon nanotube d = 6.11 Å, the energy value of Eint1 (−3.71 eV) is close to that of Eint3 (−3.75 eV), meaning 6.11 Å is the critical point for the desolvation of [Li(AN)]+ in the carboxylated carbon nanotube. When the diameter of the carboxylated carbon nanotube is less than 6.11 Å and Eint1 < Eint3 < 0, Li+ exists more stably in the pore than [Li(AN)]+. Therefore, when the pore diameter is less than 6.11 Å, [Li(AN)]+ can undergo complete desolvation in the carboxylated carbon nanotube. When the diameter of the hydroxylated carbon nanotube is greater than 6.11 Å, Eint3 drops rapidly to the minimum value, which indicates that no desolvation occurs in the system, and [Li(AN)]+ exists more stably in the pore. When the diameter of the carbon nanotube (d) is 5.91 Å, the energy value of Eint4 (−2.98 eV) is close to that of Eint5 (−3.06 eV). Therefore, 5.91 Å is the critical point for the desolvation of [Li(AN)]+ in the carbon nanotube. When the diameter of the carbon nanotube is less than 5.91 Å and Eint4 < Eint5 < 0, Li+ exists more stably in the pore than [Li(AN)]+. Thus, when the pore diameter is less than 5.91 Å, [Li(AN)]+ can undergo complete desolvation in the carbon nanotube. The results show that, compared with carbon nanotubes, Li+ and [Li(AN)]+ require less energy to enter carboxylated carbon nanotubes and are more easily incorporated. Like the previous two functional groups, carboxyl groups also improve the adsorption capacity of carbon nanotubes for Li+ and enhance the ability of carbon nanotubes to store Li+.
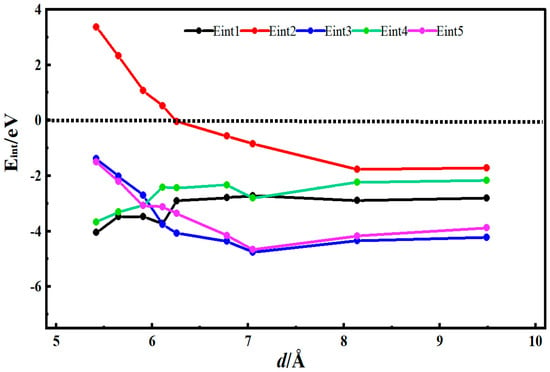
Figure 4.
Reaction energy curves of Li+, AN, [Li(AN)]+ in carboxylated functional carbon nanotubes and Li+ and [Li(AN)]+ in carbon nanotubes.
3.3. Analysis of the Effect of Carbon Nanotubes Functionalized with Different Functional Groups on the Desolvation Size of Li+
To clarify the critical desolvation size of [Li(AN)]+ complexes and the underlying atomic-scale mechanism, Figure 5 illustrates the relationship between the desolvation size of Li+ complexes and CNTs modified with -OH, -C=O, and -COOH groups (pristine CNTs as the control) in acetonitrile solvent. The simulation results show that the complete desolvation sizes of [Li(AN)]+ are 5.91 Å for pristine CNTs, 6.26 Å for hydroxylated CNTs (HCNT), and 6.11 Å for both carbonylated CNTs (CNCNT) and carboxylated CNTs (CXCNT).The differences in desolvation size arise from the distinct atomic-scale interactions between functional groups and [Li(AN)]+ complexes, which modulate the stability of the solvated complex and the energy barrier for desolvation:
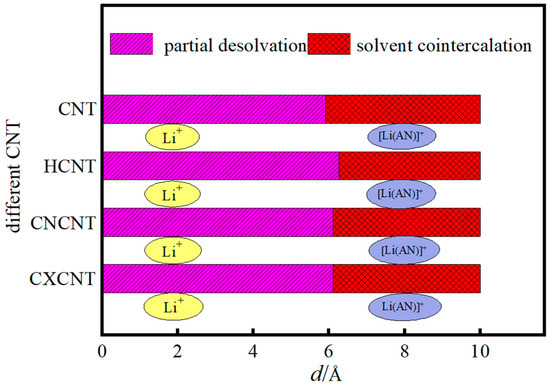
Figure 5.
Relationship between the desolvation size of Li+ complex and carbon nanotube with different functional groups.
Hydroxyl groups (-OH): The -OH group has a strong polar O-H bond and a small steric volume. At the atomic level, the electronegative O atom in -OH forms a stable double hydrogen bond with the H atoms in the AN molecule of [Li(AN)]+ and a coordination bond with Li+. This dual interaction strengthens the adsorption of [Li(AN)]+ on HCNT surfaces, reducing the energy required for AN molecules to detach from Li+. Consequently, HCNT can accommodate larger [Li(AN)]+ complexes before complete desolvation, resulting in the largest critical size (6.26 Å).
Carbonyl (-C=O) and carboxyl (-COOH) groups: The -C=O group only forms a single coordination bond with Li+ due to the lack of H atoms for hydrogen bonding, leading to weaker adsorption of [Li(AN)]+ compared to -OH. For -COOH, although it contains an O-H bond, the additional -C=O group in its structure increases steric hindrance. This steric effect weakens the hydrogen bond between -COOH and AN and offsets the coordination effect of the two O atoms, making the overall interaction strength of -COOH with [Li(AN)]+ comparable to that of -C=O. Thus, CNCNT and CXCNT exhibit the same intermediate desolvation size (6.11 Å).
Pristine CNTs: Without polar functional groups, pristine CNTs only interact with [Li(AN)]+ through weak van der Waals forces. This weak adsorption fails to stabilize [Li(AN)]+ effectively, requiring AN molecules to desorb at a smaller complex size to maintain system stability, which is why they have the smallest critical size (5.91 Å).
After functionalization, all three types of oxygen-containing groups enhance the Li+ storage capacity of CNT cylindrical pores by strengthening interactions with [Li(AN)]+, which directly contributes to the improved capacitance of supercapacitors [29].
3.4. Analysis of Relative Capacitance for Li+ Intercalation into Carbon Nanotubes with Different Functionalizations
It can be seen from Figure 6, Figure 7, Figure 8 and Figure 9 that when the pore diameters of the optimized carbon nanotubes and functionalized carbon nanotubes are 5.42 Å and 5.65 Å, Li+ is intercalated into the system at the center of the pores. With the increase in pore diameter, Li+ gradually moves closer to the pore walls, and the distance between Li+ and the pore walls shows a tendency to decrease.
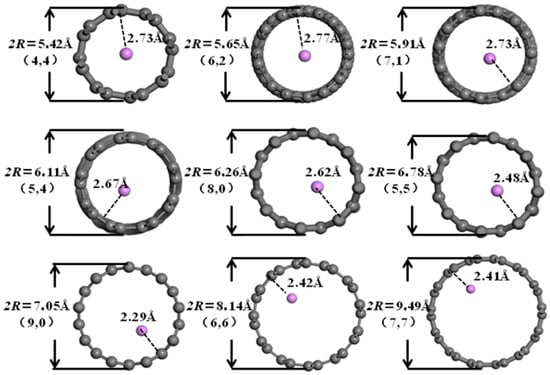
Figure 6.
The optimized structures of CNT-Li+.
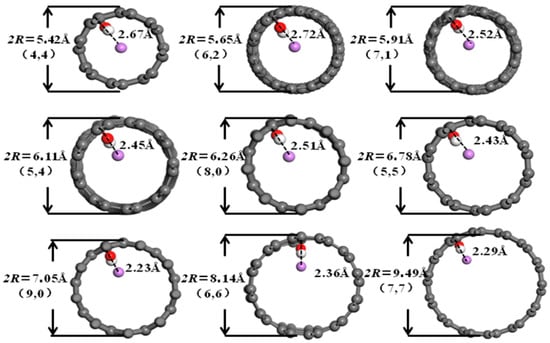
Figure 7.
The optimized structures of HCNT-Li+.
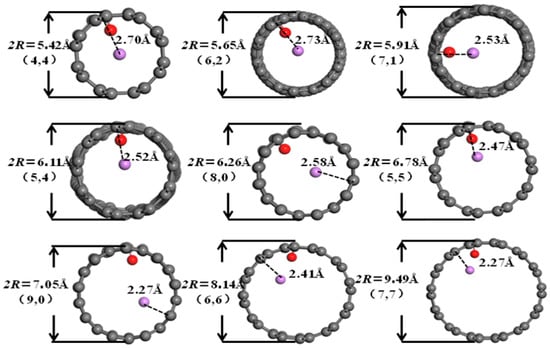
Figure 8.
The optimized structures of CNCNT-Li+.
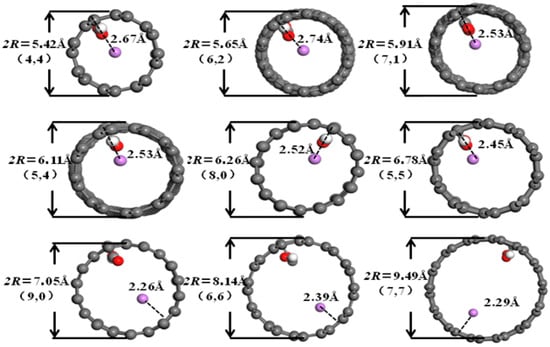
Figure 9.
The optimized structures of CXCNT-Li+.
The capacitance calculation formula [30,31] of electric double-layer capacitors (EDLCs) is
In the formula, CEDLC represents the capacitance of EDLCs, ε is the local dielectric constant of the electrolyte, A is the surface area, d1 is the radius of Li+, and R is the radius of carbon nanotubes and functionalized carbon nanotubes. In this paper, the capacitance generated by [Li(AN)]+ after removing solvent molecules is compared through the ratio of dLi+-C (i.e., relative capacitance) when Li+ is intercalated into the system of carbon nanotubes and functionalized carbon nanotubes. To study and compare the differences in dLi+-C among different structures, the relevant data are presented in Figure 10a. To ensure data consistency, this paper measures distances within the structure, using the shorter distance between Li+ and the walls of carbon nanotubes and functionalized carbon nanotubes. It can be seen from the data in Figure 10b that the value of relative capacitance is closely related to the Li+ concentration and the size of the pore diameter in which it is located.
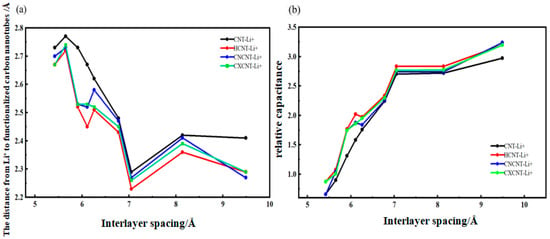
Figure 10.
(a) dLi+-C and (b) relative capacitance of Li+ embedded into CNT and functionalized CNT.
From the relative capacitance values in Figure 10b, it can be observed that under the same pore diameter, the relative capacitance generated when Li+ is intercalated into the three types of oxygen-containing functionalized carbon nanotubes is significantly higher than that of the intrinsic system. This is because the previously measured complete desolvation size of Li+ complexes is 5.91 Å for carbon nanotubes (CNT); 6.26 Å for hydroxylated carbon nanotubes (HCNT); and 6.11 Å for both carbonylated carbon nanotubes (CNCNT) and carboxylated carbon nanotubes (CXCNT). After desolvation occurs, the distance from Li+ to the three types of oxygen-containing functionalized carbon nanotubes is significantly smaller than that from Li+ to intrinsic carbon nanotubes, meaning that the relative capacitance of the three oxygen-containing functionalized carbon nanotube systems is significantly higher than that of intrinsic carbon nanotubes after desolvation. It can be seen from Table 2 that the relative capacitance generated when Li+ undergoes desolvation in hydroxylated carbon nanotubes is more than 1.2 times, with a maximum of 1.4 times; the relative capacitance generated when Li+ undergoes desolvation in carbonylated carbon nanotubes is more than 1.1 times, with a maximum of 1.3 times; and the relative capacitance generated when Li+ undergoes desolvation in carboxylated carbon nanotubes is more than 1.2 times, with a maximum of 1.3 times. It can be concluded that the larger the desolvation size of Li+ complexes, the greater the relative capacitance provided by the oxygen-containing functionalized carbon nanotube system, and the more significant the improvement in relative capacitance after desolvation of Li+ complexes. As reported by Chen et al. (2023) [32], the specific capacitance of the carboxylated graphene (GO-COOH)-based electrode increased by 32.7% (approximately 1.33 times). This value is close to the theoretical increase of 1.3 times for CXCNT in this study, which further confirms the universality of modifying carbon materials with oxygen-containing functional groups to enhance their capacitive performance [32].
Table 2.
The relative capacitance parameters of optimized functionalization CNT-Li+.
3.5. Density of States Analysis of Li+ After Desolvation in Functionalized Carbon Nanotubes
To reveal the regulatory mechanism of different oxygen-containing functional groups on the electronic structure of carbon nanotubes, this study calculated the Density of States (DOSs) for the hydroxylated (HCNT), carbonylated (CNCNT), and carboxylated (CXCNT) carbon nanotube-Li+ systems under the critical desolvation size of Li+. Furthermore, by combining the electron transfer rules and the changes in the Fermi level, the structure–activity relationship between the type of functional groups and the electronic properties of the systems was analyzed in depth. The results are shown in Figure 11, Figure 12 and Figure 13.
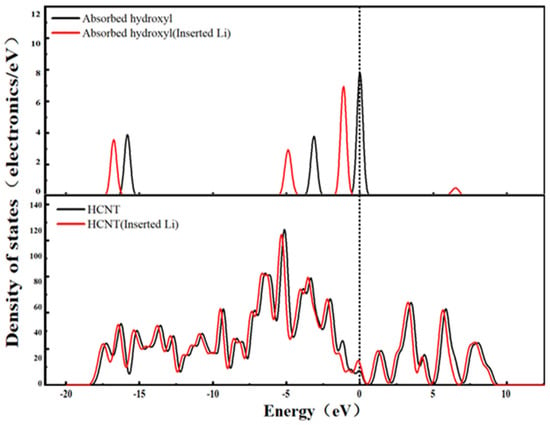
Figure 11.
State density of the HCNT system.
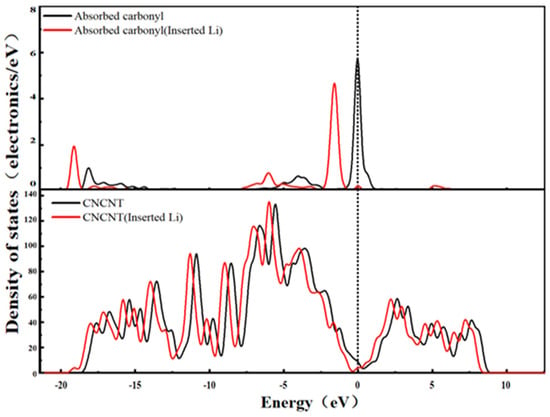
Figure 12.
State density of the CNCNT system.
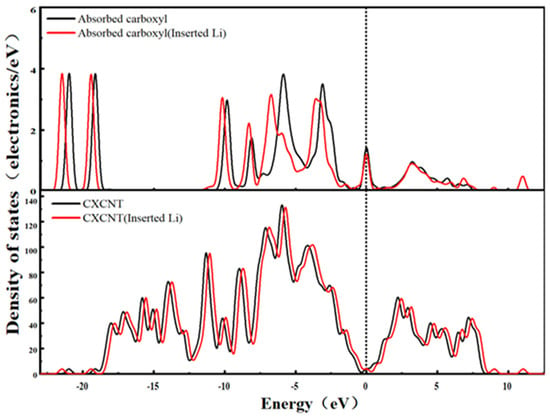
Figure 13.
State density of the CXCNT system.
3.5.1. DOS Characteristics of the Hydroxylated Carbon Nanotube (HCNT)-Li+ System
Figure 11 shows the DOS spectrum of the HCNT-Li+ system. It can be seen that after Li+ is embedded into the pores of hydroxylated carbon nanotubes, it preferentially forms a coordination interaction with the oxygen atom in the hydroxyl group (-OH): the 2p orbital electron cloud of the hydroxyl oxygen atom partially overlaps with the empty orbital of Li+, prompting the hydroxyl group to transfer a small amount of electrons to Li+ (manifested as a slight overall shift in the hydroxyl characteristic peak toward the lower binding energy direction in the DOS spectrum). This electron transfer does not cause damage to the conjugated structure of the carbon nanotube main body; on the contrary, due to the charge compensation effect of Li+, the electron delocalization of the carbon nanotube π orbital is enhanced, which is specifically reflected in the significant increase in the DOS value at the Fermi level and a slight shift in the Fermi level toward the higher-binding-energy direction. From the perspective of the electron transport mechanism, the enhanced electron delocalization of the π orbital reduces the transport resistance of electrons within the carbon nanotube framework. Meanwhile, the weak coordination interaction between the hydroxyl group and Li+ does not form an electron transport barrier. Therefore, both the metallicity (reflected by the DOS value at the Fermi level) and conductivity (reflected by electron transport resistance) of the HCNT-Li+ system show an increasing trend. This characteristic is closely related to the strong polarity of the hydroxyl group (enhancing Li+ adsorption capacity) and weak electron trapping capacity (avoiding electron localization) [33].
3.5.2. DOS Characteristics of the Carbonylated Carbon Nanotube (CNCNT)-Li+ System
Figure 12 shows the DOS spectrum of the CNCNT-Li+ system. Unlike hydroxyl groups, the oxygen atom in the carbonyl group (-C=O) has higher electronegativity. After Li+ intercalation, more electrons are transferred from the carbonyl oxygen atom to Li+—this is manifested as a significant shift in the characteristic peak of the π antibonding orbital of the C=O bond in the carbonyl group (in the high-binding-energy region) toward the low-binding-energy direction in the DOS spectrum, with a notable increase in peak intensity. This indicates that a large number of electrons are trapped in the localized orbitals of the carbonyl group. This strong electron-trapping effect has two aspects of influence. On the one hand, after Li+ gains electrons, its positive charge density decreases, and the electrostatic repulsion between Li+ and the carbon nanotube framework is weakened. This promotes the expansion of the delocalization range of electrons in the π orbitals of carbon nanotubes, which is reflected by an increase in the DOS value at the Fermi level (enhanced metallicity). On the other hand, the electrons trapped by the carbonyl group form a localized electron cloud, which hinders the long-range transport of electrons within the conjugated carbon nanotube framework. Meanwhile, the Fermi level shifts toward the low-binding-energy direction, further increasing the difficulty for electrons to cross the energy barrier and ultimately leading to a decrease in the system’s electrical conductivity.
This unique phenomenon of “enhanced metallicity yet reduced electrical conductivity” originates from the dual-action mechanism of the carbonyl group, i.e., “strong electron trapping and weak destruction of framework conjugation.” It is directly related to the double-bond structure of the carbonyl group (which enhances electron-trapping ability) and its moderate polarity (which balances Li+ adsorption and interaction with the framework).
3.5.3. DOS Characteristics of the Carboxylated Carbon Nanotube (CXCNT)-Li+ System
Figure 13 shows the DOS spectrum of the CXCNT-Li+ system. The carboxyl group (-COOH) consists of a hydroxyl group (-OH) and a carbonyl group (-C=O), but its electronic effect is not a simple superposition of the two. After Li+ intercalation, the oxygen atoms in the carboxyl group (derived from both -OH and -C=O) transfer electrons to Li+; however, the overall electron delocalization of the carboxyl group (resulting from O-C-O conjugation within -COOH) buffers this electron transfer. This is manifested in the DOS spectrum as a shift in the carboxyl characteristic peak toward the low-binding-energy direction, with the shift extent falling between that of the hydroxyl group and the carbonyl group, and no obvious localized peak is observed.
Specifically, the -O-C-O conjugated structure of the carboxyl group can disperse the locally transferred electrons to the entire carboxyl group, avoiding the hindrance to conductivity caused by electron localization. At the same time, the high polarity of the carboxyl group (higher than that of the hydroxyl group and carbonyl group) enhances the adsorption interaction between Li+ and carbon nanotubes, leading to a slight reduction in the delocalization range of electrons in the π orbitals of carbon nanotubes. This is reflected by a slight decrease in the DOS value at the Fermi level (weakened metallicity).
In addition, the electron dispersion effect of the carboxyl group maintains the integrity of the electron transport path in the system, and the Fermi level position remains largely unchanged; thus, the conductivity changes minimally. This characteristic of “weakened metallicity yet stable conductivity” is the result of the synergistic effect of the carboxyl group’s “moderate electron-trapping ability and strong electron-dispersing ability,” which demonstrates the unique advantage of the carboxyl group in balancing Li+ adsorption and electron transport.
4. Conclusions
This study employed first-principles calculations to systematically investigate the desolvation behavior of lithium-ion (Li+) complexes, relative capacitance characteristics, and post-desolvation density of states (DOSs) of pristine carbon nanotubes (CNTs) and CNTs functionalized with hydroxyl (-OH), carbonyl (-C=O), and carboxyl (-COOH) groups, using acetonitrile as the solvent. Key research findings are summarized as follows:
- •
- Functionalization of CNTs exerts a significant regulatory effect on the complete desolvation size of [Li(AN)]+ complexes. Among the systems studied, hydroxyl-modified CNTs (HCNT) exhibit the largest desolvation size, followed by carbonyl-modified CNTs (CNCNT) and carboxyl-modified CNTs (CXCNT) (which show identical desolvation sizes), while pristine CNTs present the smallest size. This result confirms that the introduction of oxygen-containing functional groups modifies the pore microenvironment of CNTs, thereby directly regulating the desolvation process of Li+ complexes.
- •
- All three types of oxygen-containing functional groups contribute to the enhancement of CNTs’ relative capacitance, with hydroxyl modification yielding the most notable improvement (a maximum enhancement of ~1.4-fold compared to pristine CNTs), and carbonyl/carboxyl modifications achieving a maximum enhancement of ~1.3-fold. The underlying mechanism for this capacitance improvement lies in the fact that functionalization strengthens the Li+ storage capacity of the cylindrical pores in CNTs, which in turn enhances the charge storage capability of the electrode material.
- •
- Post-desolvation DOS analysis reveals distinct variations in the electronic properties of CNT-Li+ systems depending on the type of functional group: the HCNT-Li+ system exhibits simultaneous enhancement in metallicity and conductivity; the CNCNT-Li+ system shows increased metallicity but a reduction in conductivity; and the CXCNT-Li+ system displays decreased metallicity while maintaining nearly unchanged conductivity. These functional group-dependent electronic property differences provide targeted theoretical guidance for selecting functionalized CNTs tailored to the specific electrical performance requirements of supercapacitors.
Overall, this study clarifies the regulatory mechanism of oxygen-containing functional groups on the desolvation behavior and electrochemical performance of CNTs. It provides a theoretical basis for optimizing the design of high-performance CNT-based supercapacitor electrodes, offering technical support for enhancing the energy density and cycle stability of supercapacitor devices.
Author Contributions
Conceptualization, F.L. and S.L.; Methodology, F.L.; Software, F.L. and W.Z.; Validation, M.Z.; Formal analysis, M.Z. and B.L.; Data curation, B.L.; Funding acquisition, F.L. and S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Fundamental Scientific Research Program of the Department of Education of Liaoning Province (Project Nos.: 2024JYTQN-15, LJ212511430005) and the Doctoral Startup Fund of Liaoning Institute of Science and Technology, China (Project No.: 2505B06).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lawal, T.A. Recent application of carbon nanotubes in energy storage and conversion devices. Carbon Trends 2025, 19, 100470. [Google Scholar] [CrossRef]
- Scarpa, D.; Cirillo, C.; Ponticorvo, E.; Cirillo, C.; Attanasio, C.; Iuliano, M.; Sarno, M. Iron Selenide Particles for High-Performance Supercapacitors. Materials 2023, 16, 5309. [Google Scholar] [CrossRef]
- Liu, F.; Yang, S.; Zhang, X.; Tang, S.; Xia, Y. Insight into the Desolvation of Quaternary Ammonium Cation with Acetonitrile as a Solvent in Hydroxyl-Flat Pores: A First-Principles Calculation. Materials 2023, 16, 3858. [Google Scholar] [CrossRef] [PubMed]
- Simoes, G.R.D.; Pequeno, H.O.D.; Montes, C.I.C. Supercapacitors and triboelectric nanogenerators based on electrodes of greener iron nanoparticles/carbon nanotubes composites. Sci. Rep. 2024, 14, 11555. [Google Scholar] [CrossRef]
- Keller, V.N.; Nikolkin, N.V.; Butakov, S.D. Optimization of the Technology for Manufacturing the Electrodes for Self-Charging Supercapacitors from Carbon Nanotubes. Russ. J. Electrochem. 2024, 60, 526–531. [Google Scholar] [CrossRef]
- An, K.H.; Kim, W.S.; Park, Y.S. Supercapacitor using single-walled carbon nanotubes electrodes. Adv. Mater. 2001, 13, 497–500. [Google Scholar] [CrossRef]
- Chen, Q.L.; Xue, K.H.; Shen, W. Fabrication and electrochemical properties of carbon nanotubes array electrode for supercapacitor. Electrochim. Acta 2004, 49, 4157–4161. [Google Scholar] [CrossRef]
- Chen, J.H.; Li, W.Z.; Wang, D.Z.; Yang, S.X.; Wen, J.G.; Ren, Z.F. Electrochemical characterization of carbon nanotubes as electrode in electrochemical double-layer capacitor. Carbon 2002, 40, 1193–1197. [Google Scholar] [CrossRef]
- Yoon, B.J.; Jeong, S.H.; Lee, K.H.; Kim, H.S.; Park, C.G.; Han, J.H. Electrical properties of electrical doublelayer capacitors with integrated carbon nanotubes electrodes. Chem. Phys. Lett. 2004, 388, 170–174. [Google Scholar] [CrossRef]
- Liu, D.; Shi, L.; Dai, Q.; Lin, X.; Mehmood, R.; Gu, Z.; Dai, L. Functionalization of carbon nanotubes for multifunctional applications. Trends Chem. 2024, 6, 186–210. [Google Scholar] [CrossRef]
- Kim, M.G.; Lee, B.; Li, M.; Noda, S.; Kim, C.; Kim, J.; Song, W.J.; Lee, S.W.; Brand, O. All-Soft Supercapacitors Based on Liquid Metal Electrodes with Integrated Functionalized Carbon Nanotubes. ACS Nano 2020, 14, 5659–5667. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, S.B.; Shan, X.Y. Insights into the effect of the interlayer spacings of bilayer graphene on the desolvation of H+, Li+, Na+, and K+ ions with water as a solvent: A first-principles study. Phys. Chem. 2019, 21, 23697–23704. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Schultz, P.A.; Desjarlais, M.P.; Mattsson, T.R.; Leung, K. Designing meaningful density functional theory calculations in materials science-a primer. Model. Simul. Mater. Sci. Eng. 2004, 13, R1. [Google Scholar] [CrossRef]
- Seifert, G.; Porezag, D.; Frauenheim, T. Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA Scheme. Int. J. Quantum Chem. 1996, 58, 185–192. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.T.; Frauenheim, S.; Su, H.; Seifert, G. Self-consistent charge density functional tight-binding method for simulation of complex material properties. Phys. Rev. B 1998, 58, 7260. [Google Scholar] [CrossRef]
- Seifert, G. Tight-binding density functional theory: An approximate Kohn-Sham DFT scheme. J. Phys. Chem. A 2007, 111, 5609–5613. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Perdew, J.P. Rehabilitation of the Perdew-Burke-Ernzerhof generalized gradient approximation for layered materials. Phys. Rev. B 2017, 95, 081105. [Google Scholar] [CrossRef]
- Irelan, R.M.; Henderson, T.M.; Scuseria, G.E. Long-range-corrected hybrids using a range-separated Perdew-Burke-Ernzerhof functional and random phase approximation correlation. J. Chem. Phys. 2011, 135, 94105–94116. [Google Scholar] [CrossRef]
- Segall, D.M.; Lindan, D.J.P.; Probert, J.M.; Pickard, J.C.; Hasnip, J.P.; Clark, J.S.; Payne, C.M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Yang, S.B.; Liu, X.L.; Zhang, X.; Tang, S.W. Insights into the effect of hydroxyl-, epoxy-, and carboxyl-pores on the desolvation of K+ with water as a solvent: A first-principles study. J. Phys. Condens. Matter 2021, 33, 445201. [Google Scholar] [CrossRef]
- Wu, S.; Yan, G.; Cheng, X. First-principles investigation of the superconducting properties of thallium sulfide. Phys. C Supercond. Its Appl. 2019, 562, 1–6. [Google Scholar] [CrossRef]
- Schira, R.; Latouche, C. DFT and hybrid-DFT calculations on the electronic Properties of vanadate materials: Theory meets experiments. New J. Chem. 2020, 44, 11602–11607. [Google Scholar] [CrossRef]
- Zhao, P.; Su, Y.; Zhang, Y.; Li, S.J.; Chen, G. CO catalytic oxidation on iron-embedded hexagonal boron nitride sheets. Chem. Phys. Lett. 2011, 515, 159–162. [Google Scholar] [CrossRef]
- Floris, A.; Timrov, I.; Himmetoglu, B.; Marzari, N.; de Gironcoli, S.; Cococcioni, M. Hubbard-corrected density functional perturbation theory with ultrasoft pseudopotentials. Phys. Rev. B 2020, 101, 064305. [Google Scholar] [CrossRef]
- Monkhorst, J.H.; Pack, D.J. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Tonel, M.Z.; Zanella, I.; Fagan, S.B. Theoretical study of small aromatic molecules adsorbed in pristineand functionalized graphene. J. Mol. Model. 2021, 27, 193. [Google Scholar] [CrossRef]
- Calborean, A.; Buimaga-Iarinca, L.; Graur, F. DFT charge transfer of hybrid molecular ferrocene/Si structures. Phys. Scr. 2015, 90, 055803. [Google Scholar] [CrossRef]
- Rabone, J.; Uffelen, P.V. DFT-based Metadynamics simulation of proton diffusion in tetragonal zirconia at 1500 K. J. Nucl. Mater. 2015, 459, 30–36. [Google Scholar] [CrossRef]
- Janes, A.; Lust, E. Use of organic esters as co-solvents for electrical double layer capacitors with low temperature performance. J. Electroanal. Chem. 2006, 588, 285–295. [Google Scholar] [CrossRef]
- Huang, J.; Sumpter, B.G.; Meunier, V. A Universal Model for Nanoporous Carbon Supercapacitors Applicable to Diverse Pore Regimes, Carbon Materials, and Electrolytes. Chem.-Eur. J. 2008, 14, 6614–6626. [Google Scholar] [CrossRef]
- Huang, J.; Sumpter, B.G.; Meunier, V. Theoretical Model for Nanoporous Carbon Supercapacitors. Angew. Chem. Int. Ed. 2008, 120, 530–534. [Google Scholar] [CrossRef]
- Chen, Z.Z.; Pei, X.X.; Song, J.L.; Song, W.Y.; Shi, Z.F.; Onditi, K.O.; Li, Q.; Jiang, X.L. Systematics and evolutionary history of the genus Micromys (Mammalia: Rodentia: Muridae). Mamm. Biol. 2023, 103, 389–403. [Google Scholar] [CrossRef]
- Yang, S.B.; Shan, X.Y.; Li, S.N. Insight into the Adsorption and Diffusion Behaviors of Na on XC3 (X = B, N and P) Doping Graphene Surfaces: A First Principle Study. Mater. Rep. B Res. Pap. 2019, 33, 1640–1645. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).