Abstract
Background/Objectives: Multidrug resistant (MDR) Salmonella represents a major global public health challenge within the One Health interface. This study aimed to characterize the genomic epidemiology of Salmonella isolates from Colombia and resolve the genetic architecture of novel MDR plasmids identified in foodborne strains. Methods: A total of 90 Salmonella isolates collected between 2002 and 2009 from various food sources and food-producing animals in Colombia were analyzed using whole-genome sequencing (WGS). Bioinformatics tools were employed for serotype prediction, multi-locus sequence typing (MLST), and resistome/virulome profiling. Long-read sequencing was utilized to close the complete sequences of representative MDR plasmids. Results: 45.6% of isolates exhibited antimicrobial resistance, with seven being classified as MDR. The major serotypes identified were Uganda (n = 20), Newport (n = 10), and Braenderup (n = 10). We characterized a novel 229,037 bp IncHI1 plasmid (pCFS0255-1) harboring a copper homeostasis and silver resistance island (CHASRI) integrated with tetracycline and macrolide resistance clusters. Additionally, a 99,288 bp IncI1 plasmid (pCFS0255-2) carrying a unique aminoglycoside resistance module was resolved. Conclusions: Our findings highlight the persistence of specific Salmonella lineages in the Colombian food chain and the role of hybrid plasmids in the co-selection of metal and antibiotic resistance. The study underscores the necessity of implementing WGS-based surveillance to track emerging MDR threats.
1. Introduction
Salmonella, a Gram-negative bacterium, is one of the most widely known foodborne pathogens, often involved in food safety incidents worldwide and which poses a challenge to public health, especially in developing countries [1].
Antimicrobial resistance (AMR) is a major global societal threat of growing concern to One Health. It also has an impact on food security as well as the economic well-being of millions of people. Among other zoonotic infectious diseases, antibiotic-resistant Salmonella in food chains associated with livestock are of concern [2]. Mobile genetic elements (MGE) such as plasmids, transposons, and insertion sequence elements can mediate the spread of not only AMR genes but also heavy metal- and biocidal resistance-encoding genes, thus limiting the treatment and control of Salmonella [3].
Whole-genome sequencing is more widely used in foodborne pathogen risk assessment [4]. It enables a variety of diagnostic applications using a streamlined pipeline, including pathogen subtyping, source tracking, and virulence gene identification, along with prediction of AMR genotypes [5].
Due to the existence of transnational food trade, the evolution of foodborne disease threats requires cross-border coordination to ensure that local food safety risks are controlled before they become international issues [6]. Within the One Health framework, some countries and regions have attempted to establish molecular tracing networks for foodborne pathogens. For example, the GenomeTrakr tracing network, led by the U.S. FDA (Food and Drug Administration), was initiated in 2012 and has since developed into a WGS (whole-genome sequencing)-based regulatory network that includes cooperation from 40 U.S. states and 30 international laboratories [7]. In China, the National Foodborne Disease Surveillance Molecular Tracing Network (TraNet) was initiated in 2013 to conduct precise tracing of various foodborne pathogens, including Salmonella, using both PFGE (Pulsed-Field Gel Electrophoresis) technology and cgMLST (core-genome multi-locus sequence typing)-based WGS classification [8,9]. In 2021, the ECDC (European Centre for Disease Prevention and Control) introduced the EpiPulse online collaboration framework, aiming to integrate the foodborne disease surveillance networks of European countries to strengthen cross-border tracing cooperation [10]. These molecular tracing networks have effectively established a nationwide and cross-border surveillance system across the collaboration public health laboratories. However, the threat of foodborne diseases may originate from regions and pathogens that are not yet included in surveillance networks. The cgMLST tracing method provides stable allele-based typing, but interpretation depends on the availability of contextual genomic datasets, presenting certain limitations when identifying strains that belong to newly emerging pathogenic species or those that are distantly related to known types. For countries or regions not included in the regulatory network, the inability to submit WGS data for centralized monitoring and analysis further limits the effectiveness of cgMLST-based tracing methods. Without access to centralized surveillance networks, these areas face challenges in identifying and classifying strains accurately through cgMLST.
Therefore, it is essential to develop a standardized WGS tracing and analysis workflow that does not rely on existing databases to address emerging and unknown food safety risks, particularly those originating from regions not connected to regulatory networks or involving pathogens that are not included in current databases.
In this study, we developed a strain tracing workflow using core-genome SNPs for phylogenetic analysis, which includes the identification of antimicrobial resistance (AMR) genes and Salmonella pathogenicity islands (SPIs). Based on this workflow, WGS was applied to 90 Salmonella species cultured from sources representing the One Health continuum in Colombia. Genetic relationships among these bacterial isolates were investigated, as well as their AMR genotypes. Specifically, for a tetracycline–aminoglycoside–gentamicin multidrug resistance (MDR) module identified, long-read sequencing was performed to close the genome and analyze the nature of these MDR plasmids.
2. Results
2.1. Geno- and Phenotypic Characterization of the 90 Salmonella Collection
2.1.1. Metadata, AST Phenotype, Serotyping, and Phylogenetic Tree
A total of 90 Salmonella isolates were cultured from food and food-producing and other animals in Colombia between 2002 and 2009. Majority of these sources included meat such as pork, chicken, beef, and their products. Non-meat products such as flour mandioka powder and a mixture of corn and egg were also included. Exotic animal sources included Iguana iguana, Hydrochoerus hydrochaeris, and Trachemys scripta callirostris.
AST results for a panel of 12 antibiotics that were tested demonstrated that there are 41 AMR-expressing isolates, including seven classified as multidrug resistant (MDR), among the collection. In addition, except for nine completely susceptible isolates, all other isolates expressed an intermediate resistance phenotype, as determined by the disk diffusion method used.
The serotype prediction results based on the whole-genome sequence indicated that there were 21 serotypes identified, of which Uganda (n = 20), Newport (n = 10), Braenderup (n = 10) and Anatum (n = 9) were in the majorities. The predicted results of the in silico Multi-locus sequence typing (MLST) corresponded with the presentation of serotypes, as shown in Figure 1. We observed discrepancies in the predicted serotypes from WGS and the laboratory-determined serotypes for 20 isolates. The detailed comparison is provided in the metadata table (Appendix A Table A1).
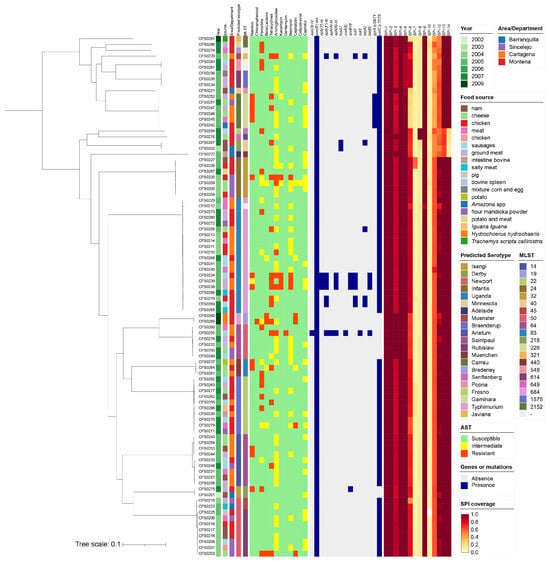
Figure 1.
Source information, phenotype, and genotype of 90 Salmonella isolates from Colombia. Information from the metadata included year of isolation, food source, geographical region/department, AST result, and presence/absence of resistance genes or site mutations. Gradient colors are used to represent SPI coverage.
Comparative genomic analysis of the 90 Salmonella isolates identified a total of 3274 core genes. The recombination-filtered core genome alignment spanned 3,126,423 bp. From this alignment, a total of 110,546 informative SNPs were extracted to construct a high-resolution maximum-likelihood phylogenetic tree.
2.1.2. AMR Genotypes in Isolates of the Collection
In silico identification of AMR genes and point mutations indicated that some individual isolates had several different antibiotic resistance-encoding genes representing different drug classes, while aac(6′)Iaa was detected in all 90 isolates. CFS0228 had a relatively unique aph(3″)Ib-sul2-tet(A)-aph(6)Id AMR genotype module, and this corresponded to the phenotype aminoglycoside, sulfonamide, and tetracycline resistance, with intermediate resistance to ceftiofur and neomycin.
CFS0129 and CFS0269 were determined to have similar AMR genotypes to CFS0228, except for aph(6)Id. However, their resistance phenotype was relatively simple, expressing resistance to tetracycline only. The lack of sulfonamide and aminoglycoside resistance, despite the presence of sul2 and aph(3″)Ib, suggests that these genes might be non-functional. The years in which these two were recovered were quite different. CFS0219 was isolated in 2004 from chicken and CFS0269 in 2007 from sausage.
CFS0224, CFS0238, and CFS0239 had the same AMR genotype: aph(3″)Ib-sul2-aph(6)Id-aph(3′)Ia-qnrB19-tet(B). At the same time, they expressed a similar resistance phenotype to kanamycin, neomycin, and tetracycline. CFS0224 and CFS0238 also expressed resistance to aminoglycosides and nalidixic acid, while CFS0238 and CFS0239 was intermediately resistant to ceftiofur. These three isolates were all recovered in 2005. CFS0238 and CFS0239 were cultured from ground meat samples, and CFS0224 was recovered from sausage. Further, the three isolates were same serotype and MLST profile, and were very close in evolutionary distance, although CFS0224 was derived from Monteria rather than Cartagena, the location for the other two isolates.
CFS0255 expressed a novel resistance genotype: tet(B)-aph(3″)Ib-aph(6)Id-aph(4)Ia-aac(3)IV-mef(B)-sul3, corresponding with its MDR phenotype aminoglycoside, gentamicin, and tetracycline.
2.1.3. SPI Genotypes Identified in the Isolate Collection
The study collection was also assessed for the presence of Salmonella Genomic Islands (SGI) by in silico analysis. Among all the 90 isolates, SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, and SPI-9 were determined to have a high coverage rate (>80%). SPI-6 has partial coverage in all isolates except Javiana, Minnesota, and Carrau serotypes. Similarly, SPI-7 was detected in CFS0226 and CFS0227 only, and were identified as serovar Infantis. SPI-8 was identified in CFS0276 and CFS0284, which were identified as the Fresno and Senftenberg serotypes, respectively. SPI-11 to SPI-14 were partially found in most isolates, while isolates CFS0202, CFS0222, CFS0267, CFS0276, and CFS0284 lacked SPI-14. SPI-10 was absent in all isolates, as shown in Figure 1.
2.2. Comparative Genomic Analysis of AMR Gene-Encoding Regions in WGS Assemblies
To better understand the distribution of AMR genes together with the upstream and downstream sequences, each of the AMR gene sequences in contigs mentioned above were studied to describe the nature of their AMR-encoding loci in addition to their upstream and downstream loci (to 5 kbp respectively).
Reference sequences from the NCBI NR/NT database of AMR regions with high identity and coverage, obtained via online BLASTN searches, are listed in Table 1.

Table 1.
Reference sequences for AMR gene regions in WGS assemblies.
The pattern of distribution of antibiotic resistance genes was found to be highly consistent among isolates CFS0224, CFS0238, and CFS0239 (Figure 2A). The sul2 resistance gene, the qnrB19 resistance gene, the tetR-tetB-tetC cluster, and the aph(3″)Ib-aph(6)Id-aph(3)Ia resistance gene modules were located on four different contigs respectively. These contigs also shared high similarities among the three isolates. Sequence comparison showed that the contigs containing sul2 were highly homologous to the reference sequences from the chromosomes of Escherichia coli FDAARGOS_1285 and Salmonella enterica subsp. enterica serovar Agona CVM N18S0017. Gene annotation indicated that there were multiple mobile genetic elements. ISSod4, ISEc25, IS629, and ISVsa3 insertion sequences were found upstream and downstream of sul2.
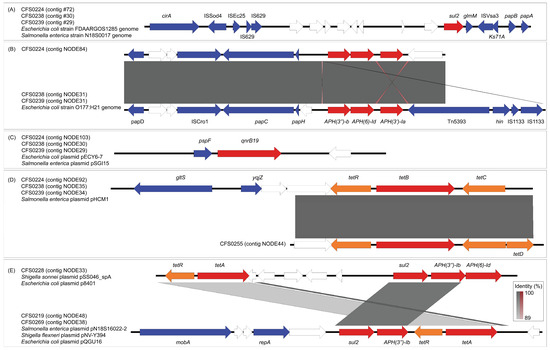
Figure 2.
Genetic structures of the antibiotic resistance genes harboring contigs and comparisons against reference sequences. CDSs are shown as arrows with different colors, wherein AMR genes were colored red, AMR-associated genes were colored orange, and other known genes in blue. Blank arrows represent hypothetical genes. Shaded areas between each of two sequences are represented by direct (gray) or inverted (red) nucleotide identity regions between loci. Isolates and contig number are listed with related reference sequences. (A) Genetic structure of sul2 flanking regions. (B) Genetic structure of aph(3″)Ib-aph(6)Id-aph(3)Ia flanking regions. (C) Genetic structure of qnrB19 flanking regions. (D) Genetic structure of tetB flanking regions. (E) Genetic structure of sul2-aph(3″)Ib-tetR-tetA flanking region in CFS0219 and CFS0269, compared with tetR-tetA-sul2-aph(3″)Ib-aph(6)Id flanking region in CFS0228.
Meanwhile, the contigs containing the aph(3″)Ib-aph(6)Id-aph(3)Ia gene cluster were highly identical to part of the chromosome of Escherichia coli O177:H21 (Figure 2B). In the upstream and downstream sequences of the aph(3″)Ib-aph(6)Id-aph(3)Ia module, a transposon Tn5393 and two copies of its related insertion sequence IS1333 were identified.
The contigs harboring qnrB19 (Figure 2C) were found to be identical with the Escherichia coli plasmid pECY6-7, which is a 2.7-kbp ColE-like small plasmid with only three CDSs. The plasmid was also found in Salmonella, and designated plasmid pSGI15.
In isolates CFS0224, CFS0238, and CFS0239, we observed a tetracycline resistance gene cluster, tetRBC, which is highly homologous to the Salmonella plasmid pHCM1 (Figure 2D). In the contigs from the second-generation sequencing of CFS0255, we identified another part of this gene cluster. Although it does not match the upstream region of the initial cluster, it contains a more complete tetracycline resistance gene cluster, tetRBCD. This contig in CFS0255 was later confirmed to be a partial plasmid pCFS0255-1 through long-read sequencing analysis.
Isolate CFS0219 and CFS0269 each had a contig with the sul2-aph(3″)Ib-tetR-tetA module (Figure 2E). This region was identical to one located on three plasmids from different bacteria, including Salmonella plasmid pN18S1602-2, Shigella flexneri plasmid pNV-Y394, and Escherichia coli plasmid pQGU16. The flanking regions in this case are different when compared with one identified in the CFS0228 tetR-tetA-sul2-aph(3″)Ib-aph(6)Id gene cluster, although they share the same four resistance genes, while the latter mentioned is identical with referenced Shigella sonnei plasmid pSS046_spA and Escherichia coli strain plasmid p8401.
2.3. Genetic Synteny and Sequence Comparison of a Novel Multidrug Resistance IncHI1 Plasmid pCFS0255-1
Two plasmids were identified in the MDR isolate CFS0255 complete genome sequence. Results of the long- and short-read sequencing and hybrid assembly confirmed these as closed circular DNA sequences. Plasmid pCFS0255-1 is 229,037 bp in length, with 46.53% in GC content, while plasmid pCFS0255-2 is 99,288 bp in length, with 50.38% in GC content.
Results obtained from annotation of these episomes using PlasmidFinder indicated that plasmid pCFS0255-1 was a IncHI1-like plasmid (Table 2). The genetic backbone structure of plasmid pCFS0255-1 was found to be highly identical to several reference plasmid complete genome sequences in NCBI GenBank database, including Escherichia coli 4M9F plasmid p4M9F (accession number MN256759.1) [11], Klebsiella pneumoniae GD21SC417 plasmid pHNGS471-1 (accession number: CP089510), Escherichia coli SD134209 plasmid pSD134209-1 (accession number: CP029690), Salmonella enterica strain CVM N18S0993 plasmid pN18S0993-1 (accession number: CP082572), Salmonella Typhi plasmid R27 (accession number: NC_002305), and Klebsiella pneumoniae strain CRE301 plasmid pKP301-1 (accession number: CP166314) [12]. Three resistance gene clusters were noted on plasmid pCFS0255-1, including the tetracycline resistance gene cluster (TET cluster), the copper homeostasis and silver resistance island (CHASRI), and the sulfonamide–macrolide–mercury resistance gene cluster (SUL-MEF-MER cluster) [13]. No other plasmids in the reference database were found containing all of these three gene islands mapped to one complete episome. Meanwhile, in the flanking region of each of the resistance gene islands, multiple copies of insertion sequences were found (Figure 3).
Table 2.
PlasmidFinder results for CFS0255 complete genome.
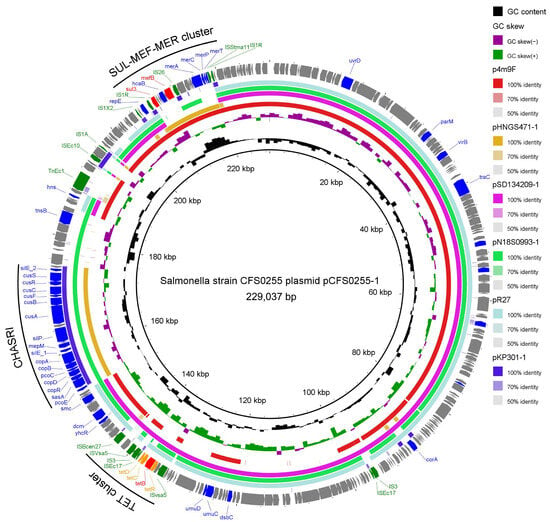
Figure 3.
Circular schematic map of the novel multidrug resistance IncH1-like plasmid pCFS0255-1, showing the genetic structure of the plasmid. CDSs were shown as arrows. ARG, regulation genes of ARG genes, and insertion sequences were highlighted in green. Other annotated genes were denoted by blue colors. Colored arcs are used to show regions of homology between the plasmid sequence and its four reference sequences, including plasmid pHNGS471-1, plasmid pSD134209-1, plasmid pN18S0993-1, and plasmid R27. The inner two rings represented GC Skew [(G − C)/(G + C), G and C represents guanine content cytosine content] and GC content. Regions of the three antibiotic resistance and metal tolerance gene clusters were identified, including the TET cluster, the CHASRI, and the SUL-MEF-MER cluster.
2.4. Genetic Synteny and Sequence Comparison of a Novel Aminoglycoside Resistance IncI1 Family Plasmid pCFS0255-2
Plasmid annotation of plasmid pCFS0255-2 suggested that it is a IncI-type plasmid. The basic backbone is highly identical to Salmonella Typhimurium 9134 plasmid p9134 (accession Number: KF705205.1) [14], Salmonella Typhimurium plasmid R64 (accession number: AP005147.1) [15], Escherichia coli 94EC plasmid p94EC-2 (accession number: CP047578.1) [16], and Salmonella Typhimurium plasmid pST1030-1C (accession number: MT507879.1) [17]. Plasmid pCFS0255-2 harbors an aph(6)Id-aph(3″)Ib-aac(3)IVa-aph(4)Ia aminoglycoside resistance-containing module, interspersed with insertion sequences IS1133, ISRle7, and IS26, which is not repeated in its entirety in the reference plasmid sequences. Smaller parts of the resistance gene cluster were found to be identical to a streptomycin resistance transposon, Tn5393 (accession number: M96392.1), described in 1993 in Erwinia amylovora [18]. All of the four reference plasmid sequences above contain a tetracycline resistance gene cluster, which is absent in plasmid pCFS0255-2 (Figure 4).
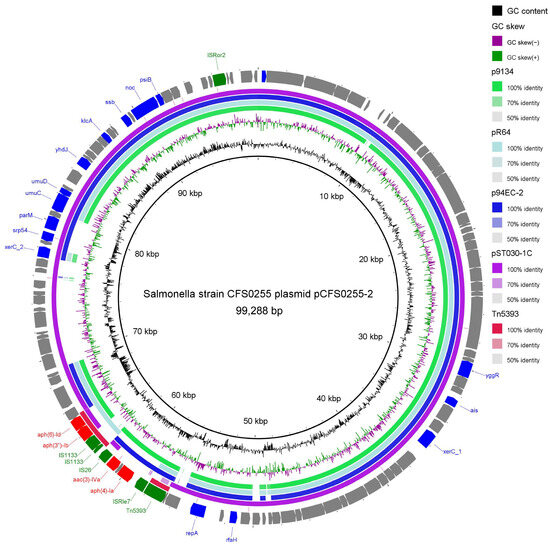
Figure 4.
Circular schematic map of the novel multidrug resistance IncI1 family plasmid pCFS0255-2, displaying the genetic structure of the plasmid. CDSs were shown as arrows. Antibiotic resistance genes are highlighted in red. Insertion sequences were highlighted in green. Other annotated genes were denoted by a blue color. Colored arcs are used to show regions of homology between the plasmid sequence and its four reference sequences, including plasmid p9134, plasmid R64, plasmid p94EC-2, and transposon Tn5393. The inner two rings represent GC Skew and GC content.
3. Discussion
3.1. Superiority of WGS for Serotyping Historical Isolates
The failure of traditional phenotypic serotyping (e.g., slide agglutination) in these older isolates can likely be attributed to several well-documented biological and technical limitations. These include the loss of surface antigens (such as the flagellar H-antigen) during prolonged storage, the transition to rough colony variants (resulting in the loss of the O-antigen), weak or non-specific auto-agglutination, and the potential use of incomplete antisera panels at the time of historical testing [19]. In contrast, in silico serotyping directly targets the underlying genetic determinants, providing a highly robust and objective prediction [20]. Importantly, the WGS-predicted serotypes are completely consistent with the clustering observed in the core-genome SNP phylogenetic tree, further confirming the superior accuracy of the genomic approach over historical laboratory records. This robust in silico confirmation establishes a reliable foundation for the subsequent genomic epidemiological analyses.
3.2. Evolutionary Insights and Mosaic Architecture of the MDR Plasmid pCFS0255-1
Comparative genomic analysis using the NCBI NR/NT database revealed that pCFS0255-1 possesses a unique assemblage of three resistance gene islands—TET, CHASRI, and SUL-MEF-MER—a combination not previously reported in a single closed plasmid sequence. Detailed synteny analysis with closely related replicons highlights the mosaic nature of this episome. While Salmonella enterica plasmid pN18S0993-1 shares high homology in the TET and CHASRI regions, it lacks the SUL-MEF-MER cluster. Conversely, plasmids from other genera, such as Klebsiella pneumoniae (pHNGS471-1) and Escherichia coli (p4M9F), harbor the SUL-MEF-MER module but diverge significantly in their backbone or other accessory repertoires. Interestingly, the pCFS0255-1 backbone exhibits the highest structural similarity to Escherichia coli plasmid pSD134209-1 and the ancestral S. Typhi R27 plasmid, yet these lack both the metal resistance and the broader antimicrobial resistance islands.
Notably, a specific subsequence (positions 194,016–196,464) flanked by insertion sequences ISEc10 and IS1A was uniquely identified in K. pneumoniae plasmids (e.g., pKP301-1), suggesting a recent acquisition from this genus. Collectively, these findings support a hypothesis of modular evolution through multiple recombination events. It is plausible that these mobile genetic elements (MGEs) have facilitated the integration of diverse resistance determinants from across the Enterobacteriaceae family, including Escherichia, Klebsiella, and Salmonella. Such interspecies horizontal gene transfer (HGT) underscores the emergence of complex “super-plasmids” in the food production chain, presenting a fortified vehicle for the co-dissemination of multidrug and heavy metal resistance.
To address whether the modular structure of pCFS0255-1 represents a persistently circulating threat, we queried its complete sequence against the NCBI nt_core database (version 20250319). Notably, filtering for high homology (coverage ≥ 80%, weighted ANI ≥ 95%) yielded multiple highly similar plasmid records isolated predominantly from recent years (2013–2023). These plasmids were identified across a global geographic distribution, including the USA, China, South Korea, Thailand, Japan, and Vietnam (Supplementary Materials Table S1). However, the complete genetic architecture of this MDR plasmid has not been fully explored or discussed until now.
Furthermore, consistent with the One Health paradigm, these structurally conserved plasmids are actively circulating among diverse hosts and environments, including food-producing animals (swine and poultry), retail meats, and human clinical fecal samples. Notably, the backbone was also identified in other Enterobacteriaceae species, such as Escherichia coli (e.g., CP064017.1, isolated from pigs in Thailand, 2018) and Klebsiella pneumoniae (e.g., CP072461.1, isolated from swine in China, 2019). The extensive recent prevalence of this plasmid architecture globally underscores that the historical pCFS0255-1 identified in our 2005 Colombian isolate is not an evolutionary dead-end. Instead, it serves as a crucial historical baseline to track the long-term persistence, global dissemination, and cross-species horizontal transfer of this MDR ‘super-plasmid’ over nearly two decades.
3.3. The CHASRI Gene Cluster
The copper homeostasis and silver resistance island (CHASRI) was first described in 2016 [15]. This 19-gene metal resistance cluster is believed to have originated in Enterobacter cloacae, and has spread across the Enterobacteriaceae family through horizontal gene transfer. CHASRI underwent sequential assembly of two gene modules: cus (copper-sensing copper efflux system) and pco (plasmid-encoded copper resistance system), facilitating its dissemination. The presence of CHASRI alongside tetracycline and sulfonamide-macrolide resistance genes on a single plasmid suggests a significant evolutionary advantage in agricultural settings. In food production systems, heavy metals such as copper are frequently utilized as supra-nutritional feed additives for growth promotion, while silver is increasingly employed in antimicrobial applications, including facility disinfection and as emerging nano-based growth enhancers [21,22]. This environmental pressure can drive the persistence of multidrug-resistant (MDR) plasmids even in the absence of direct antibiotic use [23]. Through a process of co-selection, the exposure to heavy metals selects for the metal resistance island, which indirectly maintains the linked AMR genes within the bacterial population. This ‘hitchhiking’ mechanism ensures that the plasmid remains stable, effectively creating a reservoir of antibiotic resistance that persists despite reduced antibiotic consumption.
A study by the US-FDA investigated the prevalence of CHASRI in Salmonella, conducting phylogenetic clustering analysis along with source and serotype statistics on 4954 CHASRI-containing Salmonella deposited in the NCBI database [24]. The study identified poultry, pork, and environmental sources as the primary origins of CHASRI-containing Salmonella. CHASRI exhibits diverse forms in Salmonella. One identified form is a chromosomal mobile genetic island known as SGI-3, found in Salmonella 4 [5], 12:i:–. SGI-3 contains a CHASRI, in addition to an arsenic resistance operon, and genes associated with conjugative transfer and DNA replication or partitioning [25]. CHASRI was also found to be present in the large genomic island denoted as SGI-4, identified in Salmonella enterica serovar Monophasi [26]. The CHASRI identified in this study represented a previously unreported form in Salmonella. It exists within a multidrug-resistant plasmid alongside a tetracycline resistance gene cluster and a sulfonamide-macrolide-mercury resistance gene cluster. This co-occurrence suggests that the integration of CHASRI into this plasmid may have been driven by complex co-selection and cross-selection pressures during its evolutionary history.
3.4. Limitations of the Study
Several limitations of the current study should be acknowledged. Primarily, the Salmonella isolates analyzed were recovered between 2002 and 2009. While this historical collection serves as a crucial baseline to track the long-term persistence and evolutionary origins of complex MDR plasmids, such as pCFS0255-1, it may not accurately reflect the current epidemiological landscape, circulating serotypes, or contemporary AMR profiles of foodborne Salmonella in Colombia. Furthermore, the collection comprises a relatively modest sample size (n = 90), geographically restricted to specific departments in northern Colombia (Montería, Cartagena, Sincelejo, and Barranquilla), which may limit the broader generalizability of the findings across the entire country. Finally, although our sampling spans various food sources and food-producing animals within the One Health continuum, the absence of corresponding human clinical isolates from the same timeframe restricts the ability to definitively reconstruct the transmission dynamics of these resistant lineages from the food chain to human infections. Despite these limitations, the genomic characterization presented here offers valuable insights into the historical AMR phenotypes and genotypes and reinforces the necessity of implementing continuous, centralized WGS-based surveillance to track emerging MDR threats.
4. Materials and Methods
4.1. Bacterial Isolates
A collection of 90 Salmonella species recovered between 2002 and 2009 from a variety of food products and food-producing animals in Colombia was obtained from the University of Cordoba (Colombia). All bacterial isolates were streaked on XLD medium (Oxoid, Basingstoke, England) to check for purity and confirmed as Salmonella using a Salmonella latex test (Oxoid). These were then stored at −80 °C in glycerol stocks. The serotype data for laboratory testing were provided by the University of Cordoba (Colombia). Metadata including sources, years, and locations of all isolates and the corresponding lab-tested serotypes are shown in Appendix A Table A1.
4.2. Antibiotic Susceptibility Testing (AST)
Susceptibilities to 15 antimicrobial compounds were determined by disk diffusion and interpreted according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (2007) [27]. Escherichia coli ATCC® 25922 (American Type Culture Collection, Manassas, VA, USA) was included as a control.
Minimum inhibitory concentrations (MIC) for nalidixic acid (Sigma-Aldrich, Arklow, Ireland) and ciprofloxacin (Sigma-Aldrich) were determined by broth microdilution, in the absence and presence of 40 µg/mL phenyl-arginine-ß-naphthylamide (PAßN) (Sigma-Aldrich), to assess the presence of active efflux mechanisms.
4.3. Whole-Genome Sequencing and Data Analysis
Short-read sequences were obtained using Illumina HiSeqX platform (Illumina, San Diego, CA, USA) and the 150 bp paired-end approach, from libraries with an average insert size of 350 bp. Raw sequences were initially trimmed using Trimmomatic (v0.39) [28] followed by de novo assembly for complete genomes performed using SPAdes (v3.15.3) [29]. A subset of the isolate collection was re-sequenced using long-read sequencing methods on a MinION platform. Assembly was performed using Canu (v2.2) with hybrid assembly method to NGS reads [30] and sequences were polished using NextPolish (v1.4.1-0) [31]. Salmonella genome assemblies were annotated with the prokaryotic genome annotation tool Prokka (v1.14.6) [32]. Antibiotic resistance genes were predicted using the ABRicate (v1.0.0) software package, combining three reference databases, CARD (v3.2.9) [33], ResFinder (v4.5.0) [34], and NCBI AMRFinderPlus (v2024-05-02.2) [35]. Gene names were unified to the NCBI AMRFinderPlus references and all resistance genes were screened using the BLASTN algorithm, with minimum nucleotide identity and alignment length coverage of 80%.
Plasmids markers were predicted using PlasmidFinder (v2.1.6) [36], and the sequences were validated using BLAST (BLASTN). Sequence hits against reference sequences with the best coverage and identity scores were recorded. Since any single reference sequence could not completely cover the plasmids found in this study, partially covered reference sequences with high identity were also selected, including a transposon sequence, to ensure complete coverage of the plasmids in this study. Other mobile genetic elements were annotated using MGEFinder (v1.0.6) [37]. Sequence structure and genes of each plasmid were visualized using Brig (v0.95) and compared against reference sequences using BLASTN [38]. Partial sequence comparisons for the MDR gene islands and up-/down-streams were visualized using EasyFig (version 2.2.3) [39].
Serotypes of these isolates were predicted in silico from WGS assemblies using SeqSero2 (version 1.0.1) [40], and multi-locus sequence typing (MLST) of the isolates was predicted with reference to the PubMLST database using the MLST tool (v 2.20.0) [41]. The Salmonella pathogenicity islands (SPIs) were identified by PAIDB (v2.0), using the BLASTN method [42], and a threshold and coverage percentage of 90 was used for comparison with the reference sequences.
Core genomes were calculated using Roary (v3.13.0) from all 90 WGS assemblies [43]. Snippy (v4.6.0) was applied to clean up core genome alignment [44]. Gubbins (v3.2.1) and SNP-sites (v2.3.1) were used to filter the single nucleotide polymorphism alignment from the cleaned core genome alignment [45,46]. A maximum-likelihood phylogenetic tree based on the extracted core-genome SNPs was constructed using FastTree (v2.1.10) with the JC + CAT model, and the root of the tree was adjusted to its midpoint [47]. The final outputs were used for the illustration of tree and performed using Biopython (version 1.79).
5. Conclusions
In conclusion, this study developed a whole-genome analysis workflow based on core-genome SNPs for monitoring global foodborne pathogen transmission risks. This workflow was applied to assess and predict the resistance and pathogenicity of 90 Salmonella from Colombia. The study bacterial samples were identified by these methods to be of different serotypes, capable of predicting AMR-encoding genes and SPIs. A multidrug-resistant isolate CFS0255, characterized by two resistance-encoding plasmids, was described using a combination of short- and long-read sequencing. Furthermore, the emergence of plasmid pCFS0255-1 was hypothesized based on a multi-stepped integration process involving three resistance gene clusters, including: the TET operon, a CHASRI, and the SUL-MEF-MER cluster.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/antibiotics15050511/s1, Supplementary Table S1: BLASTN search results and metadata of highly similar plasmids to pCFS0255-1.
Author Contributions
Conceptualization, M.L. and S.F.; methodology, M.L. and G.M.; software, M.L.; validation, M.L., S.F. and L.B.; formal analysis, M.L. and G.M.; resources, S.M.; data curation, G.M., S.F. and S.M.; writing—original draft preparation, M.L.; writing—review and editing, S.F. and G.M.; visualization, M.L.; supervision, S.F., G.M. and L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Metadata of the strains used in this study is listed in Appendix A. WGS data is available upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AMR | Antimicrobial Resistance |
| ANI | Average Nucleotide Identity |
| AST | Antimicrobial Susceptibility Testing |
| CARD | Comprehensive Antibiotic Resistance Database |
| CHASRI | Copper Homeostasis and Silver Resistance Island |
| CLSI | Clinical and Laboratory Standards Institute |
| COIPARS | Colombian Integrated Program for Antimicrobial Resistance Surveillance |
| HGT | Horizontal Gene Transfer |
| Inc | Incompatibility Group |
| IS | Insertion Sequence |
| MDR | Multidrug Resistant/Multidrug Resistance |
| MGE | Mobile Genetic Element |
| MIC | Minimum Inhibitory Concentration |
| MLST | Multi-Locus Sequence Typing |
| NGS | Next-Generation Sequencing |
| NTS | Non-Typhoidal Salmonella |
| PAßN | Phenyl-Arginine-ß-Naphthylamide |
| SNP | Single Nucleotide Polymorphism |
| SPI | Salmonella Pathogenicity Island |
| ST | Sequence Type |
| WGS | Whole-Genome Sequencing |
| XLD | Xylose Lysine Deoxycholate |
Appendix A
Table A1.
Metadata of isolates in this study.
References
- Loredana, P.G.; Papa, M.I. Salmonella spp. Infection-a Continuous Threat Worldwide. Germs 2021, 11, 88. [Google Scholar]
- Mthembu, T.P.; Zishiri, O.T.; El Zowalaty, M.E. Genomic Characterization of Antimicrobial Resistance in Food Chain and Livestock-Associated Salmonella Species. Animals 2021, 11, 872. [Google Scholar] [CrossRef]
- Eng, S.K.; Pusparajah, P.; Mutalib, N.S.A.; Ser, H.L.; Chan, K.G.; Lee, L.H. Salmonella: A Review on Pathogenesis, Epidemiology and Antibiotic Resistance. Front. Life Sci. 2015, 8, 284–293. [Google Scholar] [CrossRef]
- Rantsiou, K.; Kathariou, S.; Winkler, A.; Skandamis, P.; Saint-Cyr, M.J.; Rouzeau-Szynalski, K.; Amézquita, A. Next Generation Microbiological Risk Assessment: Opportunities of Whole Genome Sequencing (Wgs) for Foodborne Pathogen Surveillance, Source Tracking and Risk Assessment. Int. J. Food Microbiol. 2018, 287, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.W.; Bell, R.; Ferreira, C.M.; Gonzalez-Escalona, N.; Hoffmann, M.; Muruvanda, T.; Ottesen, A.; Ramachandran, P.; Reed, E.; Sharma, S.; et al. Genomics of Foodborne Pathogens for Microbial Food Safety. Curr. Opin. Biotechnol. 2018, 49, 224–229. [Google Scholar] [CrossRef]
- Savelli, C.J.; Acevedo, R.F.G.; Simpson, J.; Mateus, C. The Utilisation of Tools to Facilitate Cross-Border Communication During International Food Safety Events, 1995–2020: A Realist Synthesis. Glob. Health 2021, 17, 65. [Google Scholar] [CrossRef]
- Allard Marc, W.; Strain, E.; Melka, D.; Bunning, K.; Steven, M.M.; Eric, W.B.; Timme, R. Practical Value of Food Pathogen Traceability through Building a Whole-Genome Sequencing Network and Database. J. Clin. Microbiol. 2016, 54, 1975–1983. [Google Scholar] [CrossRef]
- Li, W.; Wu, S.; Fu, P.; Liu, J.; Han, H.; Bai, L.; Pei, X.; Li, N.; Liu, X.; Guo, Y. National Molecular Tracing Network for Foodborne Disease Surveillance in China. Food Control 2018, 88, 28–32. [Google Scholar] [CrossRef]
- Li, W.; Cui, Q.; Bai, L.; Fu, P.; Han, H.; Liu, J.; Guo, Y. Application of Whole-Genome Sequencing in the National Molecular Tracing Network for Foodborne Disease Surveillance in China. Foodborne Pathog. Dis. 2021, 18, 538–546. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC). Epipulse: The European Surveillance Portal for Infectious Diseases; European Centre for Disease Prevention and Control: Solna, Sweden, 2021. [Google Scholar]
- Lv, L.-C.; Lu, Y.-Y.; Gao, X.; He, W.-Y.; Gao, M.-Y.; Mo, K.-B.; Liu, J.-H. Characterization of Ndm-5-Producing Enterobacteriaceae Isolates from Retail Grass Carp (Ctenopharyngodon Idella) and Evidence of Blandm-5-Bearing Inchi2 Plasmid Transfer between Ducks and Fish. Zool. Res. 2022, 43, 255. [Google Scholar] [CrossRef] [PubMed]
- Sherburne, C.K.; Lawley, T.D.; Gilmour, M.W.; Blattner, F.R.; Burland, V.; Grotbeck, E.; Rose, D.J.; Taylor, D.E. The Complete DNA Sequence and Analysis of R27, a Large Inchi Plasmid from Salmonella Typhi That Is Temperature Sensitive for Transfer. Nucleic Acids Res. 2000, 28, 2177–2186. [Google Scholar] [CrossRef]
- Staehlin, B.M.; Gibbons, J.G.; Rokas, A.; O’hAlloran, T.V.; Slot, J.C. Evolution of a Heavy Metal Homeostasis/Resistance Island Reflects Increasing Copper Stress in Enterobacteria. Genome Biol. Evol. 2016, 8, 811–826. [Google Scholar] [CrossRef]
- Kubasova, T.; Matiasovicova, J.; Rychlik, I.; Juricova, H. Complete Sequence of Multidrug Resistance P9134 Plasmid and Its Variants Including Natural Recombinant with the Virulence Plasmid of Salmonella Serovar Typhimurium. Plasmid 2014, 76, 8–14. [Google Scholar] [CrossRef]
- Sampei, G.-I.; Furuya, N.; Tachibana, K.; Saitou, Y.; Suzuki, T.; Mizobuchi, K.; Komano, T. Complete Genome Sequence of the Incompatibility Group I1 Plasmid R64. Plasmid 2010, 64, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Saw, W.-Y.; Tan, L.W.L.; Moong, D.K.N.; Nagarajan, N.; Teo, Y.Y.; Seedorf, H. Emergence of Tigecycline-and Eravacycline-Resistant Tet (X4)-Producing Enterobacteriaceae in the Gut Microbiota of Healthy Singaporeans. J. Antimicrob. Chemother. 2020, 75, 3480–3484. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.; Calia, C.; Ferrara, M.; D’ADdabbo, P.; Scrascia, M.; Mulè, G.; Monno, R.; Pazzani, C. Antimicrobial Resistance Gene Shuffling and a Three-Element Mobilisation System in the Monophasic Salmonella Typhimurium Strain St1030. Plasmid 2020, 111, 102532. [Google Scholar] [CrossRef]
- Chiou, C.S.; Jones, A.L. Nucleotide Sequence Analysis of a Transposon (Tn5393) Carrying Streptomycin Resistance Genes in Erwinia amylovora and Other Gram-Negative Bacteria. J. Bacteriol. 1993, 175, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.; Barretto, C.; Portmann, A.-C.; Fournier, C.; Karczmarek, A.; Voets, G.; Li, S.; Deng, X.; Klijn, A. Salmonella Serotyping; Comparison of the Traditional Method to a Microarray-Based Method and an in silico Platform Using Whole Genome Sequencing Data. Front. Microbiol. 2019, 10, 2554. [Google Scholar] [CrossRef]
- Uelze, L.; Borowiak, M.; Deneke, C.; Szabó, I.; Fischer, J.; Tausch, S.H.; Malorny, B. Performance and Accuracy of Four Open-Source Tools for In Silico Serotyping of Salmonella spp. Based on Whole-Genome Short-Read Sequencing Data. Appl. Environ. Microbiol. 2020, 86, e02265-19. [Google Scholar] [CrossRef]
- Frippiat, T.; Art, T.; Delguste, C. Silver Nanoparticles as Antimicrobial Agents in Veterinary Medicine: Current Applications and Future Perspectives. Nanomaterials 2025, 15, 202. [Google Scholar] [CrossRef]
- Kim, B.; Jeong, J.Y.; Park, S.H.; Jung, H.; Kim, M. Effects of Dietary Copper Sources and Levels on Growth Performance, Copper Digestibility, Fecal and Serum Mineral Characteristics in Growing Pigs. J. Anim. Sci. Technol. 2022, 64, 885–896. [Google Scholar] [CrossRef]
- Haendiges, J.; Brown, E.W.; Ferreira, C.; Hoffmann, M.; Reed, E.; Zheng, J.; Tikekar, R. Exploring the Genetic Landscape of the Copper Homeostasis and Silver Resistance Island (Chasri) in Salmonella Enterica. PLoS ONE 2025, 20, e0334908. [Google Scholar] [CrossRef]
- Julie, H.; Brown, E.W.; Tikekar, R.; Hoffmann, M. Comprehensive Study of the Copper Homeostasis and Silver Resistance Island (CHASRI) in Salmonella enterica. Presented at the FDA Science Forum, Virtual, 13–14 June 2023. [Google Scholar]
- Arai, N.; Sekizuka, T.; Tamamura, Y.; Kusumoto, M.; Hinenoya, A.; Yamasaki, S.; Iwata, T.; Watanabe-Yanai, A.; Kuroda, M.; Akiba, M. Salmonella Genomic Island 3 Is an Integrative and Conjugative Element and Contributes to Copper and Arsenic Tolerance of Salmonella enterica. Antimicrob. Agents Chemother. 2019, 63, e00429-19. [Google Scholar] [CrossRef]
- Branchu, P.; Charity, O.J.; Bawn, M.; Thilliez, G.; Dallman, T.J.; Petrovska, L.; Kingsley, R.A. Sgi-4 in Monophasic Salmonella Typhimurium St34 Is a Novel Ice That Enhances Resistance to Copper. Front. Microbiol. 2019, 10, 1118. [Google Scholar] [CrossRef]
- CLSI M100-S17; Performance Standards for Antimicrobial Susceptibility Testing; Seventeenth Informational Supplement. CLSI: Wayne, PA, USA, 2007.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. Spades: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly Via Adaptive K-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. Nextpolish: A Fast and Efficient Genome Polishing Tool for Long-Read Assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Valeria, B.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. Resfinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. Amrfinderplus and the Reference Gene Catalog Facilitate Examination of the Genomic Links among Antimicrobial Resistance, Stress Response, and Virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef]
- Carattoli, A.; Hasman, H. Plasmidfinder and in Silico Pmlst: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (Wgs). In Horizontal Gene Transfer; Springer: Berlin/Heidelberg, Germany, 2020; pp. 285–294. [Google Scholar]
- Durrant, M.G.; Li, M.M.; Siranosian, B.A.; Montgomery, S.B.; Bhatt, A.S. A Bioinformatic Analysis of Integrative Mobile Genetic Elements Highlights Their Role in Bacterial Adaptation. Cell Host Microbe 2020, 27, 140–153.e9. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. Blast Ring Image Generator (Brig): Simple Prokaryote Genome Comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bakker, H.C.D.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. Seqsero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data. Appl. Environ. Microbiol. 2019, 85, e01746-19. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: Bigsdb Software, the Pubmlst. Org Website and Their Applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Yoon, S.H.; Park, Y.-K.; Kim, J.F. Paidb V2. 0: Exploration and Analysis of Pathogenicity and Resistance Islands. Nucleic Acids Res. 2015, 43, D624–D630. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Seemann, T. Snippy: Rapid Haploid Variant Calling and Core Genome Alignment. GitHub. 2019. Available online: https://github.com/tseemann/snippy (accessed on 10 February 2020).
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid Phylogenetic Analysis of Large Samples of Recombinant Bacterial Whole Genome Sequences Using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. Snp-Sites: Rapid Efficient Extraction of Snps from Multi-Fasta Alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.