
The Premise of the Paradox: Examining the Evidence That Motivated GIPR Agonist and Antagonist Drug Development Programs

{kind=link}
{kind=link}
Abstract
1. Introduction
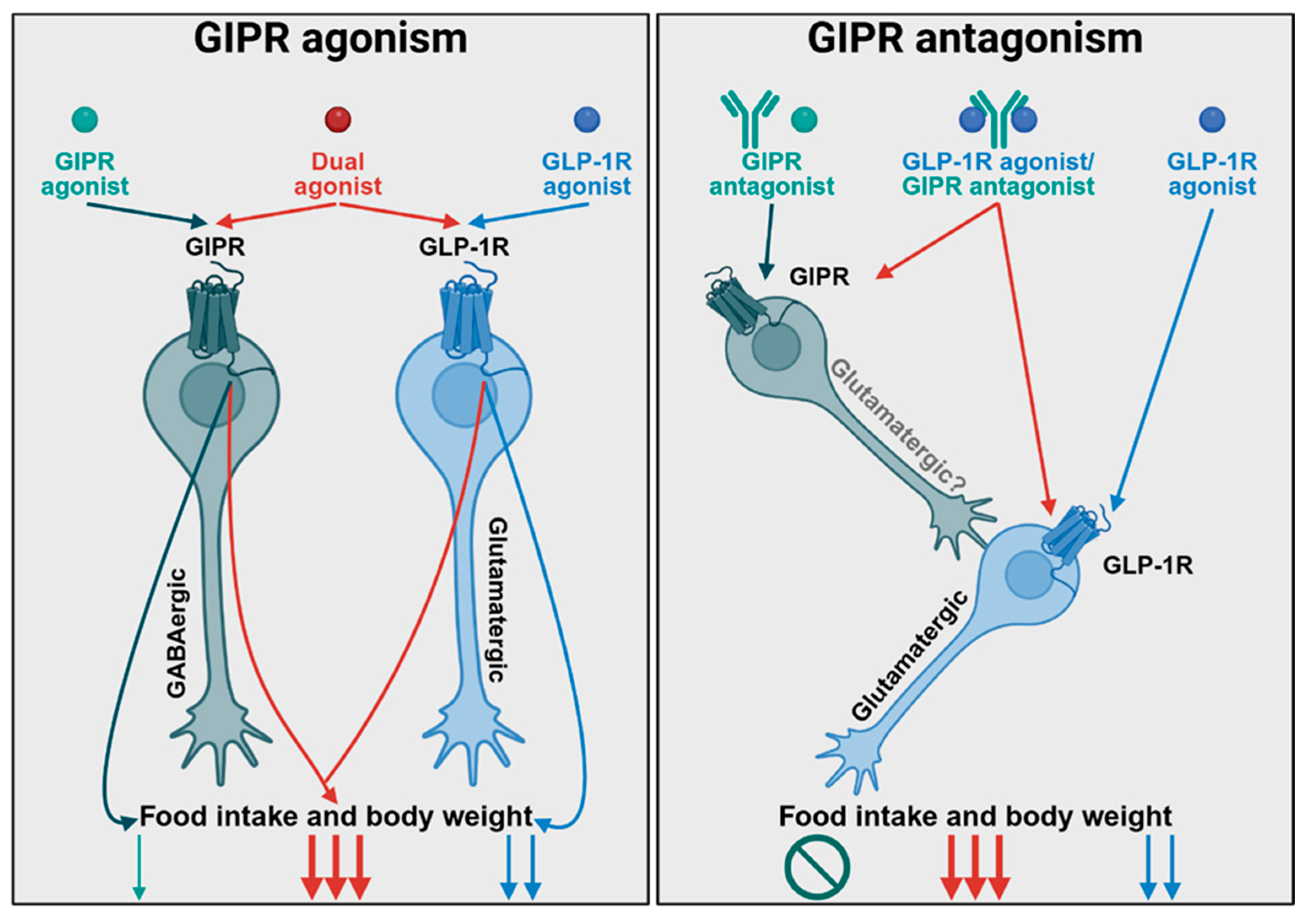
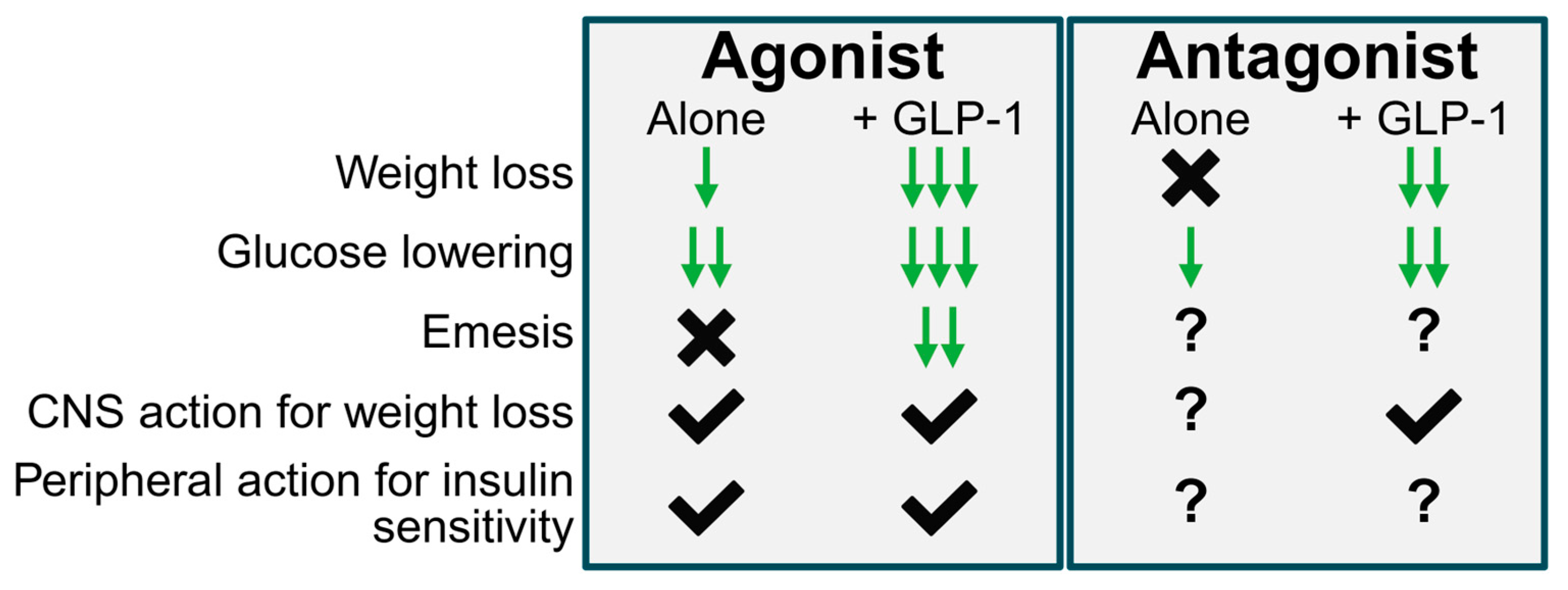
2. In Favor of Antagonism
3. Human Genetics Data in Favor of Both Agonism and Antagonism
4. In Favor of Agonism
5. Synthesis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed]
- Davies, M.; Faerch, L.; Jeppesen, O.K.; Pakseresht, A.; Pedersen, S.D.; Perreault, L.; Rosenstock, J.; Shimomura, I.; Viljoen, A.; Wadden, T.A.; et al. Semaglutide 2.4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): A randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet 2021, 397, 971–984. [Google Scholar] [CrossRef]
- Davies, M.J.; Bergenstal, R.; Bode, B.; Kushner, R.F.; Lewin, A.; Skjoth, T.V.; Andreasen, A.H.; Jensen, C.B.; DeFronzo, R.A.; Group, N.N.S. Efficacy of Liraglutide for Weight Loss Among Patients with Type 2 Diabetes: The SCALE Diabetes Randomized Clinical Trial. JAMA 2015, 314, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Sloop, K.W.; Loghin, C.; Alsina-Fernandez, J.; Urva, S.; Bokvist, K.B.; Cui, X.; Briere, D.A.; Cabrera, O.; Roell, W.C.; et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol. Metab. 2018, 18, 3–14. [Google Scholar] [CrossRef]
- Veniant, M.M.; Lu, S.C.; Atangan, L.; Komorowski, R.; Stanislaus, S.; Cheng, Y.; Wu, B.; Falsey, J.R.; Hager, T.; Thomas, V.A.; et al. A GIPR antagonist conjugated to GLP-1 analogues promotes weight loss with improved metabolic parameters in preclinical and phase 1 settings. Nat. Metab. 2024, 6, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Novikoff, A.; Muller, T.D. Antagonizing GIPR adds fire to the GLP-1R flame. Trends Endocrinol. Metab. 2024, 35, 566–568. [Google Scholar] [CrossRef]
- Muller, T.D.; Adriaenssens, A.; Ahren, B.; Bluher, M.; Birkenfeld, A.; Campbell, J.E.; Coghlan, M.P.; D’Alessio, D.; Deacon, C.F.; DelPrato, S.; et al. Glucose-dependent insulinotropic polypeptide (GIP). Mol. Metab. 2025, 95, 102118. [Google Scholar]
- Nauck, M.; Stockmann, F.; Ebert, R.; Creutzfeldt, W. Reduced incretin effect in Type 2 (non-insulin-dependent) diabetes. Diabetologia 1986, 29, 46–52. [Google Scholar] [CrossRef]
- Vilsboll, T.; Krarup, T.; Madsbad, S.; Holst, J.J. Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia 2002, 45, 1111–1119. [Google Scholar]
- Mentis, N.; Vardarli, I.; Kothe, L.D.; Holst, J.J.; Deacon, C.F.; Theodorakis, M.; Meier, J.J.; Nauck, M.A. GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes 2011, 60, 1270–1276. [Google Scholar] [CrossRef]
- Bergmann, N.C.; Lund, A.; Gasbjerg, L.S.; Meessen, E.C.E.; Andersen, M.M.; Bergmann, S.; Hartmann, B.; Holst, J.J.; Jessen, L.; Christensen, M.B.; et al. Effects of combined GIP and GLP-1 infusion on energy intake, appetite and energy expenditure in overweight/obese individuals: A randomised, crossover study. Diabetologia 2019, 62, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, N.C.; Gasbjerg, L.S.; Heimburger, S.M.; Krogh, L.S.L.; Dela, F.; Hartmann, B.; Holst, J.J.; Jessen, L.; Christensen, M.B.; Vilsboll, T.; et al. No Acute Effects of Exogenous Glucose-Dependent Insulinotropic Polypeptide on Energy Intake, Appetite, or Energy Expenditure When Added to Treatment With a Long-Acting Glucagon-Like Peptide 1 Receptor Agonist in Men With Type 2 Diabetes. Diabetes Care 2020, 43, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Borner, T.; Workinger, J.L.; Tinsley, I.C.; Fortin, S.M.; Stein, L.M.; Chepurny, O.G.; Holz, G.G.; Wierzba, A.J.; Gryko, D.; Nexo, E.; et al. Corrination of a GLP-1 Receptor Agonist for Glycemic Control without Emesis. Cell Rep. 2020, 31, 107768. [Google Scholar] [CrossRef] [PubMed]
- Borner, T.; Geisler, C.E.; Fortin, S.M.; Cosgrove, R.; Alsina-Fernandez, J.; Dogra, M.; Doebley, S.; Sanchez-Navarro, M.J.; Leon, R.M.; Gaisinsky, J.; et al. GIP Receptor Agonism Attenuates GLP-1 Receptor Agonist-Induced Nausea and Emesis in Preclinical Models. Diabetes 2021, 70, 2545–2553. [Google Scholar] [CrossRef]
- Knop, F.K.; Urva, S.; Rettiganti, M.; Benson, C.; Roell, W.; Mather, K.J.; Haupt, A.; Pratt, E.J. 56-OR: A Long-Acting Glucose-Dependent Insulinotropic Polypeptide Receptor Agonist Shows Weight Loss without Nausea or Vomiting. Diabetes 2023, 72 (Suppl. 1), 56-OR. [Google Scholar] [CrossRef]
- Yamane, S.; Harada, N.; Hamasaki, A.; Muraoka, A.; Joo, E.; Suzuki, K.; Nasteska, D.; Tanaka, D.; Ogura, M.; Harashima, S.; et al. Effects of glucose and meal ingestion on incretin secretion in Japanese subjects with normal glucose tolerance. J. Diabetes Investig. 2012, 3, 80–85. [Google Scholar] [CrossRef]
- Asmar, M.; Asmar, A.; Simonsen, L.; Gasbjerg, L.S.; Sparre-Ulrich, A.H.; Rosenkilde, M.M.; Hartmann, B.; Dela, F.; Holst, J.J.; Bulow, J. The Gluco- and Liporegulatory and Vasodilatory Effects of Glucose-Dependent Insulinotropic Polypeptide (GIP) Are Abolished by an Antagonist of the Human GIP Receptor. Diabetes 2017, 66, 2363–2371. [Google Scholar] [CrossRef]
- Thondam, S.K.; Daousi, C.; Wilding, J.P.; Holst, J.J.; Ameen, G.I.; Yang, C.; Whitmore, C.; Mora, S.; Cuthbertson, D.J. Glucose-dependent insulinotropic polypeptide promotes lipid deposition in subcutaneous adipocytes in obese type 2 diabetes patients: A maladaptive response. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E224–E233. [Google Scholar] [CrossRef]
- Moller, C.L.; Vistisen, D.; Faerch, K.; Johansen, N.B.; Witte, D.R.; Jonsson, A.; Pedersen, O.; Hansen, T.; Lauritzen, T.; Jorgensen, M.E.; et al. Glucose-Dependent Insulinotropic Polypeptide Is Associated With Lower Low-Density Lipoprotein But Unhealthy Fat Distribution, Independent of Insulin: The ADDITION-PRO Study. J. Clin. Endocrinol. Metab. 2016, 101, 485–493. [Google Scholar] [CrossRef]
- Creutzfeldt, W.; Ebert, R.; Willms, B.; Frerichs, H.; Brown, J.C. Gastric inhibitory polypeptide (GIP) and insulin in obesity: Increased response to stimulation and defective feedback control of serum levels. Diabetologia 1978, 14, 15–24. [Google Scholar] [CrossRef]
- Jones, I.R.; Owens, D.R.; Luzio, S.D.; Hayes, T.M. Obesity is associated with increased post-prandial GIP levels which are not reduced by dietary restriction and weight loss. Diabete Metab. 1989, 15, 11–22. [Google Scholar] [PubMed]
- Miyawaki, K.; Yamada, Y.; Yano, H.; Niwa, H.; Ban, N.; Ihara, Y.; Kubota, A.; Fujimoto, S.; Kajikawa, M.; Kuroe, A.; et al. Glucose intolerance caused by a defect in the entero-insular axis: A study in gastric inhibitory polypeptide receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 14843–14847. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, K.; Yamada, Y.; Ban, N.; Ihara, Y.; Tsukiyama, K.; Zhou, H.; Fujimoto, S.; Oku, A.; Tsuda, K.; Toyokuni, S.; et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 2002, 8, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Hansotia, T.; Maida, A.; Flock, G.; Yamada, Y.; Tsukiyama, K.; Seino, Y.; Drucker, D.J. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J. Clin. Investig. 2007, 117, 143–152. [Google Scholar] [CrossRef]
- Ugleholdt, R.; Pedersen, J.; Bassi, M.R.; Fuchtbauer, E.M.; Jorgensen, S.M.; Kissow, H.L.; Nytofte, N.; Poulsen, S.S.; Rosenkilde, M.M.; Seino, Y.; et al. Transgenic rescue of adipocyte glucose-dependent insulinotropic polypeptide receptor expression restores high fat diet-induced body weight gain. J. Biol. Chem. 2011, 286, 44632–44645. [Google Scholar] [CrossRef]
- Joo, E.; Harada, N.; Yamane, S.; Fukushima, T.; Taura, D.; Iwasaki, K.; Sankoda, A.; Shibue, K.; Harada, T.; Suzuki, K.; et al. Inhibition of Gastric Inhibitory Polypeptide Receptor Signaling in Adipose Tissue Reduces Insulin Resistance and Hepatic Steatosis in High-Fat Diet-Fed Mice. Diabetes 2017, 66, 868–879. [Google Scholar] [CrossRef]
- Beaudry, J.L.; Kaur, K.D.; Varin, E.M.; Baggio, L.L.; Cao, X.; Mulvihill, E.E.; Bates, H.E.; Campbell, J.E.; Drucker, D.J. Physiological roles of the GIP receptor in murine brown adipose tissue. Mol. Metab. 2019, 28, 14–25. [Google Scholar] [CrossRef]
- Campbell, J.E.; Ussher, J.R.; Mulvihill, E.E.; Kolic, J.; Baggio, L.L.; Cao, X.; Liu, Y.; Lamont, B.J.; Morii, T.; Streutker, C.J.; et al. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat. Med. 2016, 22, 84–90. [Google Scholar] [CrossRef]
- Zhang, Q.; Delessa, C.T.; Augustin, R.; Bakhti, M.; Collden, G.; Drucker, D.J.; Feuchtinger, A.; Caceres, C.G.; Grandl, G.; Harger, A.; et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 2021, 33, 833–844.e5. [Google Scholar] [CrossRef]
- Liu, C.M.; Killion, E.A.; Hammoud, R.; Lu, S.C.; Komorowski, R.; Liu, T.; Kanke, M.; Thomas, V.A.; Cook, K.; Sivits, G.N., Jr.; et al. GIPR-Ab/GLP-1 peptide-antibody conjugate requires brain GIPR and GLP-1R for additive weight loss in obese mice. Nat. Metab. 2025. [Google Scholar] [CrossRef]
- Liskiewicz, A.; Khalil, A.; Liskiewicz, D.; Novikoff, A.; Grandl, G.; Maity-Kumar, G.; Gutgesell, R.M.; Bakhti, M.; Bastidas-Ponce, A.; Czarnecki, O.; et al. Glucose-dependent insulinotropic polypeptide regulates body weight and food intake via GABAergic neurons in mice. Nat. Metab. 2023, 5, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Ayala, J.E.; Bracy, D.P.; James, F.D.; Burmeister, M.A.; Wasserman, D.H.; Drucker, D.J. Glucagon-like peptide-1 receptor knockout mice are protected from high-fat diet-induced insulin resistance. Endocrinology 2010, 151, 4678–4687. [Google Scholar] [CrossRef] [PubMed]
- Killion, E.A.; Wang, J.; Yie, J.; Shi, S.D.; Bates, D.; Min, X.; Komorowski, R.; Hager, T.; Deng, L.; Atangan, L.; et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci. Transl. Med. 2018, 10, eaat3392. [Google Scholar] [CrossRef]
- Mroz, P.A.; Finan, B.; Gelfanov, V.; Yang, B.; Tschop, M.H.; DiMarchi, R.D.; Perez-Tilve, D. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol. Metab. 2019, 20, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, B.; Capozzi, M.E.; Nui, J.; Hannou, S.A.; Finan, B.; Naylor, J.; Ravn, P.; D’Alessio, D.A.; Campbell, J.E. Pharmacological antagonism of the incretin system protects against diet-induced obesity. Mol. Metab. 2020, 32, 44–55. [Google Scholar] [CrossRef]
- Yang, B.; Gelfanov, V.M.; El, K.; Chen, A.; Rohlfs, R.; DuBois, B.; Kruse Hansen, A.M.; Perez-Tilve, D.; Knerr, P.J.; D’Alessio, D.; et al. Discovery of a potent GIPR peptide antagonist that is effective in rodent and human systems. Mol. Metab. 2022, 66, 101638. [Google Scholar] [CrossRef]
- Gutgesell, R.M.; Khalil, A.; Liskiewicz, A.; Maity-Kumar, G.; Novikoff, A.; Grandl, G.; Liskiewicz, D.; Coupland, C.; Karaoglu, E.; Akindehin, S.; et al. GIPR agonism and antagonism decrease body weight and food intake via different mechanisms in male mice. Nat. Metab. 2025. [Google Scholar] [CrossRef]
- Jensen, M.H.; Sanni, S.J.; Riber, D.; Holst, J.J.; Rosenkilde, M.M.; Sparre-Ulrich, A.H. AT-7687, a novel GIPR peptide antagonist, combined with a GLP-1 agonist, leads to enhanced weight loss and metabolic improvements in cynomolgus monkeys. Mol. Metab. 2024, 88, 102006. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Lango Allen, H.; Lindgren, C.M.; Luan, J.; Magi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef]
- Akbari, P.; Gilani, A.; Sosina, O.; Kosmicki, J.A.; Khrimian, L.; Fang, Y.Y.; Persaud, T.; Garcia, V.; Sun, D.; Li, A.; et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science 2021, 373, eabf8683. [Google Scholar] [CrossRef]
- Saxena, R.; Hivert, M.F.; Langenberg, C.; Tanaka, T.; Pankow, J.S.; Vollenweider, P.; Lyssenko, V.; Bouatia-Naji, N.; Dupuis, J.; Jackson, A.U.; et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 2010, 42, 142–148. [Google Scholar] [CrossRef]
- Karhunen, V.; Daghlas, I.; Zuber, V.; Vujkovic, M.; Olsen, A.K.; Knudsen, L.B.; Haynes, W.G.; Howson, J.M.M.; Gill, D. Leveraging human genetic data to investigate the cardiometabolic effects of glucose-dependent insulinotropic polypeptide signalling. Diabetologia 2021, 64, 2773–2778. [Google Scholar] [CrossRef]
- Gasbjerg, L.S.; Helsted, M.M.; Hartmann, B.; Jensen, M.H.; Gabe, M.B.N.; Sparre-Ulrich, A.H.; Veedfald, S.; Stensen, S.; Lanng, A.R.; Bergmann, N.C.; et al. Separate and Combined Glucometabolic Effects of Endogenous Glucose-Dependent Insulinotropic Polypeptide and Glucagon-like Peptide 1 in Healthy Individuals. Diabetes 2019, 68, 906–917. [Google Scholar] [CrossRef]
- Finan, B.; Ma, T.; Ottaway, N.; Muller, T.D.; Habegger, K.M.; Heppner, K.M.; Kirchner, H.; Holland, J.; Hembree, J.; Raver, C.; et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci. Transl. Med. 2013, 5, 209ra151. [Google Scholar] [CrossRef]
- Knerr, P.J.; Mowery, S.A.; Douros, J.D.; Premdjee, B.; Hjollund, K.R.; He, Y.; Kruse Hansen, A.M.; Olsen, A.K.; Perez-Tilve, D.; DiMarchi, R.D.; et al. Next generation GLP-1/GIP/glucagon triple agonists normalize body weight in obese mice. Mol. Metab. 2022, 63, 101533. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Urva, S.; Roell, W.C.; Qu, H.; Loghin, C.; Moyers, J.S.; O’Farrell, L.S.; Briere, D.A.; Sloop, K.W.; Thomas, M.K.; et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: From discovery to clinical proof of concept. Cell Metab. 2022, 34, 1234–1247.e9. [Google Scholar] [CrossRef] [PubMed]
- Bossart, M.; Wagner, M.; Elvert, R.; Evers, A.; Hubschle, T.; Kloeckener, T.; Lorenz, K.; Moessinger, C.; Eriksson, O.; Velikyan, I.; et al. Effects on weight loss and glycemic control with SAR441255, a potent unimolecular peptide GLP-1/GIP/GCG receptor triagonist. Cell Metab. 2022, 34, 59–74.e10. [Google Scholar] [CrossRef] [PubMed]
- Finan, B.; Douros, J.D.; Goldwater, R.; Hansen, A.M.K.; Hjerpsted, J.B.; Hjollund, K.R.; Kankam, M.K.; Knerr, P.J.; Konkar, A.; Mowery, S.A.; et al. A once-daily GLP-1/GIP/glucagon receptor tri-agonist (NN1706) lowers body weight in rodents, monkeys and humans. Mol. Metab. 2025, 96, 102129. [Google Scholar] [CrossRef]
- Wean, J.; Kowalsky, A.H.; Laker, R.; Will, S.; Drucker, D.J.; Rhodes, C.J.; Seeley, R.J. Specific loss of GIPR signaling in GABAergic neurons enhances GLP-1R agonist-induced body weight loss. Mol. Metab. 2025, 95, 102074. [Google Scholar] [CrossRef]
- Sisley, S.; Gutierrez-Aguilar, R.; Scott, M.; D’Alessio, D.A.; Sandoval, D.A.; Seeley, R.J. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J. Clin. Investig. 2014, 124, 2456–2463. [Google Scholar] [CrossRef]
- Samms, R.J.; Sloop, K.W.; Gribble, F.M.; Reimann, F.; Adriaenssens, A.E. GIPR Function in the Central Nervous System: Implications and Novel Perspectives for GIP-Based Therapies in Treating Metabolic Disorders. Diabetes 2021, 70, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Samms, R.J.; Cosgrove, R.; Snider, B.M.; Furber, E.C.; Droz, B.A.; Briere, D.A.; Dunbar, J.; Dogra, M.; Alsina-Fernandez, J.; Borner, T.; et al. GIPR Agonism Inhibits PYY-Induced Nausea-Like Behavior. Diabetes 2022, 71, 1410–1423. [Google Scholar] [CrossRef] [PubMed]
- Regmi, A.; Aihara, E.; Christe, M.E.; Varga, G.; Beyer, T.P.; Ruan, X.; Beebe, E.; O’Farrell, L.S.; Bellinger, M.A.; Austin, A.K.; et al. Tirzepatide modulates the regulation of adipocyte nutrient metabolism through long-acting activation of the GIP receptor. Cell Metab. 2024, 36, 1534–1549.e7. [Google Scholar] [CrossRef] [PubMed]
- Ravussin, E.; Sanchez-Delgado, G.; Martin, C.K.; Beyl, R.A.; Greenway, F.L.; O’Farrell, L.S.; Roell, W.C.; Qian, H.R.; Li, J.; Nishiyama, H.; et al. Tirzepatide did not impact metabolic adaptation in people with obesity, but increased fat oxidation. Cell Metab. 2025, 37, 1060–1074.e4. [Google Scholar] [CrossRef]
- Douros, J.D.; Flak, J.N.; Knerr, P.J. The agony and the efficacy: Central mechanisms of GLP-1 induced adverse events and their mitigation by GIP. Front. Endocrinol. 2025, 16, 1530985. [Google Scholar] [CrossRef]
- Willard, F.S.; Douros, J.D.; Gabe, M.B.; Showalter, A.D.; Wainscott, D.B.; Suter, T.M.; Capozzi, M.E.; van der Velden, W.J.; Stutsman, C.; Cardona, G.R. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight 2020, 5, e140532. [Google Scholar] [CrossRef]
- Douros, J.D.; Mokrosinski, J.; Finan, B. The GLP-1R as a model for understanding and exploiting biased agonism in next generation medicines. J. Endocrinol. 2024, 261, e230226. [Google Scholar] [CrossRef]
- Jones, B.; Buenaventura, T.; Kanda, N.; Chabosseau, P.; Owen, B.M.; Scott, R.; Goldin, R.; Angkathunyakul, N.; Correa, I.R., Jr.; Bosco, D.; et al. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat. Commun. 2018, 9, 1602. [Google Scholar] [CrossRef]
- Lucey, M.; Pickford, P.; Bitsi, S.; Minnion, J.; Ungewiss, J.; Schoeneberg, K.; Rutter, G.A.; Bloom, S.R.; Tomas, A.; Jones, B. Disconnect between signalling potency and in vivo efficacy of pharmacokinetically optimised biased glucagon-like peptide-1 receptor agonists. Mol. Metab. 2020, 37, 100991. [Google Scholar] [CrossRef]
- Hinds, C.E.; Peace, E.; Chen, S.; Davies, I.; El Eid, L.; Tomas, A.; Tan, T.; Minnion, J.; Jones, B.; Bloom, S.R. Abolishing beta-arrestin recruitment is necessary for the full metabolic benefits of G protein-biased glucagon-like peptide-1 receptor agonists. Diabetes Obes. Metab. 2024, 26, 65–77. [Google Scholar] [CrossRef]
- Douros, J.D.; Novikoff, A.; DuBois, B.; Rohlfs, R.; Mokrosinski, J.; Hogendorf, W.F.J.; Augustin, R.; Merkestein, M.; Egaa Martini, L.B.; Linderoth, L.; et al. A GLP-1 analogue optimized for cAMP-biased signaling improves weight loss in obese mice. Mol. Metab. 2025, 102124. [Google Scholar] [CrossRef] [PubMed]
- El, K.; Douros, J.D.; Willard, F.S.; Novikoff, A.; Sargsyan, A.; Perez-Tilve, D.; Wainscott, D.B.; Yang, B.; Chen, A.; Wothe, D.; et al. The incretin co-agonist tirzepatide requires GIPR for hormone secretion from human islets. Nat. Metab. 2023, 5, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Killion, E.A.; Chen, M.; Falsey, J.R.; Sivits, G.; Hager, T.; Atangan, L.; Helmering, J.; Lee, J.; Li, H.; Wu, B.; et al. Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat. Commun. 2020, 11, 4981. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E. Targeting the GIPR for Obesity: To Agonize or Antagonize? Potential Mechanisms. Mol. Metab. 2020, 46, 101139. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J. Biol. Chem. 1998, 273, 18677–18680. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douros, J.D.; Mowery, S.A.; Knerr, P.J. The Premise of the Paradox: Examining the Evidence That Motivated GIPR Agonist and Antagonist Drug Development Programs. J. Clin. Med. 2025, 14, 3812. https://doi.org/10.3390/jcm14113812
Douros JD, Mowery SA, Knerr PJ. The Premise of the Paradox: Examining the Evidence That Motivated GIPR Agonist and Antagonist Drug Development Programs. Journal of Clinical Medicine. 2025; 14(11):3812. https://doi.org/10.3390/jcm14113812
Chicago/Turabian StyleDouros, Jonathan D., Stephanie A. Mowery, and Patrick J. Knerr. 2025. "The Premise of the Paradox: Examining the Evidence That Motivated GIPR Agonist and Antagonist Drug Development Programs" Journal of Clinical Medicine 14, no. 11: 3812. https://doi.org/10.3390/jcm14113812
APA StyleDouros, J. D., Mowery, S. A., & Knerr, P. J. (2025). The Premise of the Paradox: Examining the Evidence That Motivated GIPR Agonist and Antagonist Drug Development Programs. Journal of Clinical Medicine, 14(11), 3812. https://doi.org/10.3390/jcm14113812