


Genes, Volume 16, Issue 2 (February 2025) – 128 articles
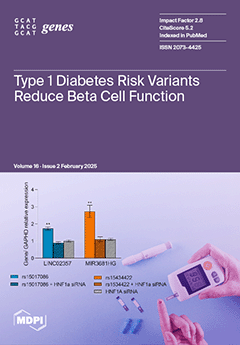
Cover Story (view full-size image):
The variants rs10517086 and rs1534422 are predictive of type 1 diabetes mellitus (T1DM) development and poor residual β cell function within the first year of diagnosis. However, the mechanism by which risk is conferred is unknown. Using CRISPR/Cas9, we show that both variants significantly reduce glucose-stimulated insulin secretion, increase the production of pro-inflammatory cytokines, and reduce the expression of several β cell markers and transcription factors. However, much of this appeared to be under the control of HNF1A. Analysis of human data revealed an association of rs10517086_A genotype with C-peptide in 153 individuals with T1DM, but not in 417 people with T2DM. Our data suggest that rs1534422 and rs10517086 exert multiple insults on the β cell through excessive upregulation of HNF1A and the induction of pro-inflammatory cytokines. View this paper
- Issues are regarded as officially published after their release is announced to the table of contents alert mailing list.
- You may sign up for e-mail alerts to receive table of contents of newly released issues.
- PDF is the official format for papers published in both, html and pdf forms. To view the papers in pdf format, click on the "PDF Full-text" link, and use the free Adobe Reader to open them.
Previous Issue
Next Issue