Expanding the Clinical Spectrum Associated with the Recurrent Arg203Trp Variant in PACS1: An Italian Cohort Study

 , , , , , ,
, , , , , , 
Abstract
1. Introduction
2. Materials and Methods
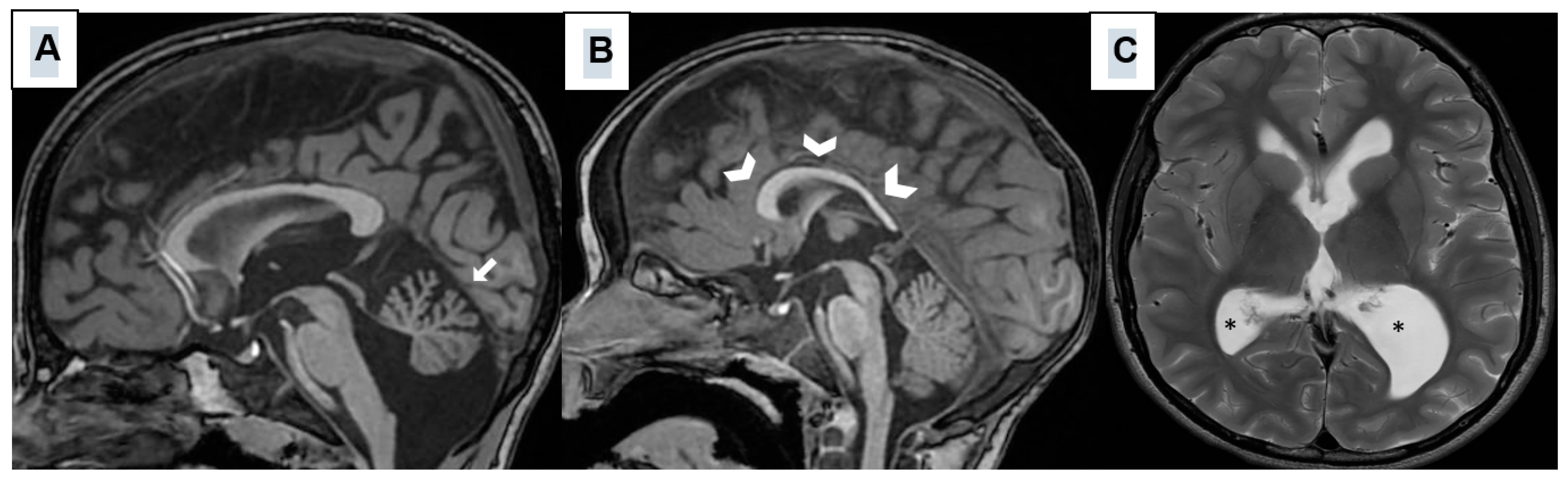
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SHMS | Schuurs–Hoeijkakers syndrome |
| ID | intellectual disability |
| ASD | autism spectrum disorder |
| HPO | human phenotype ontology |
| PCR-RFLP | polymerase chain reaction–restriction fragment length polymorphism |
| bp | base pair |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
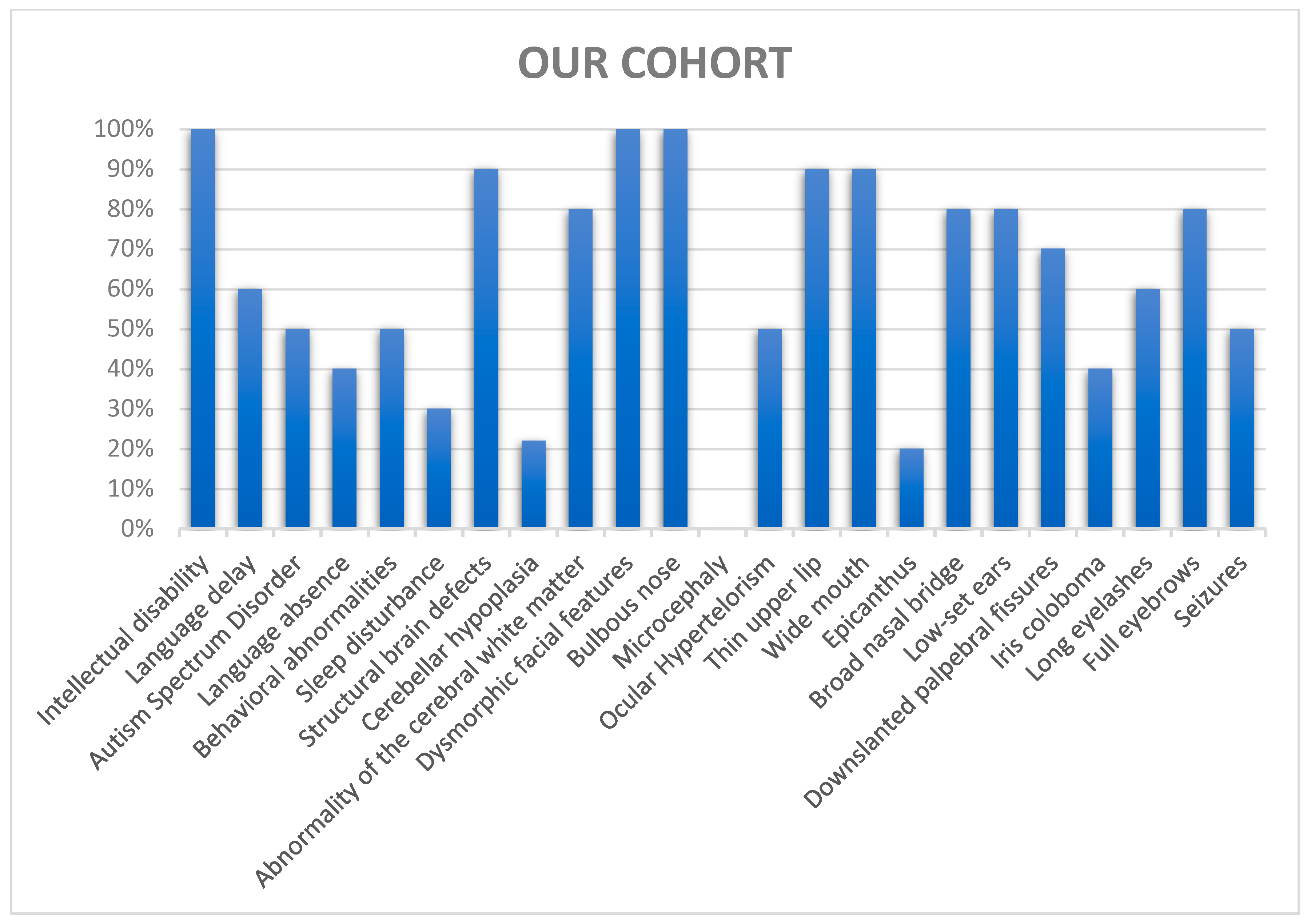
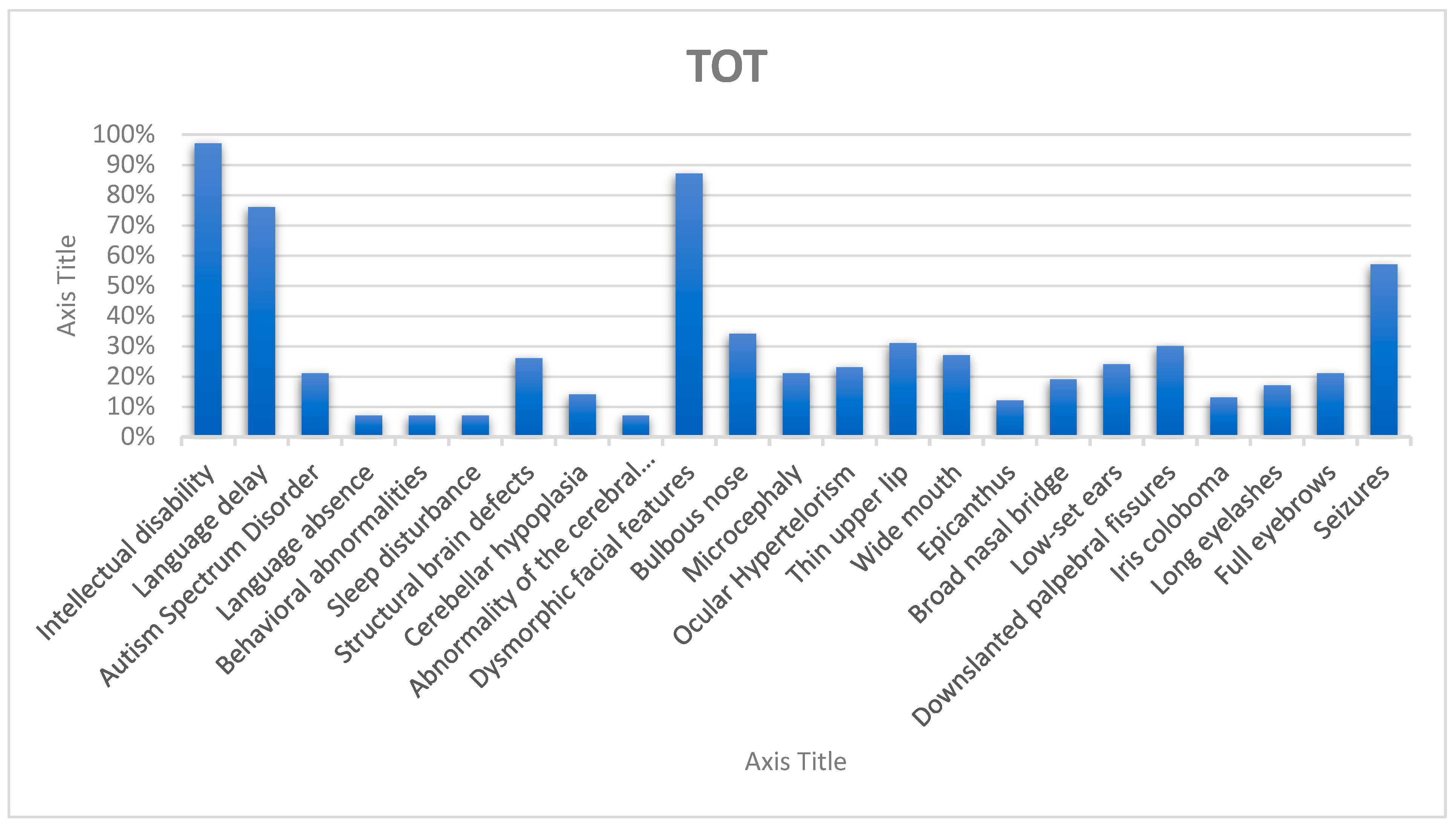
| Trait (HPO) | Present Cohort | Overall Italian Cohort | Tenorio-Castaño et al., 2021 [4] |
|---|---|---|---|
| Neurodevelopmental disorders | |||
| Intellectual disability (HP:0001249) | 10/10 100% | 12/12 100% | 56/58 97% |
| Language delay (HP:0000750) | 6/10 60% | 8/12 66% | 42/55 76% |
| Language absence (HP:0001344) | 4/10 40% | 4/12 33% | 4/56 7% |
| Autism spectrum disorder (HP:0000729) | 5/10 50% | 5/12 41% | 12/56 21% |
| Behavioral and/or emotional abnormalities (HP:0000708) | 5/10 50% | 5/12 41% | 4/56 7% |
| Sleep disturbance (HP:0002360) | 3/10 30% | 3/12 25% | 4/56 7% |
| Brain abnormalities | |||
| Structural brain defects (HP:0012443) | 9/9 100% | 10/11 90% | 13/49 26% |
| Cerebellar hypoplasia (HP:0001321) | 2/9 22% | 2/11 18% | 6/43 14% |
| Abnormalities of the cerebral white matter (HP:0002500) | 8/10 80% | 8/12 66% | 3/43 7% |
| Craniofacial features | |||
| Dysmorphic facial features (HP:0001999) | 10/10 100% | 12/12 100% | 49/60 82% |
| Bulbous nose (HP:0000414) | 10/10 100% | 12/12 100% | 13/61 21% |
| Microcephaly (HP:0000252) | 0/10 0% | 0/12 0% | 13/61 21% |
| Ocular hypertelorism (HP:0000316) | 5/10 50% | 7/12 58% | 13/61 21% |
| Thin upper lip (HP:0000219) | 9/10 90% | 11/12 91,6% | 11/61 18% |
| Wide mouth (HP:0000154) | 9/10 90% | 11/12 91,6% | 8/61 13% |
| Epicanthus (HP:0000286) | 2/10 20% | 4/12 33% | 6/61 10% |
| Broad nasal bridge (HP:0012811) | 8/10 80% | 10/12 83% | 6/61 10% |
| Low-set ears (HP:0000369) | 8/10 80% | 8/12 66% | 9/61 15% |
| Down-slanted palpebral fissures (HP:0000494) | 7/10 70% | 9/12 75% | 14/61 23% |
| Iris coloboma (HP:0000612) | 4/10 40% | 4/12 33% | 5/57 9% |
| Long eyelashes (HP:0000527) | 6/10 60% | 6/12 50% | 7/61 12% |
| Full eyebrows (HP:0004523) | 8/10 80% | 9/12 75% | 7/61 12% |
| Other | |||
| Seizures (HP:0001250) | 5/10 50% | 6/12 50% | 33/58 57% |
References
- Lusk, L.; Smith, S.; Martin, C.; Taylor, C.; Chung, W. PACS1 Neurodevelopmental disorder. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2020; pp. 1993–2024. Bookshelf ID: NBK559434. [Google Scholar] [PubMed]
- Schuurs-Hoeijmakers, J.H.; Oh, E.C.; Vissers, L.E.; Swinkels, M.E.; Gilissen, C.; Willemsen, M.A.; Holvoet, M.; Steehouwer, M.; Veltman, J.A.; de Vries, B.B.; et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am. J. Hum. Genet. 2012, 91, 1122–1127. [Google Scholar] [CrossRef]
- Schuurs-Hoeijmakers, J.H.; Landsverk, M.L.; Foulds, N.; Kukolich, M.K.; Gavrilova, R.H.; Greville-Heygate, S.; Hanson-Kahn, A.; Bernstein, J.A.; Glass, J.; Chitayat, D.; et al. Clinical delineation of the PACS1-related syndrome—Report on 19 patients. Am. J. Med. Genet. A 2016, 170, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Tenorio-Castaño, J.; Morte, B.; Nevado, J.; Martinez-Glez, V.; Santos-Simarro, F.; García-Miñaúr, S.; Palomares-Bralo, M.; Pacio-Míguez, M.; Gómez, B.; Arias, P.; et al. Schuurs-Hoeijmakers syndrome (PACS1 neurodevelopmental disorder): Seven novel patients and a review. Genes 2021, 12, 738. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bruno, L.P.; Doddato, G.; Baldassarri, M.; Rizzo, C.L.; Resciniti, S.; Bruttini, M.; Mirjam, L.; Zguro, K.; Furini, S.; Mencarelli, M.A.; et al. Expanding the clinical spectrum associated with the PACS1 p.Arg203Trp mutational hot-spot: Two additional Italian patients. Am. J. Med. Genet. A 2023, 191, 284–288. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Uzgiris, I.C.; Hunt, J.M.C.V. La valutazione nella prima infanzia. In Scale Ordinali Dello Sviluppo Psicologico; La Nuova Italia: Bari, Italy, 1999. [Google Scholar]
- Bayley, N. Bayley Scales of Infant and Toddler Development, 3rd ed.; (BAYLEY-III); Giunti Psychometrics: Florence, KY, USA, 2005. [Google Scholar]
- Griffiths, R. Griffiths Scales of Child Development, 3rd ed.; Hogrefe: Oxford, UK, 2016. [Google Scholar]
- Wechsler, D. WISC-IV. In Wechsler Intelligence Scale for Children-IV; Giunti Psychometrics: Florence, KY, USA, 2012. [Google Scholar]
- Sparrow, S.S.; Cicchetti, D.V.; Balla, D.A. Vineland-II. In Vineland Adaptive Behavior Scales-II—Second Edition—Survey Interview Form; Giunti Psychometric: Florence, KY, USA, 2016. [Google Scholar]
- Bisiacchi, P.; Cendron, M.; Gugliotta, M.; Tressoldi, P.E.; Vio, C. Neuropsychological Assessment Battery for Developmental Age—BVN 5-11; 11-18; Erickson: Trento, Italy, 2005. [Google Scholar]
- Stella, G.; Pizzoli, C.; Tressoldi, P. Peabody Picture Vocabulary Test; Omega Ed: Torino, Italy, 2000. [Google Scholar]
- Bishop, D.V.M. Test for Reception of Grammar: TROG-2; Giunti O.S: Firenze, Italy, 2009. [Google Scholar]
- Marini, A.; Marotta, L.; Bulgheroni, S.; Fabbro, F. Language Assessment Battery, BVL 4-12; Giunti O.S: Firenze, Italy, 2015. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Arnedo, M.; Ascaso, Á.; Latorre-Pellicer, A.; Lucia-Campos, C.; Gil-Salvador, M.; Ayerza-Casas, A.; Pablo, M.J.; Gómez-Puertas, P.; Ramos, F.J.; Bueno-Lozano, G.; et al. Molecular Basis of the Schuurs-Hoeijmakers Syndrome: What We Know about the Gene and the PACS-1 Protein and Novel Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 9649. [Google Scholar] [CrossRef]
- Bolke, P.; Nemani, S.P.; Mettan, O. Clinicoradiological correlation in a case of Schuurs—Hoeijmakers syndrome. Case Rep. Clin. Radiol. 2024, in press. [Google Scholar] [CrossRef]
- Available online: https://www.pacs1foundation.org/ (accessed on 10 February 2024).
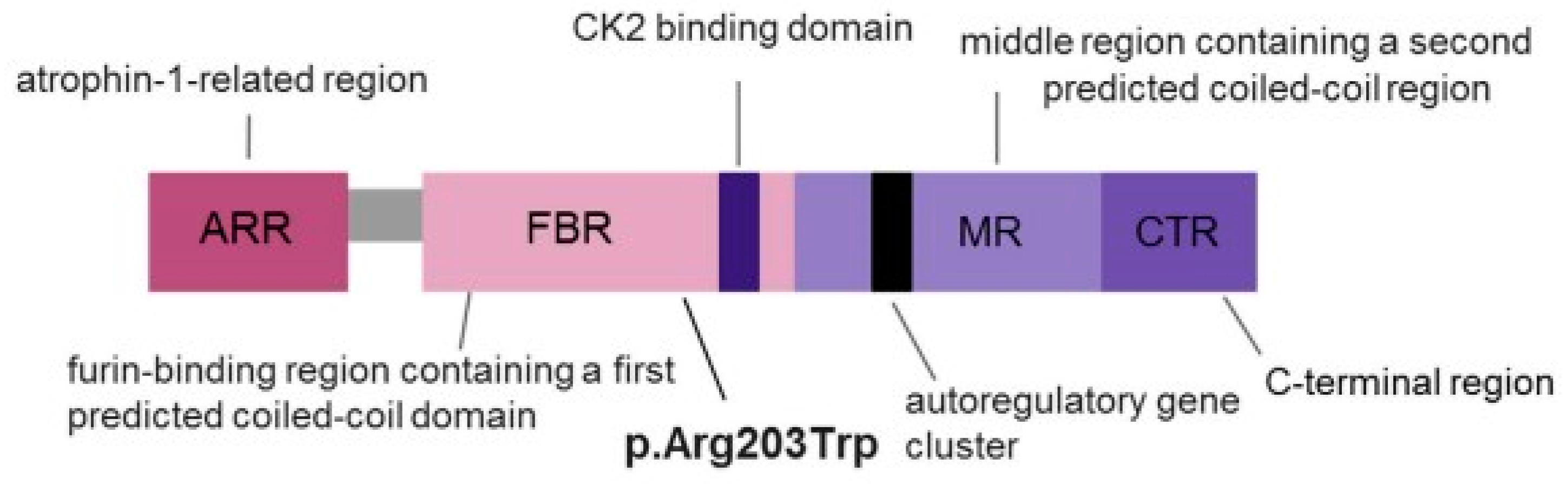
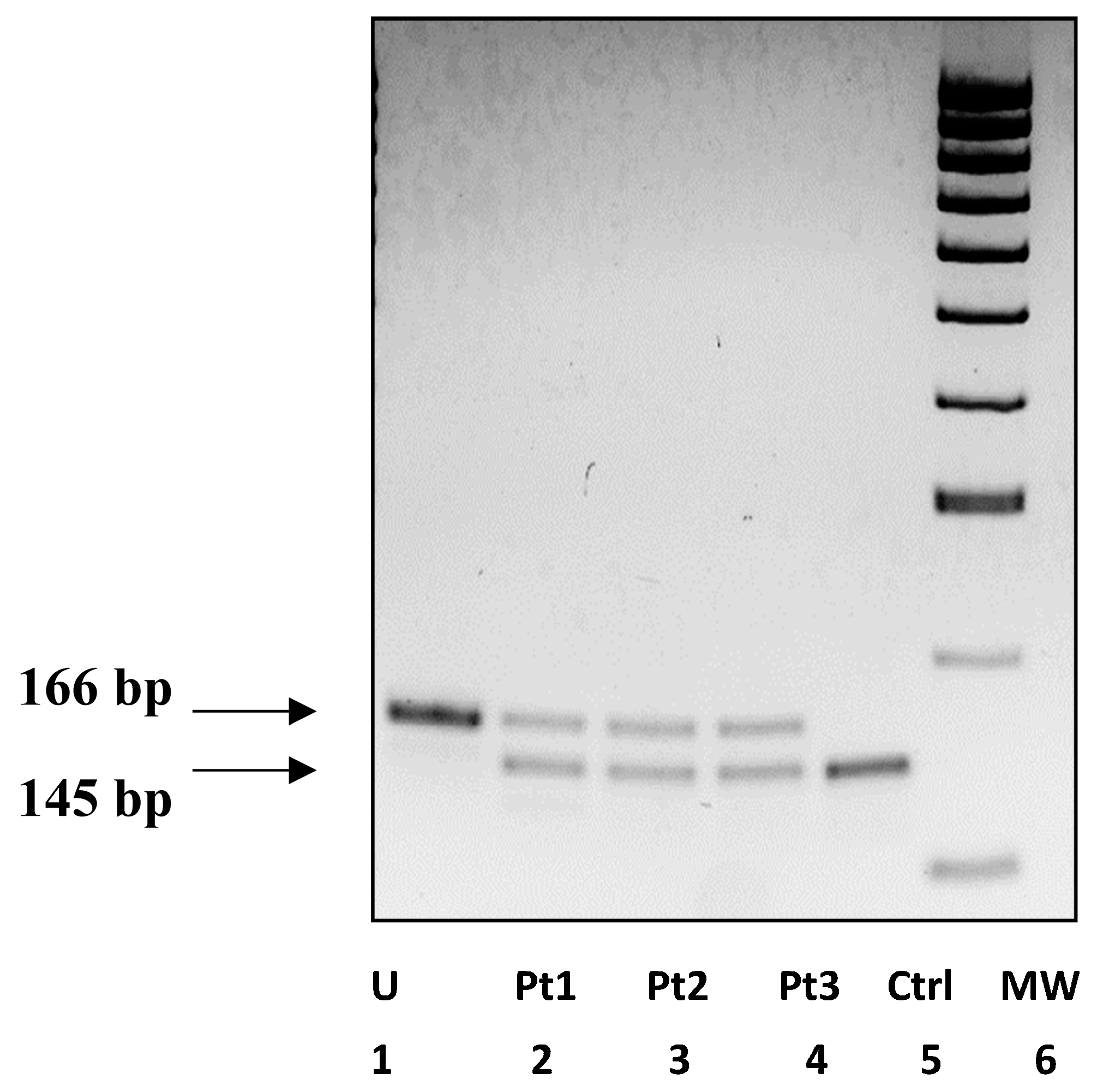

| Patient | Sex | Age | Intellectual Disability | Language | Behavioral and Socio-Relational Abnormalities |
|---|---|---|---|---|---|
| C.V. | F | 2.1 | Moderate (acquisition of sensorimotor skills, deficit in adaptive behavior) | Severely impaired (non-verbal) | Emotional–behavioral dysregulation and stereotyped movements |
| T.B. | F | 2.7 | Moderate (acquisition of sensorimotor skills, deficit in adaptive behavior) | Severely impaired (non-verbal) | Stereotyped movements |
| Q.F. | M | 2.9 | Severe (acquisition of sensorimotor skills, deficit in adaptive behavior) | Severely impaired (non-verbal) | Autistic features |
| R.E. | M | 3.7 | Moderate (acquisition of sensorimotor skills, severe deficit in adaptive behavior) | Severely impaired (non-verbal) | Autistic features and emotional–behavioral dysregulation |
| M.I. | F | 12.10 | Severe (acquisition of early visual/graphic representation skills, deficit in adaptive behavior) | Severely impaired (stereotyped sentences) | Autistic features |
| C.G. | F | 15.1 | Moderate (acquisition of early preoperational skills, severe deficit in adaptive behavior) | Moderately impaired (can pronounce simple and complex sentences) | Emotional–behavioral dysregulation |
| S.C. | F | 16 | Moderate (acquisition of preoperational skills, severe deficit in adaptive behavior) | Mildly impaired (can pronounce simple and complex sentences) | Emotional–behavioral inhibition |
| D.D.V. | M | 17.5 | Severe (acquisition of early visual/graphic representation skills and severe deficit in adaptive behavior) | Severely impaired (stereotyped sentences) | Autistic features |
| S.N. | M | 19.1 | Mild (acquisition of formal operational skills, mild deficit in adaptive behavior) | Mildly impaired (can pronounce simple and complex sentences) | Autistic features |
| S.G. | F | 22.2 | Severe (acquisition of early visual/graphic representation skills, severe deficit in adaptive behavior) | Severely impaired (presents some vowel productions and few words) | Emotional–behavioral dysregulation |
| Category | Features |
|---|---|
| Craniofacial Features | - Thick eyebrows - Down-slanting palpebral fissures - Ocular hypertelorism - Low-set ears - Wide mouth with a thin upper lip |
| Neurodevelopmental Findings | - Global developmental delay - Intellectual disability - Language delay or Language absence - Behavioral abnormalities |
| Neuroimaging Abnormalities | - Cerebellar Hypoplasia (hypoplasia of the cerebellar vermis) - Mild ventriculomegaly - Hypoplastic corpus callosum - Abnormality of cerebral white matter |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, S.; Lopergolo, D.; De Falco, A.; Meossi, C.; Satolli, S.; Pasquariello, R.; Trovato, R.; Tessa, A.; Casalini, C.; Pfanner, L.; et al. Expanding the Clinical Spectrum Associated with the Recurrent Arg203Trp Variant in PACS1: An Italian Cohort Study. Genes 2025, 16, 227. https://doi.org/10.3390/genes16020227
Pagano S, Lopergolo D, De Falco A, Meossi C, Satolli S, Pasquariello R, Trovato R, Tessa A, Casalini C, Pfanner L, et al. Expanding the Clinical Spectrum Associated with the Recurrent Arg203Trp Variant in PACS1: An Italian Cohort Study. Genes. 2025; 16(2):227. https://doi.org/10.3390/genes16020227
Chicago/Turabian StylePagano, Stefano, Diego Lopergolo, Alessandro De Falco, Camilla Meossi, Sara Satolli, Rosa Pasquariello, Rosanna Trovato, Alessandra Tessa, Claudia Casalini, Lucia Pfanner, and et al. 2025. "Expanding the Clinical Spectrum Associated with the Recurrent Arg203Trp Variant in PACS1: An Italian Cohort Study" Genes 16, no. 2: 227. https://doi.org/10.3390/genes16020227
APA StylePagano, S., Lopergolo, D., De Falco, A., Meossi, C., Satolli, S., Pasquariello, R., Trovato, R., Tessa, A., Casalini, C., Pfanner, L., Astrea, G., Battini, R., & Santorelli, F. M. (2025). Expanding the Clinical Spectrum Associated with the Recurrent Arg203Trp Variant in PACS1: An Italian Cohort Study. Genes, 16(2), 227. https://doi.org/10.3390/genes16020227