Whole-Genome Bisulfite Sequencing Reveals a Role for DNA Methylation in Variants from Callus Culture of Pineapple (Ananas comosus L.)


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Construction and Sequencing of DNA Libraries
2.3. Data Filtering and Read Alignment
2.4. Analysis of Methylation Level
2.5. Differentially Methylated Regions (DMR) Analysis
3. Results
3.1. Generation of Pineapple Methylation Data
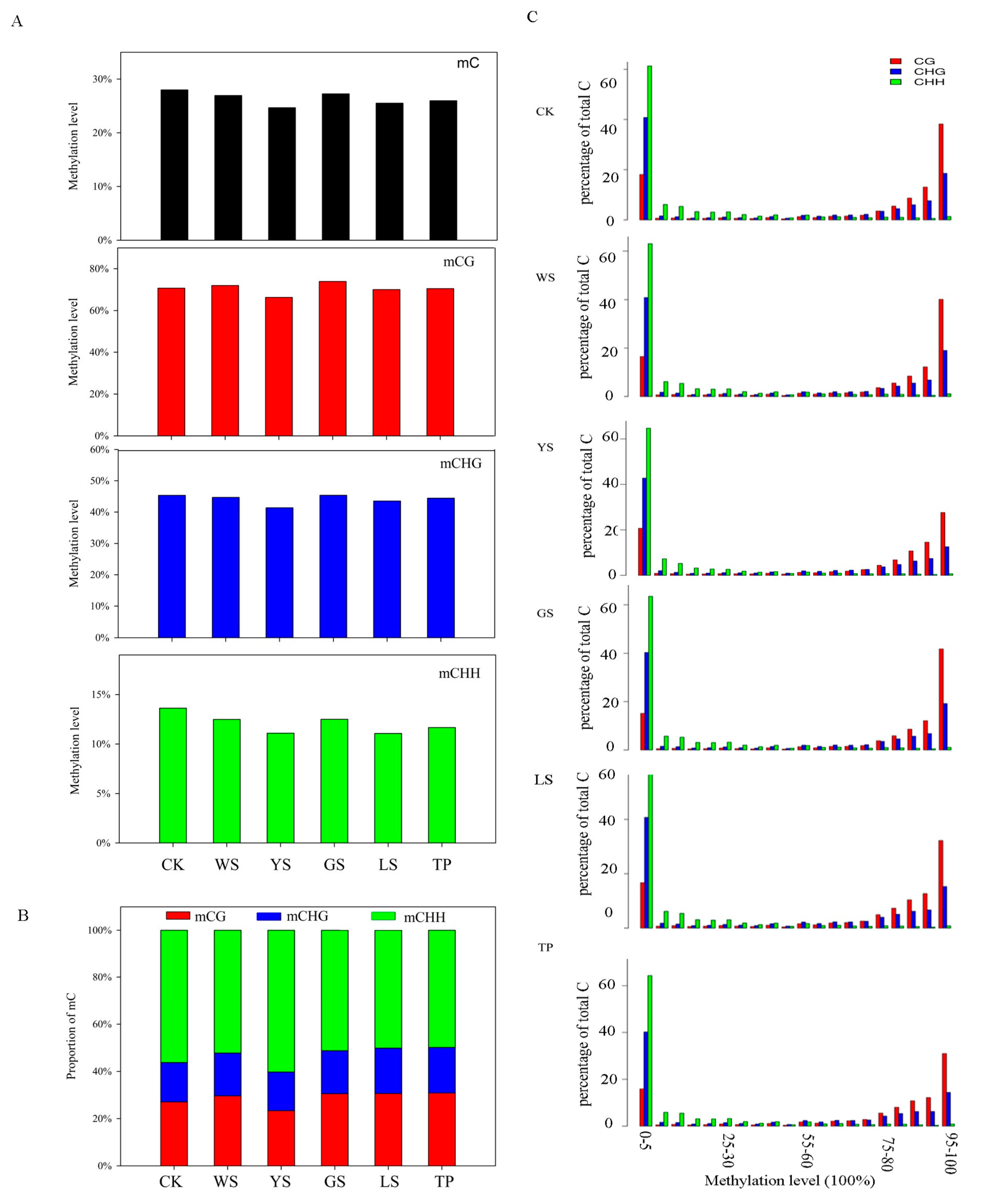
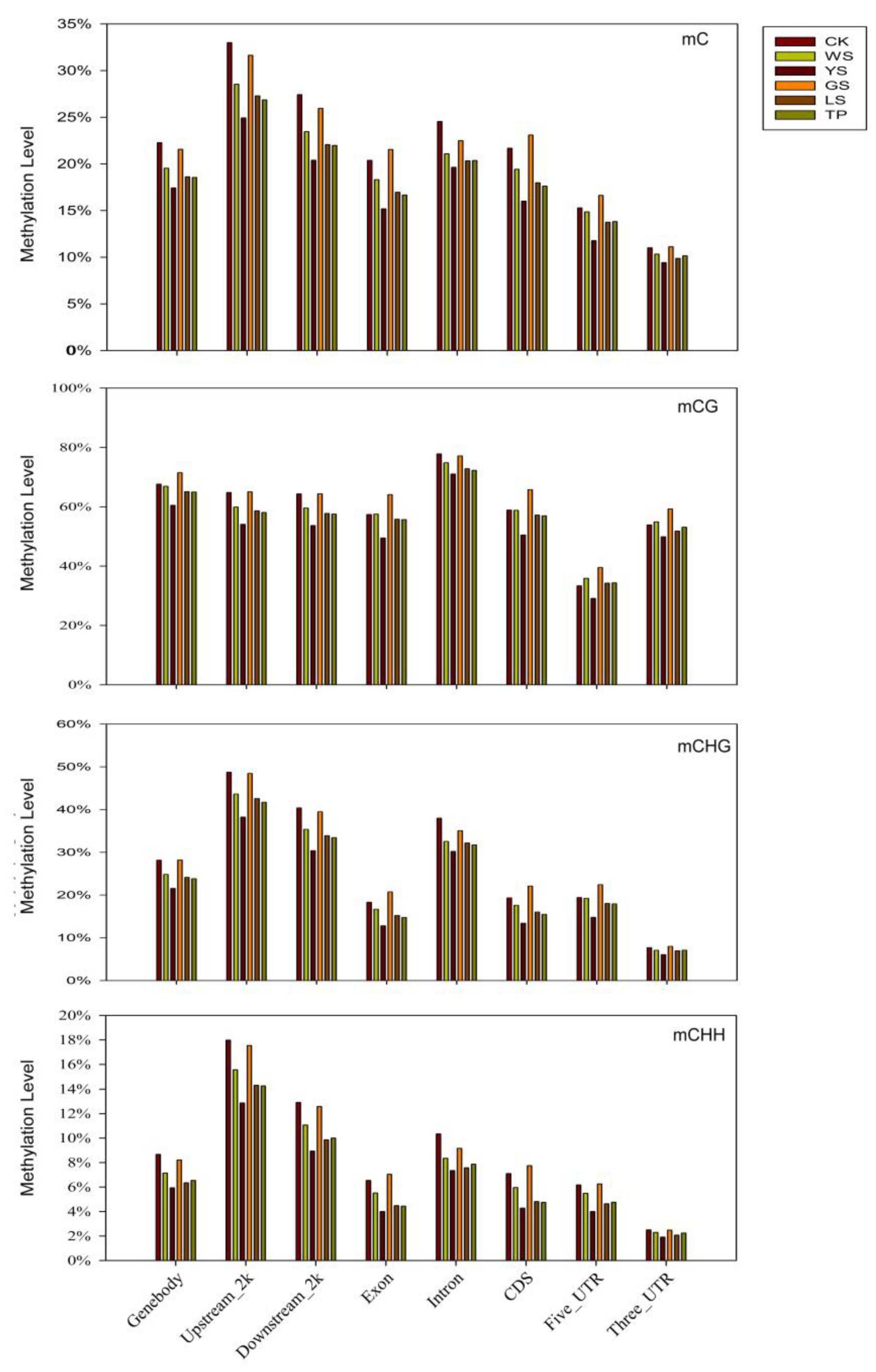
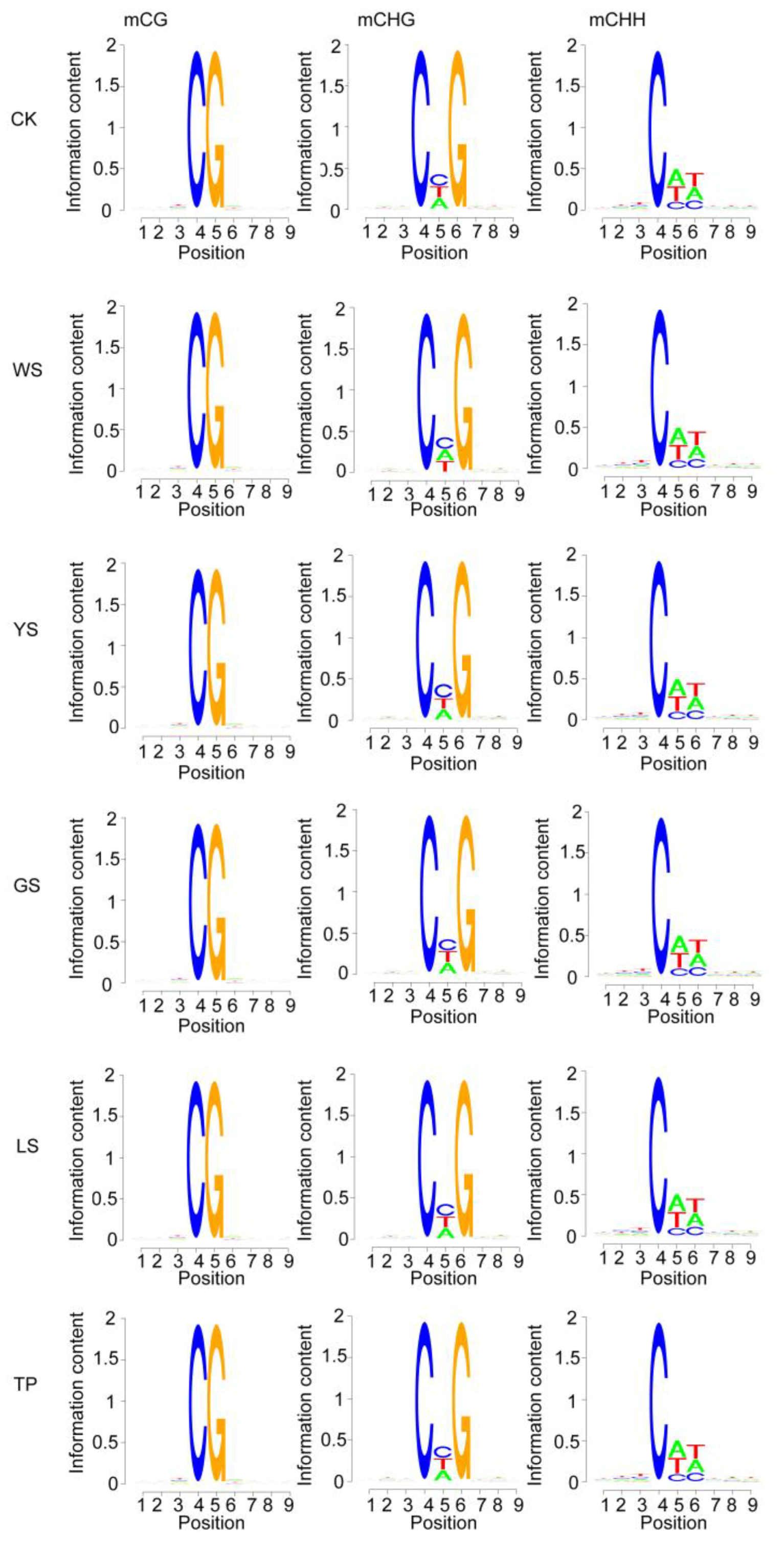
3.2. DNA Methylation Pattern Analysis
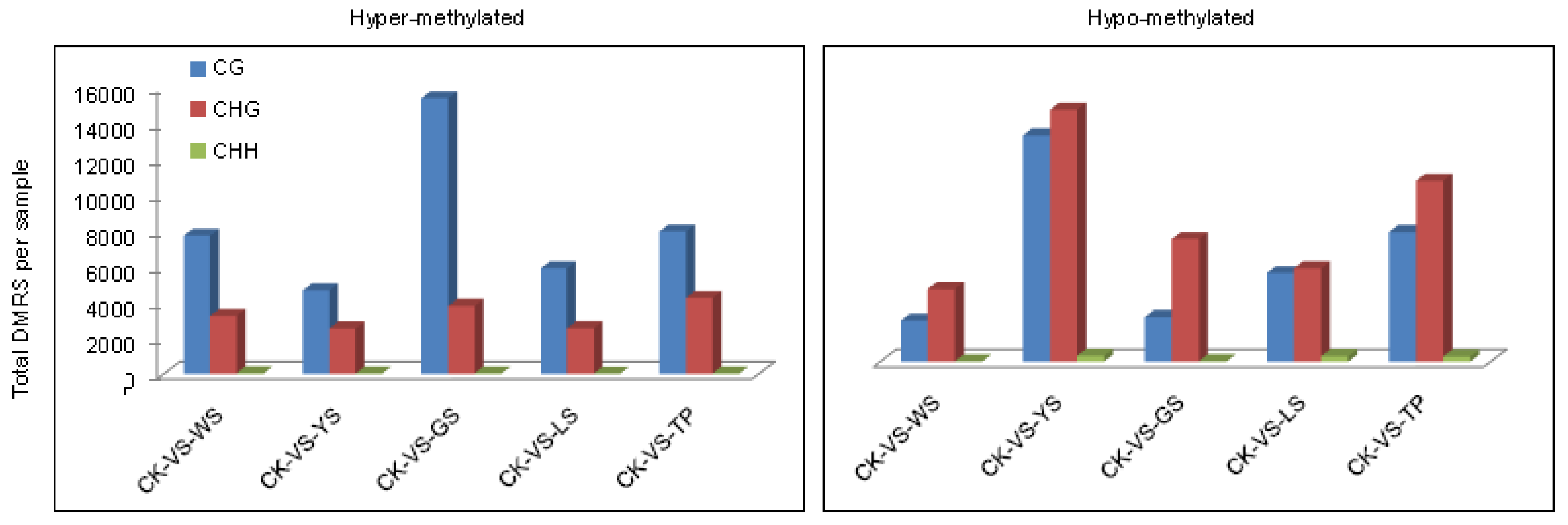
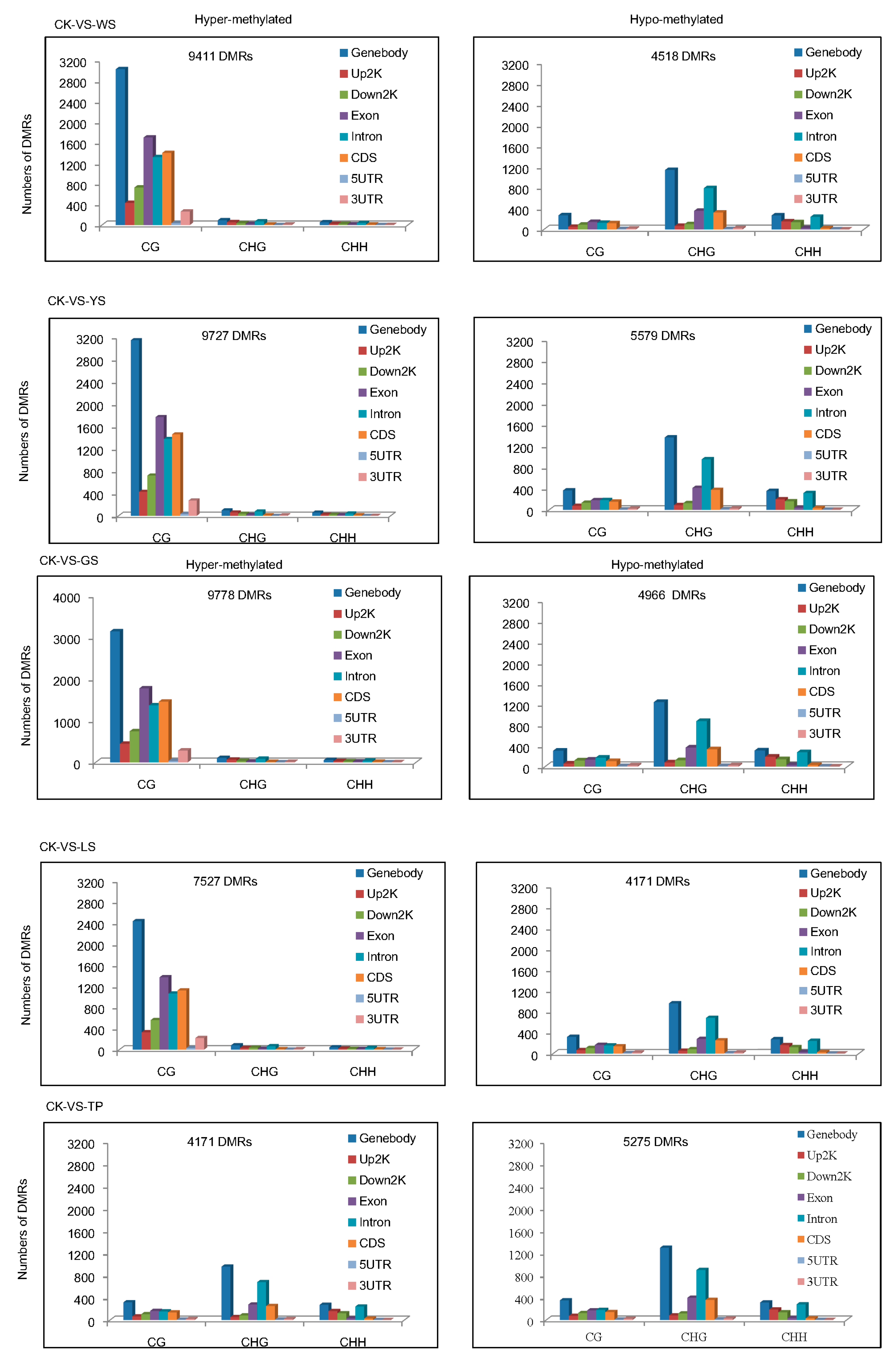
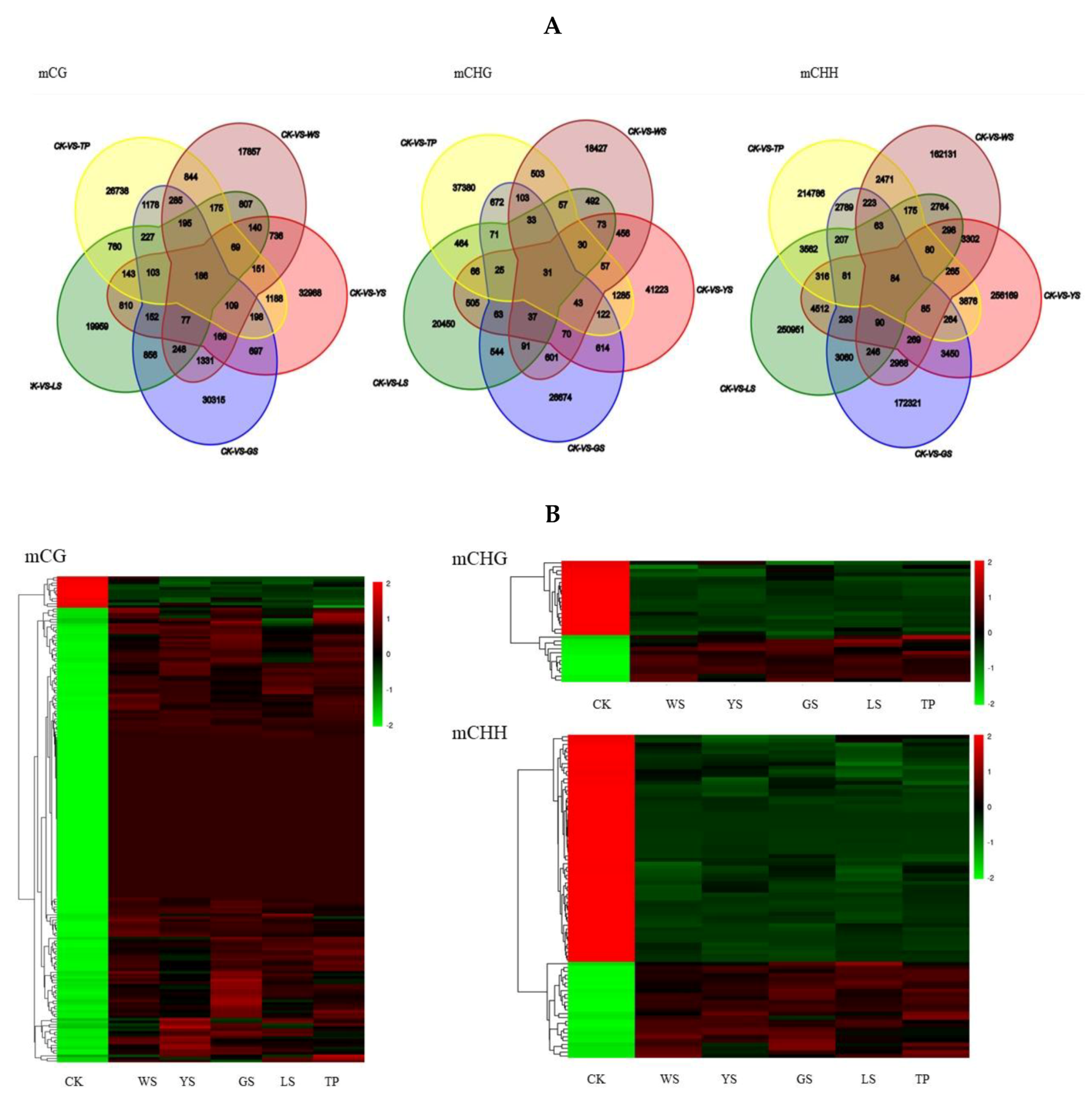
3.3. Analysis of Differentially Methylated Regions
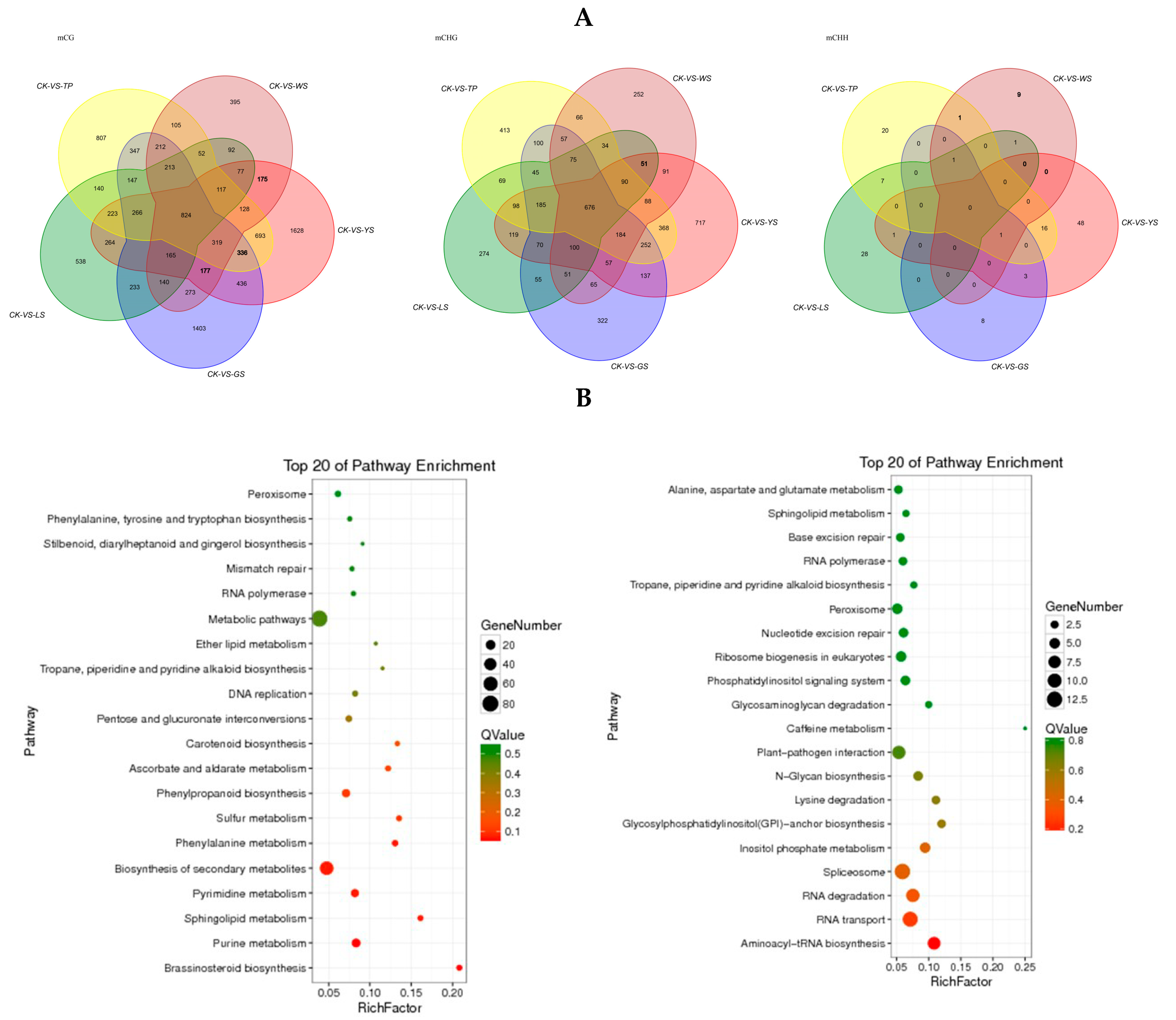
3.4. Enrichment Analysis of DMR-Related Genes
3.5. The Conserved DMR-Related Genes between the 5SV Plants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Papa, C.M.; Springer, N.M.; Muszynski, M.G.; Meeley, R.; Kaeppler, S.M. Maize Chromomethylase Zea methyltransferase2 Is Required for CpNpG Methylation. Plant Cell 2001, 13, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Quinga, L.A.P.; Fraga, H.P.D.F.; Vieira, L.D.N.; Guerra, M.P. Epigenetics of long-term somatic embryogenesis in Theobroma cacao L.: DNA methylation and recovery of embryogenic potential. Plant Cell Tissue Organ Cult. 2017, 131, 295–305. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [PubMed]
- Bardini, M.; Labra, M.; Winfield, M.; Sala, F. Antibiotic-induced DNA methylation changes in calluses of Arabidopsis thaliana. Plant Cell Tissue Organ Cult. 2003, 72, 157–162. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Boil. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Larkin, P.J.; Scowcroft, W.R. Somaclonal variation—A novel source of variability from cell cultures for plant improvement. Theor. Appl. Genet. 1981, 60, 197–214. [Google Scholar] [CrossRef]
- Jaligot, E.; Rival, A.; Beule, T.; Dussert, S.; Verdeil, J.-L. Somaclonal variation in oil palm (Elaeis guineensis Jacq.): The DNA methylation hypothesis. Plant Cell Rep. 2000, 19, 684–690. [Google Scholar] [CrossRef]
- Schellenbaum, P.; Mohler, V.; Wenzel, G.; Walter, B. Variation in DNA methylation patterns of grapevine somaclones (Vitis vinifera L.). BMC Plant Boil. 2008, 8, 78. [Google Scholar] [CrossRef]
- Kitimu, S.R.; Taylor, J.; March, T.J.; Tairo, F.; Wilkinson, M.J.; López, C.M.R. Meristem micropropagation of cassava (Manihot esculenta) evokes genome-wide changes in DNA methylation. Front. Plant Sci. 2015, 6, 00590. [Google Scholar] [CrossRef]
- Landey, R.B.; Cenci, A.; Guyot, R.; Bertrand, B.; Georget, F.; Dechamp, E.; Herrera, J.-C.; Aribi, J.; Lashermes, P.; Etienne, H. Assessment of genetic and epigenetic changes during cell culture ageing and relations with somaclonal variation in Coffea arabica. Plant Cell Tissue Organ Cult. 2015, 122, 517–531. [Google Scholar] [CrossRef]
- Machczyńska, J.; Orłowska, R.; Mańkowski, D.R.; Zimny, J.; Bednarek, P.T. DNA methylation changes in triticale due to in vitro culture plant regeneration and consecutive reproduction. Plant Cell Tissue Organ Cult. 2014, 119, 289–299. [Google Scholar] [CrossRef]
- González, A.I.; Saiz, A.; Acedo, A.; Ruiz, M.L.; Polanco, C. Analysis of genomic DNA methylation patterns in regenerated and control plants of rye (Secale cereale L.). Plant Growth Regul. 2013, 70, 227–236. [Google Scholar] [CrossRef]
- Meilina, O.; Ordway, J.M.; Jiang, N. Loss of Karma transposon methylation underliesthe mantled somaclonal variant of oil palm. Nature 2015. [Google Scholar] [CrossRef]
- Wakasa, K. Vaeiation in the plants differentiated from the tissue cuture of pineapple. Jpn. J. Breed. 1979, 29, 13–22. [Google Scholar] [CrossRef]
- Roostika, I.; Khumaida, N.; Ardie, S.W. RAPD Analysis to Detect Somaclonal Variation of Pineapple In Vitro Cultures during Micropropagation. J. BIOTROPIA 2015, 22, 109–119. [Google Scholar]
- Kohpaii, F.N.; Farahani, F.; Noormohammadi, Z. Somaclonal variation in the in vitro regenerated pineapple (Ananas comosus): Investigation of the cellular characteristics biochemical specificities and ISSR markers. Phytol. Balc. 2017, 23, 73–83. [Google Scholar]
- Soneji, J.R.; Rao, P.S.; Mhatre, M. Somaclonal variation in micropropagated dormant axillary buds of pineapple (Ananas comosus L., Merr.). J. Hortic. Sci. Biotechnol. 2002, 77, 28–32. [Google Scholar] [CrossRef]
- Pérez, G.; Yanez, E.; Mbogholi, A.; Valle, B.; Sagarra, F.; Yabor, L.; Aragon, C.; Gonzalez, J.; Isidron, M.; Lorenzo, J.C. New pineapple somaclonal variants: P3R5 and Dwarf. Am. J. Plant Sci. 2012, 3, 1–11. [Google Scholar] [CrossRef]
- Scherer, R.F.; Fraga, H.P.D.F.; Klabunde, G.F.; Da Silva, D.A.; Guerra, M.P. Global DNA Methylation Levels During the Development of Nodule Cluster Cultures and Assessment of Genetic Fidelity of In Vitro-Regenerated Pineapple Plants (Ananas comosus var. comosus). J. Plant Growth Regul. 2015, 34, 677–683. [Google Scholar] [CrossRef]
- Halim, N.A.A.; Tan, B.C.; Midin, M.R.; Madon, M.; Khalid, N.; Yaacob, J.S. Abscisic acid and salinity stress induced somaclonal variationand increased histone deacetylase (HDAC) activity in Ananas comosus var. MD2. Plant Cell Tissue Organ Cult. 2018, 133, 123–135. [Google Scholar] [CrossRef]
- Coronel, C.J.; González, A.I.; Ruiz, M.L.; Polanco, C. Analysis of somaclonal variation in transgenic and regenerated plants of arabidopsis thaliana using methylation related metaflp and tmd markers. Plant Cell Rep. 2018, 37, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Igamberdiev, A.U.; Debnath, S.C. Detection of DNA methylation pattern in thidiazuron-induced blueberry callus using methylation-sensitive amplification polymor-phism. Biol. Plant. 2017, 61, 511–519. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.X.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Kaeppler, S.M.; Phillips, R.L. Tissue culture-induced DNA methylation variation in maize. Proc. Natl. Acad. Sci. USA 1993, 90, 8773–8776. [Google Scholar] [CrossRef] [PubMed]
- Matthes, M.; Singh, R.; Cheah, S.-C.; Karp, A. Variation in oil palm (Elaeis guineensis Jacq.) tissue culture-derived regenerants revealed by AFLPs with methylation-sensitive enzymes. Theor. Appl. Genet. 2001, 102, 971–979. [Google Scholar] [CrossRef]
- Joyce, S.M.; Cassells, A.C. Variation in potato microplant morphology in vitro and DNA methylation. Plant Cell Tissue Organ Cult. 2002, 70, 125–137. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing; maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204. [Google Scholar] [CrossRef]
- Chakrabarty, D.; Yu, K.; Paek, K. Detection of DNA methylation changes during somatic embryogenesis of Siberian ginseng (Eleuterococcus senticosus). Plant Sci. 2003, 165, 61–68. [Google Scholar] [CrossRef]
- Francischini, J.; Kemper, E.; Costa, J.; Manechini, J.; Pinto, L. DNA methylation in sugarcane somaclonal variants assessed through methylation-sensitive amplified polymorphism. Genet. Mol. Res. 2017, 16. [Google Scholar] [CrossRef]
- Stroud, H.; Ding, B.; Simon, S.A.; Feng, S.; Bellizzi, M.; Pellegrini, M.; Wang, G.-L.; Meyers, B.C.; Jacobsen, S.E. Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.-M.; Gohain, M.; Allishe, A.; Huang, Y.-J.; Liao, J.-L.; Kuang, L.-Y.; Chen, P.-Y. Dynamics of the Methylome and Transcriptome during the Regeneration of Rice. Epigenomes 2018, 2, 14. [Google Scholar] [CrossRef]
- Han, Z.; Crisp, P.A.; Stelpflug, S.; Kaeppler, S.M.; Li, Q.; Springer, N.M. Heritable Epigenomic Changes to the Maize Methylome Resulting from Tissue Culture. Genetics 2018, 209, 983–995. [Google Scholar] [CrossRef] [PubMed]
- West, P.T.; Li, Q.; Ji, L.; Eichten, S.R.; Song, J.; Vaughn, M.W.; Schmitz, R.J.; Springer, N.M. Genomic Distribution of H3K9me2 and DNA Methylation in a Maize Genome. PLoS ONE 2014, 9, e105267. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Context of mC | Rare DMRs | Consistent DMRs | ||
|---|---|---|---|---|
| Hypermethylated | Hypomethylated | Hypermethylated | Hypomethylated | |
| CG | 36,475 | 29,657 | 4534 | 1366 |
| CHG | 15,085 | 40,480 | 1078 | 1890 |
| CHH | 122 | 994 | 2 | 8 |
| Total numbers | 51,682 | 71,131 | 5614 | 3264 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.; Xiao, X.; Zhang, H.; Li, Y.; Liu, S.; Sun, W.; Zhang, X.; Wu, Q. Whole-Genome Bisulfite Sequencing Reveals a Role for DNA Methylation in Variants from Callus Culture of Pineapple (Ananas comosus L.). Genes 2019, 10, 877. https://doi.org/10.3390/genes10110877
Lin W, Xiao X, Zhang H, Li Y, Liu S, Sun W, Zhang X, Wu Q. Whole-Genome Bisulfite Sequencing Reveals a Role for DNA Methylation in Variants from Callus Culture of Pineapple (Ananas comosus L.). Genes. 2019; 10(11):877. https://doi.org/10.3390/genes10110877
Chicago/Turabian StyleLin, Wenqiu, Xi’ou Xiao, Hongna Zhang, Yunhe Li, Shenghui Liu, Weisheng Sun, Xiumei Zhang, and Qingsong Wu. 2019. "Whole-Genome Bisulfite Sequencing Reveals a Role for DNA Methylation in Variants from Callus Culture of Pineapple (Ananas comosus L.)" Genes 10, no. 11: 877. https://doi.org/10.3390/genes10110877
APA StyleLin, W., Xiao, X., Zhang, H., Li, Y., Liu, S., Sun, W., Zhang, X., & Wu, Q. (2019). Whole-Genome Bisulfite Sequencing Reveals a Role for DNA Methylation in Variants from Callus Culture of Pineapple (Ananas comosus L.). Genes, 10(11), 877. https://doi.org/10.3390/genes10110877