Abstract
‘Fengtang’ plum is increasingly cultivated in southern China under mountain cultivation conditions, which involve lower temperatures, higher irradiance, and thinner soils. These conditions may uniquely shape the rhizosphere microbiome and influence tree health, yet their effects remain poorly understood. This study aimed to characterize the structural and taxonomic characteristics of the rhizosphere microbial community of ‘Fengtang’ plum grown under mountain cultivation conditions, and to identify key microbial groups associated with plant growth. We performed 16S rRNA and ITS high-throughput sequencing on rhizosphere and non-rhizosphere soil samples from ‘Fengtang’ plum orchards. Significant differences in α-diversity and β-diversity were observed between rhizosphere and non-rhizosphere communities. Dominant bacterial phyla included Pseudomonadota, Actinobacteriota, Acidobacteriota, and Chloroflexi; Ascomycota and Basidiomycota dominated in fungal communities. Dominant microbial groups were consistent across phylum, class, and order levels. Beneficial genera such as Streptomyces, Bacillus, and Rhizobium were enriched in the rhizosphere, and are considered putative core functional genera based on their known plant-growth-promoting traits. The microbial community in the rhizosphere shows distinct compositional patterns that may be linked to microecological balance. This study seeks to provide a comprehensive perspective for understanding the microbial communities associated with the plum tree rhizosphere.
1. Introduction
The ‘Fengtang’ plum (Prunus salicina Lindl. cv. ‘Fengtang’) is renowned for its honey-like sweetness. It is a high-quality plum variety indigenous to China, distinguished by its elevated sugar content and fine, juicy flesh. Originating from Guizhou Province, China, this variety has developed its unique characteristics within the region’s distinctive karst terrain environment and has been extensively promoted and cultivated in recent years. Despite the increasing market demand for ‘Fengtang’ plums, the industry is confronted with significant challenges, including issues such as obstacles to crop rotation, a decline in soil organic matter content in orchards, premature physiological fruit drop, and the prevalence of soil-borne diseases, all of which contribute to reduced fruit quality and yield [1,2,3]. Among these factors, an imbalance in the soil microecosystem is likely a key contributing factor [2].
The rhizosphere represents the most dynamic ecological niche at the plant–soil interface. Functioning as a “bridge” between plants and soil, the rhizosphere microbiome has emerged as a critical area of research for addressing cultivation challenges [4]. A robust rhizosphere microbial community is essential for sustaining soil health and creating an optimal soil environment conducive to the growth of fruit trees.
Research indicates that the rhizosphere microbiome affects host plants through multiple mechanisms [5]. Specifically, rhizosphere microorganisms are involved in the cycling and transformation of soil nutrients, thereby enhancing nutrient availability. These microorganisms can convert organic and inorganic substances into forms accessible to plants, thus facilitating plant growth. For instance, nitrogen-fixing bacteria and phosphorus-solubilizing bacteria transform atmospheric nitrogen and insoluble phosphorus into absorbable forms, thereby activating nutrients in host plants and satisfying the growth requirements of fruit trees [6,7]. In a study involving Prunus davidiana, inoculation with Bacillus strains YH-18 and YH-20 was shown to improve nutrient availability in the soil, while effectively maintaining microbial abundance and diversity for a duration of one month, thereby promoting the growth of Prunus davidiana [8].
Conversely, rhizosphere microorganisms contribute to the improvement of soil structure and enhance both soil aeration and water retention. Research indicates that within the rhizosphere of peach trees, specific microbial community structures are intricately linked to soil pH and potassium content, which are pivotal in sustaining the healthy growth of peach trees [9]. The antagonistic interactions of root-zone microorganisms with pathogens represent a key mechanism for safeguarding the health of fruit trees. Antagonistic strains suppress the growth and reproduction of pathogens by secreting substances such as antibiotics and enzymes [10]. For instance, certain bacteria, such as Burkholderia spp., isolated from the root zone of apple trees, exhibit antagonistic activity against the anthracnose pathogen, thereby delaying disease onset [11]. Volatile compounds produced by two Bacillus species isolated from the rhizosphere of peach trees demonstrate significant antagonistic activity against the brown rot pathogen, inhibiting its hyphal growth and causing cellular damage [12]. Additionally, Trichoderma strains isolated from the citrus tree rhizosphere exhibit effective antagonistic effects against post-harvest pathogens of citrus, offering potential as a partial replacement for chemical fungicides [13].
Moreover, empirical research has demonstrated that diverse bacterial strains can modulate host plant growth through hormonal pathways. For instance, bacteria that produce indole-3-acetic acid (IAA) facilitate root development [10], while strains that produce 1-aminocyclopropane-1-carboxylate (ACC) deaminase mitigate ethylene-induced stress [14]. Additionally, the inoculation of arbuscular mycorrhizal fungi has been shown to modify the composition of the rhizosphere bacterial community, promote vegetation recovery, and concurrently enhance soil chemical and biochemical properties, thereby increasing the availability of soil nutrients [15]. Arbuscular mycorrhizal fungi are also capable of inducing systemic resistance in host plants through the jasmonic acid (JA) signaling pathway, thereby augmenting their stress tolerance [16]. These findings underscore the pivotal role of root-zone microorganisms in the growth and health of fruit trees, as well as in maintaining soil health.
The rhizosphere microbial communities associated with different plant species exhibit significant variability, influenced by factors such as host genotype, root exudate composition, plant growth stage, climatic conditions, soil type, and management practices [17,18]. These variations in microbial communities are pivotal for disease resistance and environmental adaptability. Research on the rhizosphere microenvironment of eight common deciduous fruit trees in northern China revealed notable differences among tree species concerning microbial utilization, microbial diversity, enzyme activity, available minerals, and pH levels [19]. Specifically, in the rhizosphere of apple trees, the diversity of plant growth-promoting (PGP) bacteria is contingent upon the cultivation system employed [11]. The bacterial community structure in the apple tree rhizosphere undergoes gradual changes with increasing root diameter, and at a broader scale, soil heterogeneity contributes to variations in rhizosphere microbial communities among individual trees [20]. With prolonged cultivation, the diversity and structure of bacterial and fungal communities in the blueberry rhizosphere also evolve; soil pH is the primary driver shaping bacterial community structure, while available potassium is the primary driver shaping fungal community structure [21]. Therefore, optimizing the rhizosphere microbial community can enhance the yield and quality of fruit trees [22,23].
‘Fengtang’ plum is increasingly cultivated in mountainous regions of southern China due to its distinctive flavor and good adaptability to hilly terrains. However, mountain cultivation conditions—characterized by lower temperatures, higher UV irradiance, thinner topsoil, and slower organic matter turnover—impose unique environmental stresses on tree growth and root physiology. These conditions are likely to shape a distinct rhizosphere microbiome. Despite significant advancements in microbiome research concerning model plants and major crops, investigations into perennial woody fruit trees remain scarce; in particular, very little is known about the assembly, structure, and functional potential of the rhizosphere microbiome in plum trees. Lei et al. utilized a microbiome-based methodology to systematically examine the community structure and functional evolution of the ‘Fengtang’ plum’s endomicrobiome across various developmental stages [24].
Therefore, in this study, we conducted a comparative analysis of rhizosphere and non-rhizosphere samples from the ‘Fengtang’ plum tree, integrating high-throughput sequencing and bioinformatics techniques, to comprehensively characterize the rhizosphere microbiome of ‘Fengtang’ plum cultivated in mountainous karst terrains. The specific relevance of this study lies in: (i) elucidating the diversity and structural characteristics of ‘Fengtang’ plum rhizosphere microbiome through comparative analysis, identifying key biomarker taxa and core functional genera (e.g., plant-growth-promoting bacteria) that may contribute to tree health under mountain conditions, and (ii) exploring potential interaction patterns between key microbial groups in the ‘Fengtang’ plum rhizosphere and fruit tree growth, providing a baseline for the theoretical framework and future microbiome-assisted management of mountainous plum orchards.
2. Materials and Methods
2.1. Sample Collection
The sampling was conducted in Shuli Village, Xiasi Town, Kaili City, Qiandongnan Miao and Dong Autonomous Prefecture, Guizhou Province. The study area lies between approximately 26°29′–26°38′ N and 107°40′–107°50′ E, at an elevation of approximately 724 m above sea level. The ‘Fengtang’ plum trees were grown under typical mountain cultivation conditions, with terraced orchards on slopes ranging from approximately 5° to 25°.
Sample collection: We randomly selected healthy ‘Fengtang’ plum trees from the cultivation area. To minimize cross-contamination, we wiped all sampling tools with native soil collected near each sampling site. Based on the canopy projection zone (approximately 1–3 m from the trunk and 20–50 cm above ground), we identified the primary fine-root distribution area. After removing surface vegetation and debris, we gently loosened the soil with a hoe to expose fine roots (approximately 1–2 mm in diameter). The fine roots were cut using pruning shears and immediately placed into sterile sample bags. Each bag was labeled on site and stored in a biological sample collection box that had been pre-cooled with ice packs.
Collection of rhizosphere soil (RS) and non-rhizosphere soil (BS): In the laboratory, we transferred the roots into a laminar flow hood and gently shook off the bulk soil, which we collected as BS samples. The soil that remained tightly adhered to the roots (approximately 1 mm thick) was defined as rhizosphere soil (RS). We then placed the root samples into sterile 50 mL centrifuge tubes containing 20 mL of sterile 10 mM PBS solution and agitated the tubes at 120 rpm for 20 min at room temperature [25]. After removing the roots with sterile forceps, we centrifuged the suspension at 6000× g and 4 °C for 20 min to collect the RS. All soil samples were immediately flash-frozen in liquid nitrogen and stored at −80 °C until further processing.
Soil samples were systematically collected from nine ‘Fengtang’ plum trees, utilizing three sampling points per tree and obtaining three biological replicates at each sampling point, resulting in 27 samples of RS and 27 samples of BS, for a total of 54 samples.
2.2. High-Throughput 16S Ribosomal RNA and ITS Sequencing
Genomic DNA was extracted from samples utilizing the TGuide S96 Magnetic Soil/Stool DNA Kit (Tiangen Biotech, Beijing, China). The hypervariable V3-V4 region of the bacterial 16S rRNA gene was amplified using the primer pairs 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) [26]. For fungal analysis, the internal transcribed spacer (ITS) 1 region was amplified with the primer pairs F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and R (5′-GCTGCGTTCTTCATCGATGC-3′) [27]. The resulting PCR products were verified via agarose gel electrophoresis and subsequently purified using the Omega DNA purification kit (Omega Inc., Norcross, GA, USA). The purified PCR products were then subjected to paired-end sequencing (2 × 250 bp) on the Illumina NovaSeq 6000 platform(Illumina, Inc., San Diego, CA, USA).
2.3. Bioinformatic Analysis
Sequences with over 97% similarity were grouped into a single operational taxonomic unit (OTU) using USEARCH (v10.0). OTUs and amplicon sequence variants (ASVs) were taxonomically annotated using the Naive Bayes classifier in QIIME2 [28], referencing the SILVA database (release 138.1) [29] with a 70% confidence threshold. Alpha diversity was analyzed with QIIME2 to assess species diversity within samples, while beta diversity was evaluated using principal coordinate analysis (PCoA) for inter-sample diversity. A one-way analysis of variance was employed to compare bacterial abundance and diversity, and Linear Discriminant Analysis (LDA) in conjunction with effect size (LEfSe) was used to identify differentially abundant taxa.
Using the LEfSe method, co-occurrence network analyses and identification of differentially enriched microbial taxa were performed at the genus level, based on Spearman correlation with a threshold of |r| > 0.6 (p < 0.05). One-way ANOVA was used to assess significant differences in taxa relative abundance and diversity indices between groups, with significance set at p < 0.05.
3. Results
3.1. Amplicon Sequencing Results
The microbiome communities in the rhizosphere soil (RS) and bulk soil (BS) of ‘Fengtang’ plum were analyzed using high-throughput sequencing of 16S and ITS1 amplicons from extracted DNA. From 54 samples, 2,600,469 high-quality bacterial 16S rRNA reads (Supplementary Table S1) and 3,162,466 fungal ITS reads (Supplementary Table S2) were obtained via the PacBio Sequel platform, clustering into 100,580 bacterial OTUs (Supplementary Table S1) and 19,040 fungal ITS OTUs (Supplementary Table S2).
3.2. Bacterial and Fungal Community Diversity
Significant differences in alpha diversity were found in the rhizosphere and non-rhizosphere microbiome, encompassing bacterial community richness (ACE and Chao1 indices) and diversity (Shannon and Simpson indices) (Figure 1). However, fungal richness and diversity showed no significant variation between samples (Figure 1).
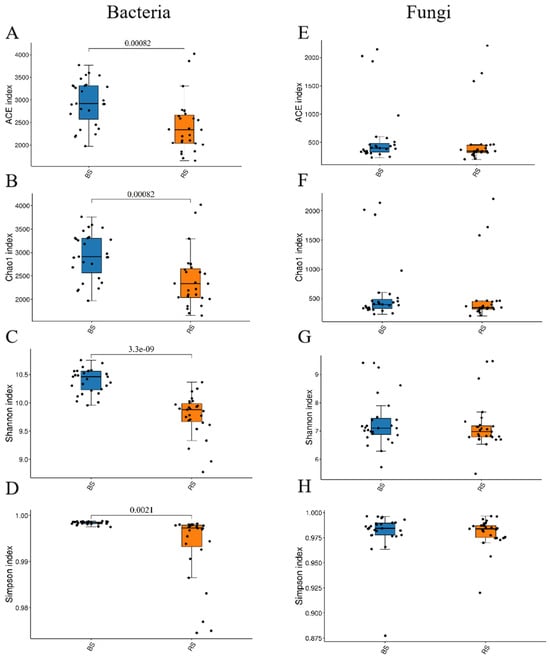
Figure 1.
Alpha diversity of bacterial (A–D) and fungal (E–H) communities in the RS and BS microbiomes of the ‘Fengtang’ plum. The Chao1 and ACE indices were employed to evaluate species richness, and the Shannon and Simpson indices were utilized to assess species diversity, p < 0.05. In the figure, blue represents the non-rhizosphere microbiome (BS), and red represents the rhizosphere microbiome (RS).
Venn diagrams illustrating the unique and shared ASVs among the different samples are presented in Figure 2. A total of 8411 (8.36%) bacterial ASVs and 2390 (12.55%) fungal ASVs were common to both RS and BS (Figure 2A,B). The number of unique bacterial (Figure 2A) and fungal (Figure 2B) ASVs of the BS microbiome is higher than that of the RS microbiome.
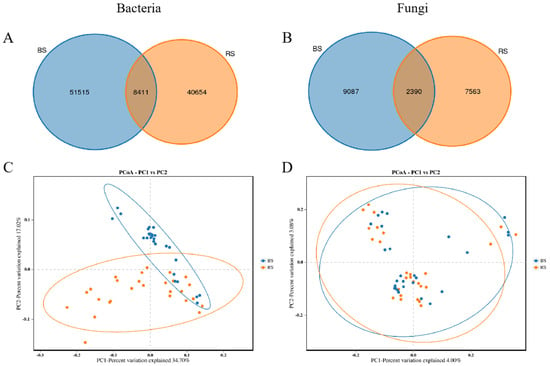
Figure 2.
The unique and shared ASVs of bacteria (A) and fungi (B) between the RS and BS microbiomes were analyzed using Bray–Curtis distances. PCoA was conducted to elucidate the bacterial (C) and fungal (D) community structures between the two groups, based on Bray–Curtis distance metrics. In the figure, blue represents the non-rhizosphere microbiome (BS), and red represents the rhizosphere microbiome (RS).
PCoA using Bray–Curtis dissimilarity was conducted to evaluate bacterial and fungal community variations across groups. Figure 2 shows significant differences in bacterial communities (Figure 2C) between RS and BS microbiomes, while fungal communities showed no significant differences (Figure 2D). Unweighted pair-group analysis results (Supplementary Figures S1 and S2) supported the PCoA findings.
3.3. Bacterial and Fungal Composition Diversity
A comprehensive analysis revealed the identification of 43 phyla, 119 classes, 363 orders, 788 families, and 1957 genera within the bacterial communities, as shown in Supplementary Table S1. Comparative analysis of the RS and BS samples demonstrated significant alterations in the taxonomic composition of the microbiomes. Notably, the relative abundance of 16 bacterial phyla exhibited significant differences between RS and BS, as illustrated in Figure 3.
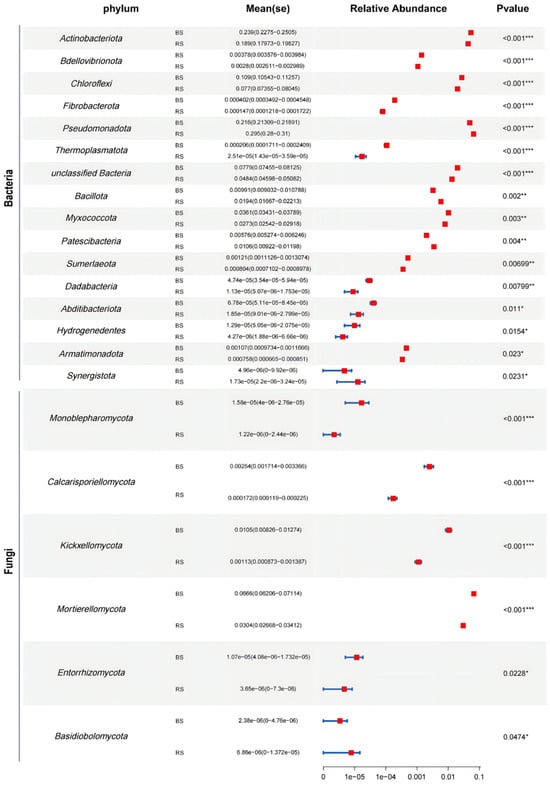
Figure 3.
Metastats statistical analysis of phyla of bacterial and fungal communities. Significance levels are denoted by asterisks: * p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001.
The relative abundances of Actinobacteriota, Bdellovibrionota, Chloroflexi, Fibrobacterota, Thermoplasmatota, Sumerlaeota, Dadabacteria, Abditibacteriota, Hydrogenedentes, Armatimonadota and Myxococcota significantly increased in BS, while the relative abundances of Pseudomonadota, Bacillota, Patescibacteria and Synergistota significantly increased in RS (Figure 3). The predominant bacterial phyla included Pseudomonadota, Acidobacteriota, Actinobacteriota and Chloroflexi, collectively relative abundance > 72.9% in each group (Figure 4A). At the genus level, the bacterial ASVs in the microbiomes were primarily classified into unclassified Vicinamibacterales, unclassified Vicinamibacteraceae, unclassified Gemmatimonadaceae, Paucibacter, unclassified Chloroflexi, Pseudolabrys and Gaiella (Figure 4C).
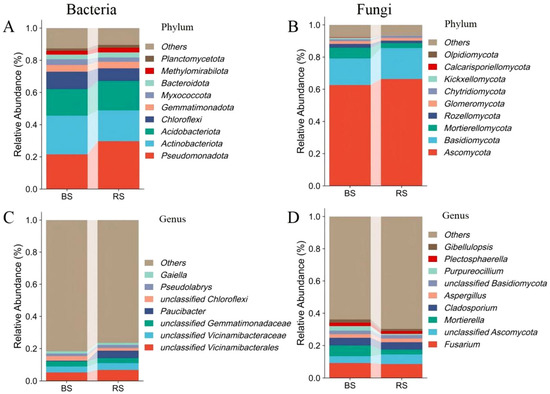
Figure 4.
The comparison of bacterial and fungal compositions between the rhizosphere and non-rhizosphere soil microbiomes at the phylum (A,B) and genus (C,D) levels.
In the fungal community, 18 phyla, 62 classes, 151 orders, 364 families, and 979 genera were identified, as documented in Supplementary Table S2. Among these, the relative abundance of 6 fungal phyla showed significant variation between RS and BS samples (Figure 3). Specifically, the relative abundances of Monoblepharomycota, Calcarisporiellomycota, Kickxellomycota, Mortierellomycota, and Entorrhizomycota were significantly elevated in BS, whereas Basidiobolomycota exhibited a significant increase in RS (Figure 3).
The dominant fungal phyla were Ascomycota, Basidiomycota and Mortierellomycota, collectively with relative abundance > 85.8% in each group (Figure 4B). At the genus level, most of the fungal ASVs were assigned to Fusarium, unclassified Ascomycota, Mortierella, Cladosporium, Aspergillus, unclassified Basidiomycota, Purpureocillium, Plectosphaerella and Gibellulopsis (Figure 4D).
3.4. Prediction of the Core Rhizosphere Microbiome of ‘Fengtang’ Plum
Through LEfSe analysis, we identified a total of 11 bacterial OTUs that were significantly different between the two groups (Figure 5). In the RS group, eight taxa were enriched, and the relative abundances of core rhizosphere microbiomes, which include plant growth-promoting rhizobacteria, are depicted in Figure 5, based on the putative functional prediction of bacterial communities. Notably, the relative abundances of Burkholderiales and Paucibacter significantly increased in the RS group, suggesting a stabilizing modulation of the rhizosphere microbiome in ‘Fengtang’ plum. However, independent experimental validation (e.g., qPCR or cultivation) is needed to confirm their status as stable core microbiome members.
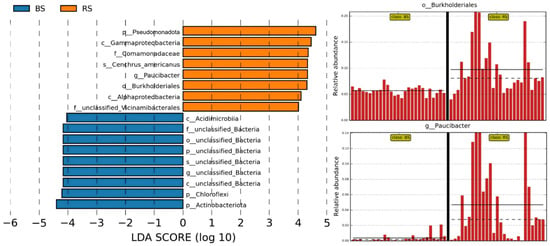
Figure 5.
LEfSe was performed to identify differentially abundant OTUs of bacteria, with an LDA threshold score of 3.0. The relative abundances of core rhizosphere microbiomes, those involved in plant growth-promoting rhizobacteria, were examined across different groups and categorized by “class.”. The solid and dashed lines identify the mean and median relative abundance values of the taxon in each group.
We conducted a detailed analysis to identify the differences in taxonomic composition between the RS and BS microbiomes. The findings revealed a significant increase in the relative abundances of the bacterial genera unclassified_Streptomycetaceae, Novosphingobium, Vicinamibacter, Rhizobium, and Paucibacter in RS rather than BS (Supplementary Figure S3).
A total of 8 significantly different fungal OTUs were found between the two groups by LEFSe analysis (Figure 6A). 2 taxa were enriched in the RS group, while the BS group exhibited enrichment of 6 taxa. The findings based on the detailed analysis of taxonomic composition revealed a significant increase in the relative abundances of the fungi genus Dactylonectria in RS rather than BS (Figure 6B). Conversely, the relative abundances of Mortierella were significantly increased in BS.
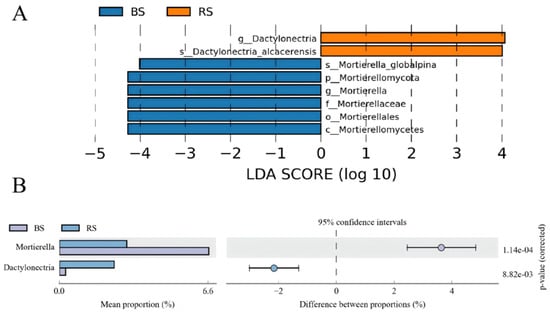
Figure 6.
LEfSe analysis was conducted to identify differentially abundant fungal OTUs (LDA threshold score 3.0) (A), and the abundance of differential fungal taxonomic compositions between RS and BS was quantified at the genus level using a two-sided t-test (B).
3.5. Correlation Analysis of the Rhizosphere Microbiome
The rhizosphere bacterial community of ‘Fengtang’ plum predominantly comprises nine major phyla. Among these, Pseudomonadota, Actinobacteriota, and Acidobacteria are the most prevalent and form the foundational structure of the rhizosphere microbial community (Figure 7). Additionally, other commonly observed bacterial groups include Chloroflexi, Firmicutes, and Gemmatimonadota. Key hub genera, such as Streptomyces, Steroidobacter, and Allorhizobium_Neorhizobium_Pararhizobium_Rhizobium, show the highest connectivity and appear to maintain extensive associations with other genera. Nitrogen-fixing rhizobia, such as Bradyrhizobium, are potentially involved in interactions with actinobacteria that may contribute to nitrogen cycling and plant growth. Furthermore, core genera with reported disease-suppression traits, such as Streptomyces and Bacillus, are potentially involved in suppressing pathogenic organisms, possibly through antibiotic production or nutrient competition. Concurrently, Chloroflexi (e.g., Kouleothrix) and Bacteroidota (e.g., unclassified Chitinophagaceae) show co-occurrence patterns that may be associated with cellulose and chitin degradation, potentially promoting organic matter turnover and contributing to plant defense mechanisms. Acidobacteria, such as Bryobacter, are oligotrophic organisms that may function as buffers during nutrient fluctuations, which could contribute to soil microecosystem stability. Additionally, taxa such as Gaiellales, Sphingomonas, and Arthrobacter display positive correlations with Acidobacteria, Actinobacteria, and Bacteroidetes, forming potentially important functional modules. The presence of unclassified connections suggests a substantial reservoir of unknown microbial resources within the rhizosphere. The compositional structure of the rhizosphere microbial community associated with the ‘Fengtang’ plum indicates high biodiversity and a complex potential for interactions.
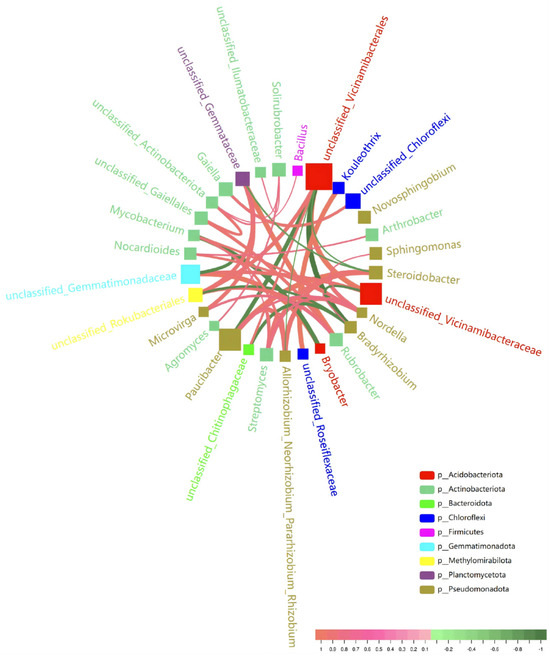
Figure 7.
The bacterial correlation network of the rhizosphere microbiome (top 30 genera). The size of the square nodes represents the abundance of each genus, while the color denotes the phylum classification. The thickness of the connecting lines illustrates the correlation strength, with red lines indicating positive correlations and green lines representing negative correlations.
The genus-level taxonomic units within the rhizosphere fungal interaction network are classified under the phyla Ascomycota, Basidiomycota, Mortierellomycota, Chytridiomycota, Glomeromycota, and unclassified fungi (Figure 8). Among these, Ascomycota and Basidiomycota are the predominant phyla, with Ascomycota being the most prevalent. Notable genera within the Ascomycota include Penicillium, Aspergillus, Fusarium, Alternaria, Clonostachys, Purpureocillium, and Talaromyces. The Basidiomycota is represented by genera such as Rhizoctonia, Trechispora, Leucoagaricus, Hannaella, Conocybe, and Agaricus. The interaction network suggests functional differentiation and ecological balance, although these interpretations are correlational. Penicillium and Aspergillus, as core saprophytic fungi, are potentially involved in organic matter degradation, and their metabolites may influence other groups. Mortierella, a representative of the Ascomycota, exhibits both saprophytic and auxiliary traits and may be potentially involved in root development, possibly through phosphorus solubilization or hormone secretion. Potential pathogens, including Fusarium, Rhizoctonia, and Alternaria, show negative co-occurrence patterns with biocontrol-associated fungi such as Clonostachys and Purpureocillium, which may be involved in maintaining a balance in disease suppression. Within Basidiomycota, taxa such as Hannaella and Agaricus are potentially involved in carbohydrate metabolism and humus transformation, while Leucoagaricus may contribute to lignin degradation. The presence of unclassified Ascomycota may indicate the existence of arbuscular mycorrhizal fungi, which could be involved in phosphorus acquisition through symbiotic associations.
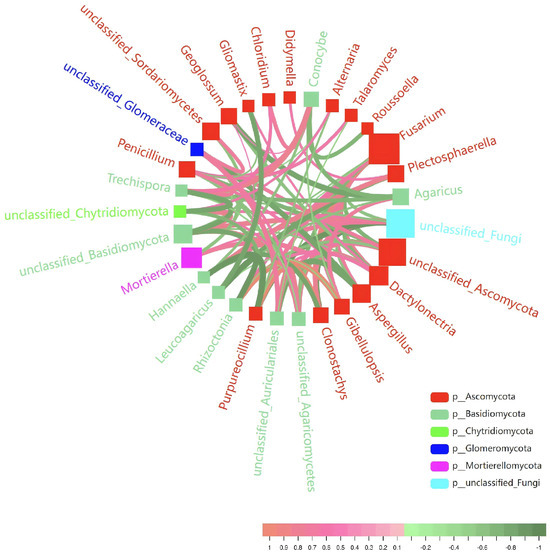
Figure 8.
The fungal correlation network of the rhizosphere microbiome (top 30 genera). The size of the square nodes represents the abundance of each genus, while the color denotes the phylum classification. The thickness of the connecting lines illustrates the correlation strength, with red lines indicating positive correlations and green lines representing negative correlations.
Collectively, the findings from the interaction network analysis indicate that the rhizosphere microbial community associated with ‘Fengtang’ plum sustains microecological balance in the rhizosphere through the differentiation and cooperation of functional groups. The prevalence of unclassified taxa highlights a rich cryptic diversity and the potential for discovering new species within the community, which may occupy critical ecological niches and function as buffers during periods of nutrient stress or environmental disturbances.
4. Discussion
The rhizosphere, serving as a distinctive ecological niche for plant–microbe interactions, promotes mutualistic relationships via root exudates that attract beneficial rhizosphere bacteria, while microorganisms simultaneously impact root growth and health [30]. The intricate interactions between plants and microorganisms are vital for sustaining the microecological equilibrium of the rhizosphere, highlighting the essential role of the rhizosphere microbial community in plant development and health. Recent advancements in high-throughput sequencing technologies have facilitated a more profound comprehension of the mechanisms through which rhizosphere microorganisms enhance plant growth and stress resilience.
This study demonstrated that, in comparison to rhizosphere soil, the non-rhizosphere soil associated with ‘Fengtang’ plum plants contained a greater number of unique bacterial and fungal ASVs, suggesting a higher level of complexity and species richness within the soil microbial community structure. Additionally, analyses of alpha and beta diversity revealed significant distinctions in the bacterial communities between the rhizosphere and non-rhizosphere soils of ‘Fengtang’ plum, with the non-rhizosphere soil displaying elevated alpha diversity, indicative of increased microbial species richness in the non-rhizosphere environment. This observed disparity in diversity is likely attributable to the distinctive microecosystem regulation and stability maintenance capabilities inherent to the rhizosphere. It is important to note that the composition and structure of microbial communities are influenced by various factors, including plant genotype, soil properties, agricultural practices, environmental conditions, and ecogeographic adaptability [31]. For example, research on the apple tree rhizosphere microbiome has demonstrated that different cultivation systems can significantly impact the diversity and functionality of rhizosphere microorganisms [11].
The primary bacterial phyla identified in the rhizosphere of the ‘Fengtang’ plum were Pseudomonadota, Acidobacteriota, Actinobacteriota, Chloroflexi, and Gemmatimonadota, with Pseudomonadota being the most prevalent. Pseudomonadota, known for its essential role in the rhizosphere of numerous plant species, encompasses Plant Growth-Promoting Rhizobacteria such as Pseudomonas and Burkholderia. These bacteria facilitate nitrogen fixation, phosphate solubilization, and siderophore production [32,33]. Actinobacteriota, particularly the genus Streptomyces, are instrumental in the suppression of soil pathogens [34,35]. Prior research has indicated that rhizosphere fungal communities are predominantly composed of Ascomycota and Basidiomycota, which are the most abundant fungal phyla in this environment [36,37].
The present study identifies Ascomycota, Basidiomycota, and Mortierellomycota as the dominant fungal phyla within the ‘Fengtang’ plum rhizosphere. This study demonstrates that the rhizosphere of the ‘Fengtang’ plum facilitates the enrichment of specific microbial communities by recruiting plant growth-promoting rhizobacteria, including Streptomyces, Rhizobium, and Bacillus. Isolates of Bacillus from the plum tree rhizosphere have been shown to enhance plant growth, improve acclimatization, increase tolerance to salt stress, and confer resistance to verticillium wilt disease [38]. Furthermore, rhizobacteria isolated from the plum rhizosphere have been reported to promote plant growth and exhibit antagonistic effects against fungal pathogens, significantly suppressing the growth of Fusarium oxysporum and Rhizoctonia solani [39]. Additionally, research indicates that Bacillus velezensis enhances the activity of resident rhizosphere Pseudomonas stutzeri through metabolic interactions, thereby promoting plant health [40].
To contextualize our findings, we compared them with previous studies on plum rhizosphere microbiomes. The resistant plum cultivar (Mihuang Plum) exhibited higher microbial diversity and richness compared with the susceptible cultivar (Pearl Plum), indicating that the microbiome is closely associated with host disease resistance [41]. The dominant bacterial phyla and fungal phyla are consistent with reports on other plum cultivars [38,42], suggesting a core microbiome shared across different Prunus species and regions. However, our study also revealed unique features: Paucibacter as a potential biomarker has not been reported in other plum rhizosphere studies, and the relative abundance of Rhizobium was markedly higher than that in lowland orchards. These distinctive patterns may reflect cultivar-specific recruitment and environmental selection under mountain conditions. Thus, while some microbial components are general for plum trees, the community composition is partly shaped by the genetic background of ‘Fengtang’ plum and the mountain habitat.
Plant roots exhibit a selective attraction towards specific microbial populations from the soil, a process characterized by successive waves of microbial colonization that culminate in the establishment of a stable rhizosphere microbial community [43]. Among the common genera of plant growth-promoting rhizobacteria are Burkholderia, Enterobacter, Streptomyces, Arthrobacter, and Pseudomonas, all of which are intricately linked to plant growth and stress resilience [32,33,44]. Studies have demonstrated that the application of Pseudomonas fluorescens to blackberries can enhance flavonoid content by modulating flavonoid metabolism, thereby improving the quality of the fruit [45]. Plant growth-promoting rhizobacteria can either compete with pathogens for scarce nutrients and space or induce systemic resistance in fruit trees, thereby bolstering their defense against pathogens [7]. The application of Streptomyces to passion fruit has been shown to increase the abundance of beneficial microorganisms, enhance the soil environment, and effectively manage Phytophthora blight [46]. The application of Bacillus or Pseudomonas to apple trees has been shown to modulate the rhizosphere microbial community, enhance phytoalexin levels in roots, and activate the plant immune system [47]. In response to pathogenic stress, fruit trees can actively recruit beneficial microorganisms through root exudates, thereby inhibiting pathogen proliferation [48]. Therefore, strategic regulation of the rhizosphere microbiome can enhance stress resilience of fruit trees and promote sustainable orchard management. Future research should establish a core microbiome database for the ‘Fengtang’ plum rhizosphere to identify beneficial microbes for developing bio-organic fertilizers and improving soil management.
Mountain cultivation conditions (lower temperature, higher diurnal variation, thinner soils, and slower organic matter turnover) are not merely geographical descriptors but key drivers of the rhizosphere microbiome assembly. The enrichment of cold-tolerant phyla (Actinobacteriota, Chloroflexi) likely results from low soil temperatures, as similar patterns are observed in high-altitude environments [49]. The higher abundance of PGPR (Bacillus, Streptomyces, Rhizobium) is consistent with adaptation to nutrient-poor and stress-prone mountain soils [50]. The functional differentiation and synergy among core genera in our co-occurrence network may reflect tighter microbial interactions under resource-limited conditions. Therefore, mountain cultivation conditions actively shape the rhizosphere microbial community and should be considered a critical factor when interpreting the results.
The LEfSe analysis and co-occurrence network analysis suggest that distinct microbial groups within the rhizosphere of the ‘Fengtang’ plum contribute to the connectivity and stability of rhizosphere network interactions by linking various functional groups through specific pathways. This microbial community is characterized by high biodiversity and a complex potential for interactions. Overall, the microbial community in the ‘Fengtang’ plum rhizosphere sustains microecological balance through the functional differentiation and synergy of core functional genera, supplemented by potential contributions from unidentified groups. This endows the ‘Fengtang’ plum rhizosphere with a stable ecosystem, marked by efficient nutrient utilization, multi-pathogen defense, and a buffering capacity for environmental adaptation. Extensive research on the rhizosphere microbiome shows that while its structure changes, the core microbiome remains abundant during plant growth, exhibiting strong root colonization, stress tolerance, and benefits to the host [51,52,53,54].
In summary, rhizosphere microorganisms are integral to the development of fruit trees. Recent advancements in various omics technologies offer novel insights and theoretical frameworks for pioneering research on fruit trees, facilitating a more comprehensive understanding of microbial functions and mechanisms. These technologies also allow for the exploration of the dynamics and regulatory mechanisms of microbial communities under diverse environmental conditions [55,56]. To optimize the use of rhizosphere microorganisms for enhancing agricultural productivity, it is imperative to improve crop stress resistance and yield through the engineering of microbial communities [57]. Such efforts will provide a scientific foundation for the effective management of the fruit tree rhizosphere environment and support the realization of sustainable agricultural production.
However, sampling was performed at only one time point (no seasonal replication) and one location, which limits the generalizability of our findings. Future studies with multi-season and multi-site sampling are needed to validate the spatiotemporal stability of the rhizosphere microbiome.
5. Conclusions
In this study, we characterized the rhizosphere and non-rhizosphere microbial communities of ‘Fengtang’ plum grown under mountain cultivation conditions using 16S rRNA and ITS sequencing. The results reveal significant differences in α-diversity and β-diversity between rhizosphere and non-rhizosphere soils, with distinct bacterial community structures. The rhizosphere of ‘Fengtang’ plum is enriched in several potentially beneficial bacterial genera, including Streptomyces, Rhizobium, Bacillus, and Burkholderia, suggesting that these taxa may serve as promising candidates for plant growth promotion in this cultivar. Although our compositional data do not allow direct inference of microbial interactions or functional activities, the observed enrichment of known plant-growth-promoting rhizobacteria provides a basis for future studies aimed at understanding the functional roles of the rhizosphere microbiome in tree nutrition and stress resistance. Overall, this study contributes foundational microbiological insights that can inform future research and soil management practices in fruit tree cultivation.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/agronomy16111084/s1, Figure S1: Unweighted pair-group analysis based on UniFrac distance for bacterial communities; Figure S2: Uweighted pair-group analysis based on UniFrac distance for fungal communities; Figure S3: Quantification of the abundance of differential bacterial taxonomic composition between the rhizosphere and non-rhizosphere soil microbiomes at the genus level; Table S1: Reads and OTUs for sequencing of the samples, and phyla, classes, orders, families, and genera identified within the bacterial communities; Table S2: Reads and OTUs for sequencing of the samples, and phyla, classes, orders, families, and genera identified within the fungal communities.
Author Contributions
Conceptualization, L.X. and Q.Y.; formal analysis, H.L.; investigation, X.P.; resources, T.L.; data curation, J.F.; writing—original draft preparation, L.X.; writing—review and editing, S.Z.; funding acquisition, Q.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This review paper was supported by Guizhou Provincial Science and Technology Projects (No. QKHJC [2025] Youth 250), the Specialized Fund for the Doctoral of Kaili University (grant No. BS20240216), the Guizhou Key Laboratory of Molecular Breeding for Characteristic Horticultural Crops [grant number Qiankehepingtai ZSYS (2025) 027], Guizhou Provincial Science and Technology Projects (Qiankehe foundation MS [2026] 722), and Provincial famous teacher Yang Qin studio (MSGZS SJ-2024002).
Data Availability Statement
The data presented in the study are deposited in the NCBI repository under accession number PRJNA1446224.
Conflicts of Interest
Tao Long is employed at Guizhou Duocaihui Agricultural Technology Co., Ltd. The remaining author declares that the research is conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Song, Q.; Wu, P.; Jiang, Y.; Chen, N.; Wang, L.; Li, Z.; Tang, B.; Chen, H.; Shen, X. PsGA2ox1-mediated gibberellin inactivation regulates hormonal reprogramming and cell wall remodelling during early fruit abscission in the ‘Fengtang’ plum. Plant Cell Physiol. 2026, pcag001. [Google Scholar] [CrossRef]
- Jiang, T.; Ren, J.; Li, D.; Luo, Y.; Huang, Y.; Gao, T.; Yang, J.; Yu, J.; Liu, L.; Yuan, H. Pseudomonas syringae exacerbates apple replant disease caused by Fusarium. Microbiol. Res. 2025, 296, 128124. [Google Scholar] [CrossRef]
- Benning, S.; Mahmoud, F.M.; Espindola-Hernandez, P.; Liu, B.; Pritsch, K.; Radl, V.; Winkler, J.B.; Winkelmann, T.; Beerhues, L.; Schloter, M. Inoculation of apple plantlets with Rhodococcus pseudokoreensis R79T enhances diversity and modulates the structure of bacterial rhizosphere communities in soil affected by apple replant disease. BMC Plant Biol. 2025, 25, 715. [Google Scholar] [CrossRef] [PubMed]
- Ge, A.-H.; Wang, E. Exploring the plant microbiome: A pathway to climate-smart crops. Cell 2025, 188, 1469–1485. [Google Scholar] [CrossRef]
- Yue, H.; Yue, W.; Jiao, S.; Kin, H.; Lee, Y.-H.; Wei, G.; Song, W.; Shu, D. Plant domestication shapes rhizosphere microbiome assembly and metabolic functions. Microbiome 2023, 11, 70. [Google Scholar] [CrossRef]
- Mus, F.; Crook, M.B.; Garcia, K.; Costas, A.G.; Geddes, B.A.; Kouri, E.D.; Paramasivan, P.; Ryu, M.-H.; Oldroyd, G.E.D.; Poole, P.S.; et al. Symbiotic nitrogen fixation and the challenges to its extension to nonlegumes. Appl. Environ. Microbiol. 2016, 82, 3698–3710. [Google Scholar] [CrossRef]
- Thepbandit, W.; Athinuwat, D. Rhizosphere Microorganisms Supply Availability of Soil Nutrients and Induce Plant Defense. Microorganisms 2024, 12, 558. [Google Scholar] [CrossRef]
- Shi, H.; Lu, L.; Ye, J.; Shi, L. Effects of Two Bacillus velezensis Microbial Inoculants on the Growth and Rhizosphere Soil Environment of Prunus davidiana. Int. J. Mol. Sci. 2022, 23, 13639. [Google Scholar] [CrossRef]
- Ren, F.; Dong, W.; Yan, D.-H. Organs, Cultivars, Soil, and Fruit Properties Affect Structure of Endophytic Mycobiota of Pinggu Peach Trees. Microorganisms 2019, 7, 322. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- dos Passos, J.F.M.; da Costa, P.B.; Costa, M.D.; Zaffari, G.R.; Nava, G.; Boneti, J.I.; de Oliveira, A.M.R.; Passaglia, L.M.P. Cultivable bacteria isolated from apple trees cultivated under different crop systems: Diversity and antagonistic activity against Colletotrichum gloeosporioides. Genet. Mol. Biol. 2014, 37, 560–572. [Google Scholar] [CrossRef]
- Liu, C.; Yin, X.; Wang, Q.; Peng, Y.; Ma, Y.; Liu, P.; Shi, J. Antagonistic activities of volatiles produced by two Bacillus strains against Monilinia fructicola in peach fruit. J. Sci. Food Agric. 2018, 98, 5756–5763. [Google Scholar] [CrossRef]
- Ferreira, F.V.; Herrmann-Andrade, A.M.; Calabrese, C.D.; Bello, F.; Vázquez, D.; Musumeci, M.A. Effectiveness of Trichoderma strains isolated from the rhizosphere of citrus tree to control Alternaria alternata, Colletotrichum gloeosporioides and Penicillium digitatum A21 resistant to pyrimethanil in post-harvest oranges (Citrus sinensis L. (Osbeck)). J. Appl. Microbiol. 2020, 129, 712–727. [Google Scholar] [CrossRef] [PubMed]
- Glick, B.R. Bacteria with ACC deaminase can promote plant growth and help to feed the world. Microbiol. Res. 2014, 169, 30–39. [Google Scholar] [CrossRef]
- Rodríguez-Caballero, G.; Caravaca, F.; Fernández-González, A.J.; Alguacil, M.M.; Fernández-López, M.; Roldán, A. Arbuscular mycorrhizal fungi inoculation mediated changes in rhizosphere bacterial community structure while promoting revegetation in a semiarid ecosystem. Sci. Total Environ. 2017, 584, 838–848. [Google Scholar] [CrossRef]
- Pieterse, C.M.J.; Zamioudis, C.; Berendsen, R.L.; Weller, D.M.; van Wees, S.C.M.; Bakker, P.A.H.M. Induced systemic resistance by beneficial microbes. Annu. Rev. Phytopathol. 2014, 52, 347–375. [Google Scholar] [CrossRef]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.K.M.; Wang, M.M.; Zhou, X.; Zhang, A.-M.; Cai, L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Z.D.; Wang, E.T.; Sun, L.Q.; Li, Y. The Effect of Climate Variables, Soil Characteristics, and Peanut Cultivars on the Rhizobial Bacteria Community. Microorganisms 2025, 13, 926. [Google Scholar] [CrossRef]
- Si, P.; Shao, W.; Yu, H.; Yang, X.; Gao, D.; Qiao, X.; Wang, Z.; Wu, G. Rhizosphere Microenvironments of Eight Common Deciduous Fruit Trees Were Shaped by Microbes in Northern China. Front. Microbiol. 2018, 9, 03147. [Google Scholar] [CrossRef]
- Becker, M.; Hellmann, M.; Knief, C. Spatio-temporal variation in the root-associated microbiota of orchard-grown apple trees. Environ. Microbiome 2022, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Wu, Y.; Yang, H.; Wang, S.; Wu, W. robial community of blueberry. Front. Plant Sci. 2022, 13, 962759. [Google Scholar]
- Liu, M.; Xue, R.; Wang, D.; Hu, Y.; Gu, K.; Yang, L.; Zhao, J.; Guan, S.; Su, J.; Jiang, Y. Variations in different preceding crops on the soil environment, bacterial community richness and diversity of tobacco-planting soil. Front. Microbiol. 2024, 15, 1389751. [Google Scholar] [CrossRef]
- Zhang, Z.; Ge, S.B.; Fan, L.-C.; Guo, S.; Hu, Q.; Ahammed, G.J.; Yan, P.; Zhang, L.-P.; Li, Z.-Z.; Zhang, J.-Y.; et al. Diversity in rhizospheric microbial communities in tea varieties at different locations and tapping potential beneficial microorganisms. Front. Microbiol. 2022, 13, 1027444. [Google Scholar] [CrossRef]
- Lei, J.; Shi, Y.; Li, H.; Wang, R. Characterizing the Endophytic Microbiome and Microbial Functional Assemblages Associated with Fengtang Plum (Prunus salicina Lindl.) Development and Resistance. Horticulturae 2025, 11, 483. [Google Scholar] [CrossRef]
- Beckers, B.; Op De Beeck, M.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.B.; Luo, Q.Q.; Zhu, Y.; Ma, J.; Cao, L.; Yang, M.; He, X.L. Microbial diversity in two traditional bacterial douchi from Gansu province in northwest China using illumina sequencing. PLoS ONE 2018, 13, e0197527. [Google Scholar]
- Orgiazzi, A.; Lumini, E.; Nilsson, R.H.; Girlanda, M.; Vizzini, A.; Bonfante, P.; Bianciotto, V. Unravelling Soil Fungal Communities from Different Mediterranean Land-Use Backgrounds. PLoS ONE 2012, 7, e34847. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, 590–596. [Google Scholar] [CrossRef]
- Feng, H.; Fu, R.; Hou, X.; Lv, Y.; Zhang, N.; Liu, Y.; Xu, Z.; Miao, Y.; Krell, T.; Shen, Q.; et al. Chemotaxis of Beneficial Rhizobacteria to Root Exudates: The First Step towards Root–Microbe Rhizosphere Interactions. Int. J. Mol. Sci. 2021, 22, 6655. [Google Scholar] [CrossRef]
- Alegria Terrazas, R.; Balbirnie-Cumming, K.; Morris, J.; Pete, E.H.; Joanne, R.; Eric, P.; Elizabeth, M.B.; Eyal, F.; Davide, B. A footprint of plant eco-geographic adaptation on the composition of the barley rhizosphere bacterial microbiota. Sci. Rep. 2020, 10, 12916. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, C.; Sui, J.; Liu, Z.; Li, Q.; Ji, C.; Song, X.; Hu, Y.; Wang, C.; Sa, R.; et al. Isolation and characteriza tion of phosphofungi, and screening of their plant growth-promoting activities. AMB Express 2018, 8, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Godínez, L.J.; Cisneros-Saguilán, P.; Toscano-Santiago, D.D.; Santiago-López, Y.E.; Fonseca-Pérez, S.N.; Ruiz-Rivas, M.; Aguirre-Noyola, J.L.; García, G. Cultivable and Non-Cultivable Approach to Bacteria from Undisturbed Soil with Plant Growth-Promoting Capacity. Microorganisms 2025, 13, 909. [Google Scholar] [CrossRef] [PubMed]
- PalaniArul, P.; Kathithachalam, A.; Muthusamy, K.; Sankarasubramanian, H.; Sivaprakasam, N.; Rangasamy, A.; Narayanan, M.B.; Mannu, J. Omics-driven perspectives elucidate the biocontrol mechanisms of actinobacteria against phytopathogens. Physiol. Mol. Plant Pathol. 2025, 140, 102970. [Google Scholar] [CrossRef]
- Maud, L.; Barakat, N.; Bornot, J.; Snini, S.P.; Mathieu, F. Biocontrol of Mycotoxigenic Fungi by Actinobacteria. J. Fungi 2025, 11, 4. [Google Scholar] [CrossRef]
- Coleman-Derr, D.; Desgarennes, D.; Fonseca-Garcia, C.; Gross, S.; Clingenpeel, S.; Woyke, T.; North, G.; Visel, A.; Partida-Martinez, L.P.; Tringe, S.G. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 2016, 209, 798–811. [Google Scholar] [CrossRef]
- Li, J.; Wu, X.; Lu, X.; Hou, D.; Liu, H.; Wang, Y.; Wu, L. Study on the Changes in the Microbial Community in Rhizosphere Soil of Blueberry Plants at Different Growth Stages. Agronomy 2024, 14, 2393. [Google Scholar] [CrossRef]
- Essalimi, B.; Esserti, S.; Rifai, L.A.; Koussa, T.; Makroum, K.; Belfaiza, M.; Rifai, S.; Venisse, J.S.; Faize, L.; Alburquerque, N.; et al. Enhancement of plant growth, acclimatization, salt stress tolerance and verticillium wilt disease resistance using plant growth-promoting rhizobacteria (PGPR) associated with plum trees (Prunus domestica). Sci. Hortic. 2022, 291, 110621. [Google Scholar] [CrossRef]
- Ali, I.; Sultan, T.; Subhan, F.; Haleem, K.S.; Sultana, N.; Tauseef, I. PGPRs of plum (Prunus domestica) rhizosphere enhance plant growth and antagonise fungal activity in vitro. Acta Agric. Scand. B Soil Plant Sci. 2018, 68, 367–378. [Google Scholar]
- Sun, X.; Xu, Z.; Xie, J.; Hesselberg-Thomsen, V.; Tan, T.; Zheng, D.; Strube, M.L.; Dragoš, A.; Shen, Q.; Zhang, R.; et al. Bacillus velezensis stimulates resident rhizosphere Pseudomonas stutzeri for plant health through metabolic interactions. ISME J. 2022, 16, 774–787. [Google Scholar] [CrossRef]
- Zhou, X.; Wei, Y.; Zhu, Y.; Li, J.; Zhou, R.; Xiao, Q.; Luo, R.; Yang, S. Integrated microbiome and metabolome approaches reveal the resistant mechanisms of leaf blight resistant plum cultivar. Chem. Biol. Technol. Agric. 2025, 12, 60. [Google Scholar] [CrossRef]
- Qian, Q.; Wang, Z.; Chen, G.; Zhang, J.; Xu, D.; Ali, H.; Wang, X. Comparison of root and inter-root soil microbial communities of plants infected with crown gall disease of Yinhong plum (Prunus salicina Lindl.) based on metagenomes. Physiol. Mol. Plant Pathol. 2025, 140, 102874. [Google Scholar] [CrossRef]
- Fracchia, F.; Mangeot-Peter, L.; Jacquot, L.; Martin, F.; Veneault-Fourrey, C.; Deveau, A. Colonization of Naive Roots from Populus tremula × alba Involves Successive Waves of Fungi and Bacteria with Different Trophic Abilities. Appl. Environ. Microbiol. 2021, 87, e02541-20. [Google Scholar] [CrossRef]
- Tian, L.; Lin, X.; Tian, J.; Ji, L.; Chen, Y.; Tran, L.-S.P.; Tian, C. Research Advances of Beneficial Microbiota Associated with Crop Plants. Int. J. Mol. Sci. 2020, 21, 1792. [Google Scholar] [CrossRef]
- Garcia-Seco, D.; Zhang, Y.; Gutierrez-Mañero, F.J.; Martin, C.; Ramos-Solano, B. Application of Pseudomonas fluorescens to Blackberry under Field Conditions Improves Fruit Quality by Modifying Flavonoid Metabolism. PLoS ONE 2015, 10, e0142639. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Sung, K.-Y.; Tuan, S.-J.; Huang, J.-W.; Lin, Y.-H.; Huang, T.-P. A Streptomyces Agent for Biocontrol of Phytophthora Blight and Its Modulation of Rhizosphere Microbiomes in Passion Fruit. Plant Dis. 2026, 110, 133–143. [Google Scholar] [CrossRef]
- Hauschild, K.; Orth, N.; Liu, B.; Giongo, A.; Gschwendtner, S.; Beerhues, L.; Schloter, M.; Vetterlein, D.; Winkelmann, T.; Smalla, K. Rhizosphere competent inoculants modulate the apple root–associated microbiome and plant phytoalexins. Appl. Microbiol. Biotechnol. 2024, 108, 344. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Muñoz, L.A.; Sánchez-García, M.E.; Calderón-Pérez, B.; De la Torre-Almaraz, R.; Ruiz-Medrano, R.; Xoconostle-Cázares, B. Metagenomic Analysis of Rhizospheric Bacterial Community of Citrus Trees Expressing Phloem-Directed Antimicrobials. Microb. Ecol. 2024, 87, 93. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.N. Microbes adapted to cold and their use as biofertilizers for mountainous regions. In Advances in Plant Microbiome and Sustainable Agriculture; Elsevier: Amsterdam, The Netherlands, 2020; Chapter 8; pp. 167–186. [Google Scholar]
- Singh, M.; Jayant, K.; Singh, D.; Suyal, D.C.; Mitra, A.; Bhutani, S. Microbial adaptations at higher altitude for sustainable development: A review. J. Appl. Pharm. Sci. 2023, 13, 1–9. [Google Scholar]
- Zhang, Y.; Wang, W.; Shen, Z.; Wang, J.; Chen, Y.; Wang, D.; Liu, G.; Han, M. Comparison and interpretation of characteristics of Rhizosphere microbiomes of three blueberry varieties. BMC Microbiol. 2021, 21, 30. [Google Scholar] [CrossRef]
- Xun, W.; Ren, Y.; Yan, H.; Ma, A.; Liu, Z.; Wang, L.; Zhang, N.; Xu, Z.; Miao, Y.; Feng, H.; et al. Sustained Inhibition of Maize Seed-Borne Fusarium Using a Bacillus-Dominated Rhizospheric Stable Core Microbiota with Unique Cooperative Patterns. Adv. Sci. 2023, 10, 2205215. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Yuan, X.; Yang, J.; Yang, Y.; Jv, H.; Li, R.; Jia, Z.; Ruan, Y. Selection of rhizosphere communities of diverse rotation crops reveals unique core microbiome associated with reduced banana Fusarium wilt disease. New Phytol. 2023, 238, 2194–2209. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, T.; Huang, Q.; Guo, H.; Zhang, H.; Xu, Q.; Shen, Q.; Ling, N. Core species impact plant health by enhancing soil microbial cooperation and network complexity during community coalescence. Soil Biol. Biochem. 2024, 188, 109231. [Google Scholar] [CrossRef]
- Jacoby, R.P.; Chen, L.; Schwier, M.; Koprivova, A.; Kopriva, S. Recent advances in the role of plant metabolites in shaping the root microbiome. F1000Research 2020, 9, 151. [Google Scholar] [CrossRef]
- Li, J.; Yan, G.; Duan, X.; Zhang, K.; Zhang, X.; Zhou, Y.; Wu, C.; Zhang, X.; Tan, S.; Hua, X.; et al. Research Progress and Trends in Metabolomics of Fruit Trees. Front. Plant Sci. 2022, 13, 881856. [Google Scholar] [CrossRef]
- Ashwani, K.; Anamika, D. Rhizosphere microbiome: Engineering bacterial competitiveness for enhancing crop production. J. Adv. Res. 2020, 24, 337–352. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.