
How Do the Surroundings of the C-NO2 Fragment Affect the Mechanical Sensitivity of Trinitroaromatic Molecules? Evidence from Crystal Structures and Ab Initio Calculations


Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Analysis of Crystal Structures of Trinitro Aromatic Molecules
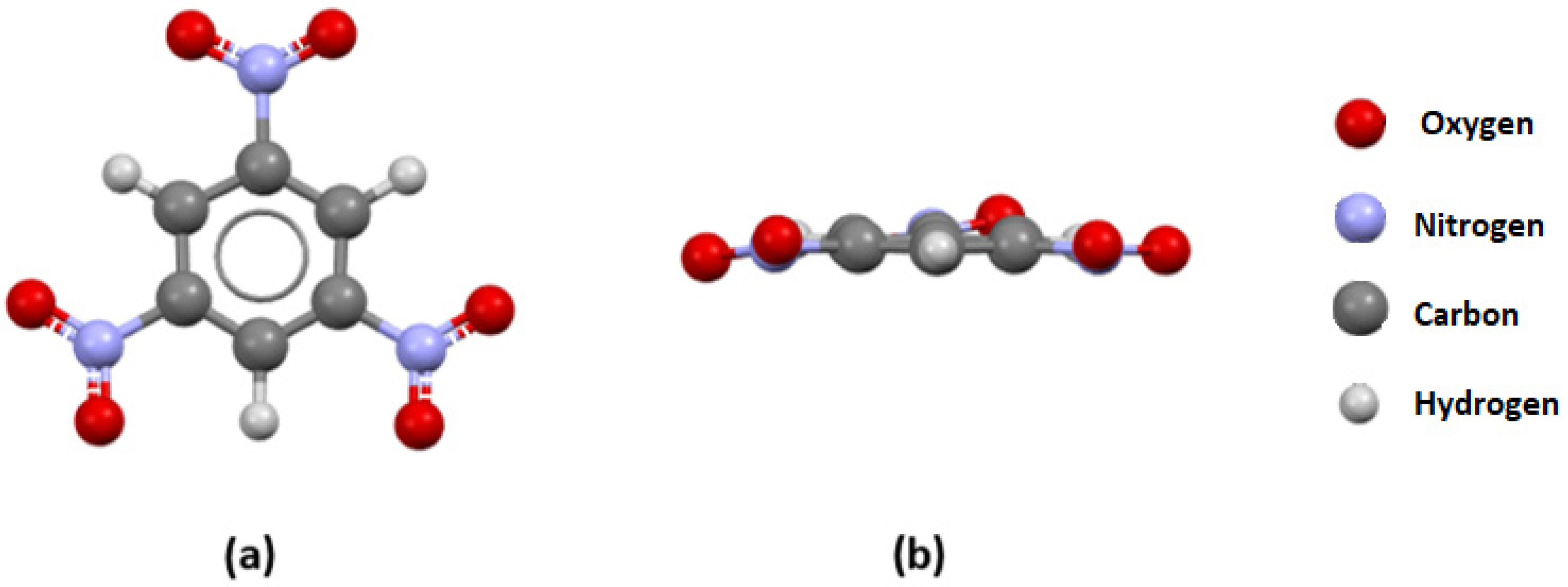
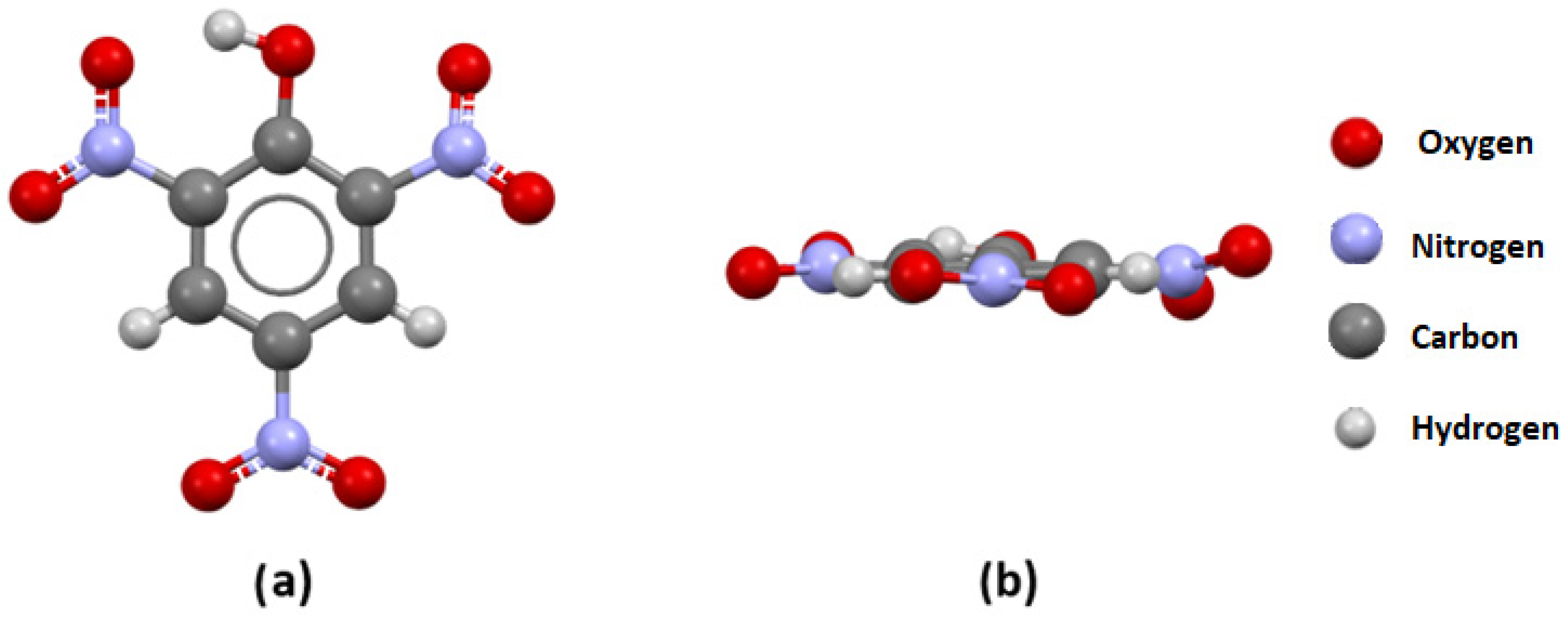
3.2. Visual Analysis of Crystal Structures
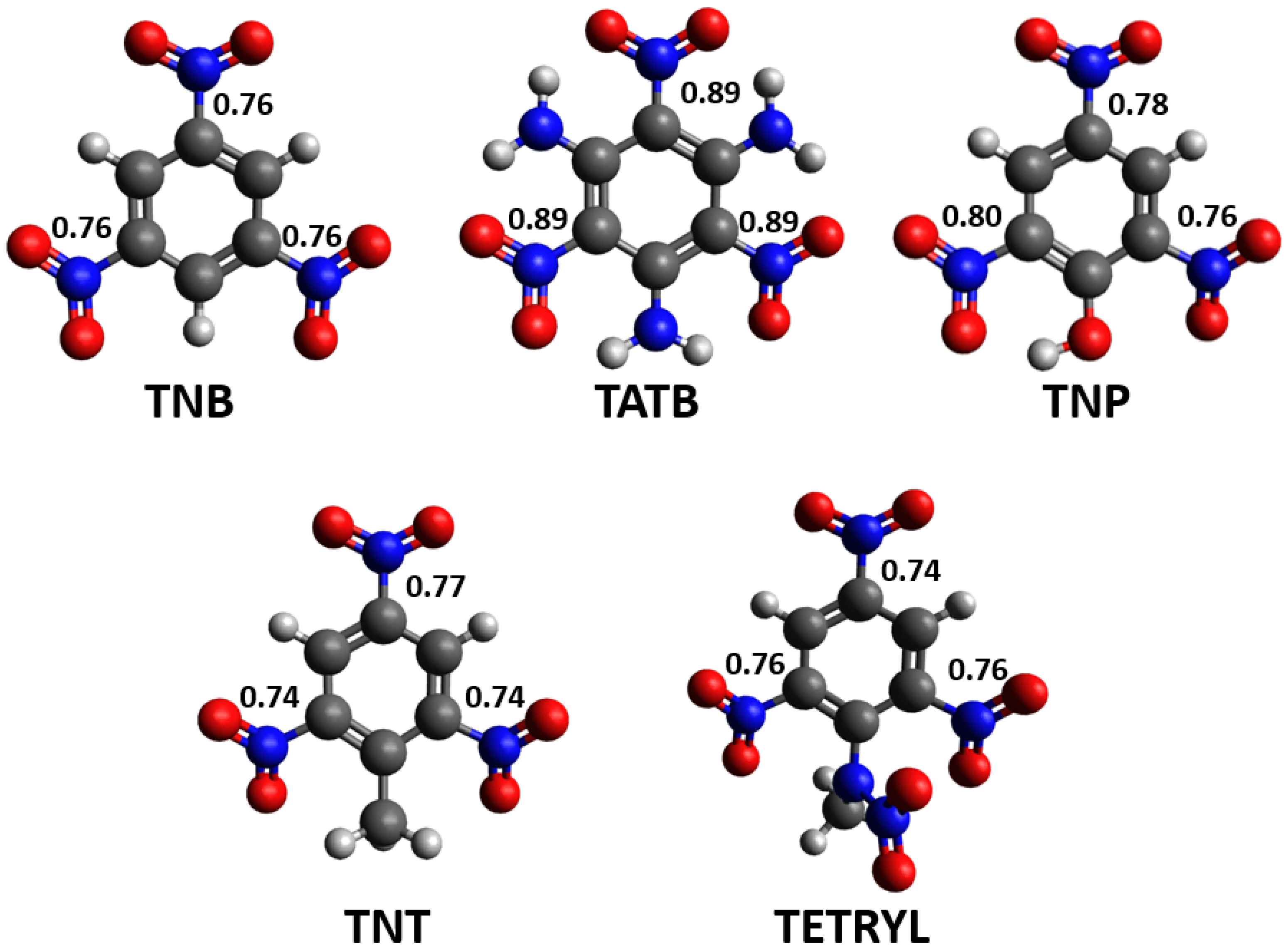
3.3. Density Functional Theory Calculations
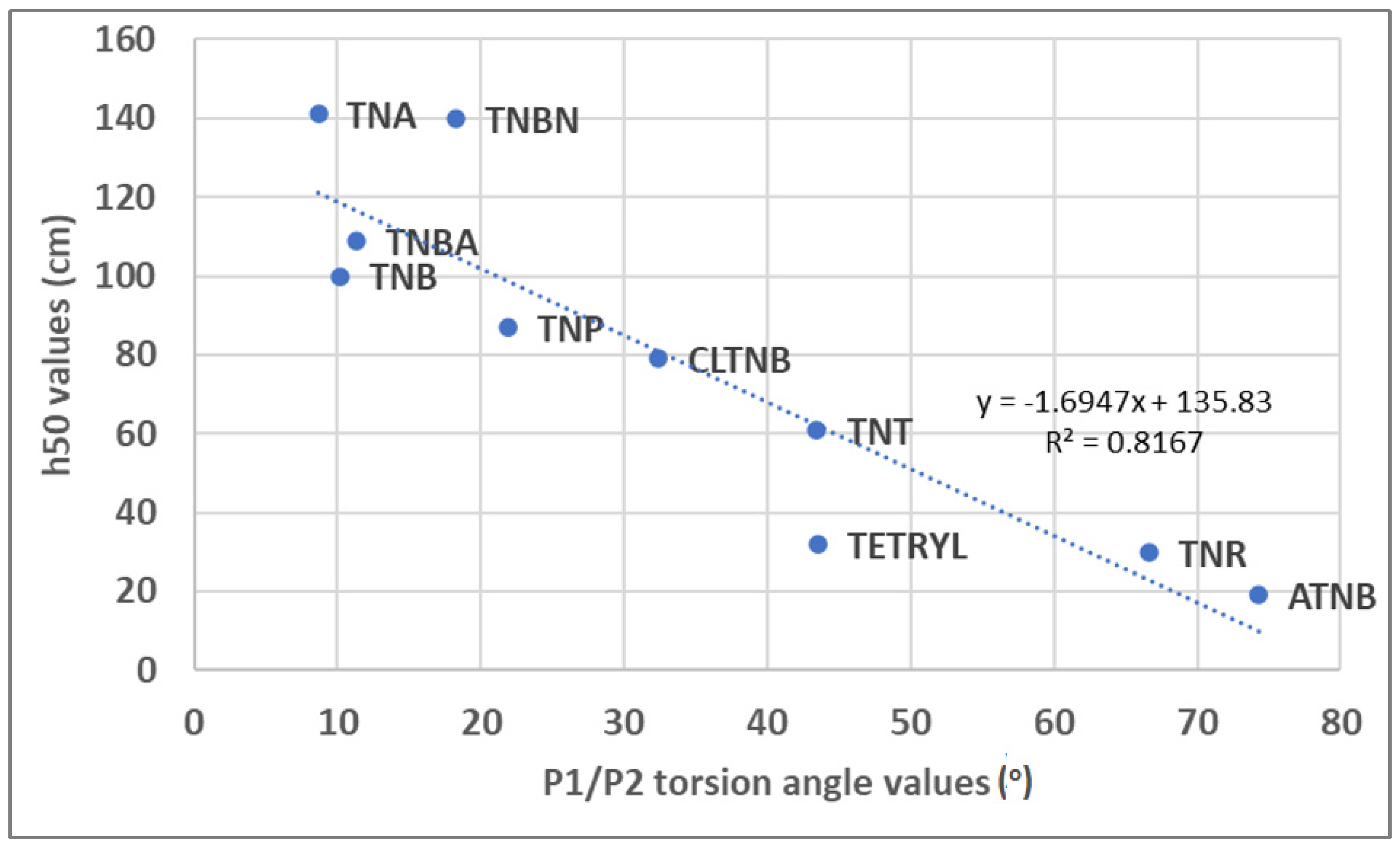
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Richardson, P.; Kitos, A.A.; Triglav, M.; Ovens, J.S.; Laroche, I.; Delisle, S.; Jolicoeur, B.; Brusso, J.L.; Murugesu, M. Synthesis and detonation performance of novel tetrazolyl–triazine nitrogen-rich energetic materials. Mater. Adv. 2023, 4, 5775–5784. [Google Scholar] [CrossRef]
- Born, M.; Plank, J.; Klapötke, T.M. Energetic Polymers: A Chance for Lightweight Reactive Structure Materials? Prop. Explos. Pyrotech. 2022, 47, e202100368. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J. Molecular dynamics simulations for the sensitivity and moisture adsorption on the surface of a novel cocrystal: CL-20/DNAN. J. Mol. Model. 2025, 31, 178. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, J.P.; Dodke, V.S. Some Novel High Energy Materials for Improved Performance. Z. Anorg. Allg. Chem. 2021, 647, 1856–1882. [Google Scholar] [CrossRef]
- Born, M.; Karaghiosoff, K.; Klapötke, T.M.; Voggenreiter, M. Oxetane Monomers Based on the Powerful Explosive LLM-116: Improved Performance, Insensitivity, and Thermostability. ChemPlusChem 2022, 87, e202200049. [Google Scholar] [CrossRef] [PubMed]
- Nešić, J.; Cvijetić, I.N.; Bogdanov, J.; Marinković, A. Synthesis and Characterization of Azido Esters as Green Energetic Plasticizers. Propellants Explos. Pyrotech. 2021, 46, 1537–1546. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Detonation Performance and Sensitivity: A Quest for Balance. In Advances in Quantum Chemistry; Academic Press: Cambridge, MA, USA, 2014; Volume 69, pp. 1–30. [Google Scholar]
- Politzer, P.; Murray, J.S. High Performance, Low Sensitivity: Conflicting or Compatible? Propellants Explos. Pyrotech. 2016, 41, 414–425. [Google Scholar] [CrossRef]
- Kretić, D.S.; Maslarević, M.I.; Veljković, D.Ž. Small Deviations in Geometries Affect Detonation Velocities and Pressures of Nitroaromatic Molecules. Organics 2025, 6, 17. [Google Scholar] [CrossRef]
- Kretić, D.S.; Veljković, I.S.; Veljković, D.Ž. Tris(3-nitropentane-2,4-dionato-κ2 O,O′) Complexes as a New Type of Highly Energetic Materials: Theoretical and Experimental Considerations. Chemistry 2023, 5, 1843–1854. [Google Scholar] [CrossRef]
- Shoaf, A.L.; Bayse, C.A. Trigger Bond Analysis of Nitroaromatic Energetic Materials Using Wiberg Bond Indices. J. Comput. Chem. 2018, 39, 1236–1248. [Google Scholar] [CrossRef]
- Aina, A.A.; Misquitta, A.J.; Phipps, M.J.S.; Price, S.L. Charge Distributions of Nitro Groups Within Organic Explosive Crystals: Effects on Sensitivity and Modeling. ACS Omega 2019, 4, 8614–8625. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wei, S.-H.; Zhang, C. Review of the Intermolecular Interactions in Energetic Molecular Cocrystals. Cryst. Growth Des. 2020, 20, 7065–7079. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualising crystal structures. Acta Crystallogr. Sect. B 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Kiselev, V.G. Comment on “Decomposition mechanisms of trinitroalkyl compounds: A theoretical study from aliphatic to aromatic nitro compounds”. Phys. Chem. Chem. Phys. 2015, 17, 10283–10284. [Google Scholar] [CrossRef]
- Storm, C.B.; Stine, J.R.; Kramer, J.F. Sensitivity Relationships in Energetic Materials. In Chemistry and Physics of Energetic Materials; NATO ASI Series; Bulusu, S.N., Ed.; Springer: Dordrecht, The Netherlands, 1990; Volume 309. [Google Scholar]
- Politzer, P.; Murray, J.S. Some molecular/crystalline factors that affect the sensitivities of energetic materials: Molecular surface electrostatic potentials, lattice free space and maximum heat of detonation per unit volume. J. Mol. Model 2015, 21, 25. [Google Scholar] [CrossRef]
- Rice, B.M.; Hare, J.J. A Quantum Mechanical Investigation of the Relation between Impact Sensitivity and the Charge Distribution in Energetic Molecules. J. Phys. Chem. A 2002, 106, 1770–1783. [Google Scholar] [CrossRef]
- Brill, T.B.; James, K.J. Thermal Decomposition of Energetic Materials. 61. Perfidy in the Amino-2,4,6-trinitrobenzene Series of Explosives. J. Phys. Chem. 1993, 97, 8152–8758. [Google Scholar] [CrossRef]
- Brill, T.B.; James, K.J. Kinetics and Mechanisms of Thermal Decomposition of Nitroaromatlc Explosives. Chem. Rev. 1999, 93, 2667–2692. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | BDE (Non-Corrected) 1 | BDE (ZPE Corrected) |
|---|---|---|
| o-iodonitrobenzene | 70.12 | 65.92 |
| m-iodonitrobenzene | 75.21 | 70.95 |
| p-iodonitrobenzene | 76.13 | 71.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretić, D.S.; Đunović, A.B.; Ninković, D.B.; Veljković, D.Ž. How Do the Surroundings of the C-NO2 Fragment Affect the Mechanical Sensitivity of Trinitroaromatic Molecules? Evidence from Crystal Structures and Ab Initio Calculations. Crystals 2025, 15, 692. https://doi.org/10.3390/cryst15080692
Kretić DS, Đunović AB, Ninković DB, Veljković DŽ. How Do the Surroundings of the C-NO2 Fragment Affect the Mechanical Sensitivity of Trinitroaromatic Molecules? Evidence from Crystal Structures and Ab Initio Calculations. Crystals. 2025; 15(8):692. https://doi.org/10.3390/cryst15080692
Chicago/Turabian StyleKretić, Danijela S., Aleksandra B. Đunović, Dragan B. Ninković, and Dušan Ž. Veljković. 2025. "How Do the Surroundings of the C-NO2 Fragment Affect the Mechanical Sensitivity of Trinitroaromatic Molecules? Evidence from Crystal Structures and Ab Initio Calculations" Crystals 15, no. 8: 692. https://doi.org/10.3390/cryst15080692
APA StyleKretić, D. S., Đunović, A. B., Ninković, D. B., & Veljković, D. Ž. (2025). How Do the Surroundings of the C-NO2 Fragment Affect the Mechanical Sensitivity of Trinitroaromatic Molecules? Evidence from Crystal Structures and Ab Initio Calculations. Crystals, 15(8), 692. https://doi.org/10.3390/cryst15080692