Abstract
Podocyte injury is a central event in the pathogenesis of diabetic nephropathy (DN). We conducted a systematic review across four major databases, identifying 7769 records and including 130 studies that met predefined eligibility criteria. Methodological quality was assessed with Joanna Briggs Institute tools, yielding a mean score of 81.3%, indicating overall moderate-to-high rigor despite design-contingent limitations. Publication activity was sparse until 2018 but increased markedly thereafter, with more than 80% of studies published between 2019 and 2025. Temporal analyses confirmed a strong positive trend (p = 0.86, p < 0.0001), reflecting the rapid expansion of this field. Study designs evolved from early human-only descriptions to integrated multi-model approaches combining human tissue, animal experiments, and in vitro systems, thus balancing clinical relevance with mechanistic exploration. Geographically, Asia emerged as the leading contributor, complemented by increasing multinational collaborations. Mechanistic synthesis highlighted five reproducible pillars of podocyte injury: slit-diaphragm and adhesion failure, mTOR–autophagy–ER stress disequilibrium, mitochondrial and lipid-driven oxidative injury, immune, complement, and inflammasome activation, and epigenetic and transcriptomic reprogramming. Collectively, these findings underscore a convergent mechanistic cascade driving podocyte dysfunction, while also providing a framework for therapeutic interventions aimed at restoring barrier integrity, metabolic balance, and immune regulation in DN.
1. Introduction
Podocytes are highly specialized epithelial cells that cover the external surface of glomerular capillaries along the glomerular basement membrane (GBM), forming the outer layer of the glomerular filtration barrier (GFB) and preserving its integrity through a complex cytoskeleton, intercellular junctions, and interdigitating foot processes [1]. Under pathological conditions such as diabetic nephropathy (DN), terminally differentiated podocytes may undergo epithelial–mesenchymal transition (EMT), a reprogramming linked to slit-diaphragm instability, loss of adhesion, cytoskeletal disorganization, and barrier failure [2,3]. Disruption of these structural and molecular elements impairs glomerular function and promotes proteinuria and renal disease progression, underscoring the central role of podocytes in GFB homeostasis [1].
Diabetes mellitus (DM) is expanding globally, and diabetic kidney disease (DKD/DN) remains one of its most common and costly complications, with prevalence estimates of ~30–50% among people with type 2 diabetes across studied populations and a substantial impact on morbidity, mortality, and health-care expenditure [4,5,6]. Clinically, older age, albuminuria, and reduced estimated glomerular filtration rate (eGFR) mark a trajectory from early podocyte stress to kidney failure, reinforcing the need for timely detection and intervention [5,6].
Histopathologically, DN features GBM thickening, mesangial expansion, and progressive glomerulosclerosis, with Kimmelstiel–Wilson nodules in advanced stages and tubulointerstitial injury comprising tubular atrophy, interstitial fibrosis, and arteriolosclerosis, all converging on loss of renal function [7,8]. Human and experimental evidence indicates that podocyte injury, foot-process effacement, slit-diaphragm disruption, and progressive depletion, emerges early and predicts the transition to albuminuria and eGFR decline, positioning the podocyte as both a mechanistic hub and a therapeutic target [1,9].
At the mechanistic level, chronic hyperglycemia activates interlocking metabolic, hemodynamic, and inflammatory pathways, including the polyol and hexosamine routes, protein kinase C, the renin–angiotensin–aldosterone system, and advanced glycation end products, that culminate in oxidative stress, mitochondrial dysfunction, cytoskeletal remodeling, and slit-diaphragm failure [10,11,12]. In podocytes, recurrent molecular targets include mTOR signaling, autophagy and endoplasmic-reticulum stress, and transcriptional axes such as TXNIP–mTOR and Wnt/β-catenin, alongside alterations of structural and adhesion proteins (nephrin/podocin, integrins), which together drive detachment, effacement, and apoptosis [3,13,14]. These pathways connect to clinical readouts through conventional biomarkers (albuminuria, eGFR) and, increasingly, through omics signatures that may refine risk stratification and support precision medicine [6,7,15].
Therapeutically, rigorous glycemic and blood-pressure control remains foundational, while sodium glucose cotransporter-2 inhibitors (SGLT2i) reduce albuminuria, modulate metabolic and hemodynamic stressors, and slow DN progression, with growing interest in combination strategies involving GLP-1 receptor agonists and mineralocorticoid receptor antagonists [6,16,17]. Nevertheless, the persistent global burden and interindividual heterogeneity highlight knowledge gaps in how systemic drivers integrate with intracellular hubs and organellar/structural targets within podocytes along the disease continuum [5,6].
Given the high prevalence and impact of DN in type 2 diabetes, the centrality of podocyte injury, and the multiplicity of implicated pathways, there is a need for an integrative synthesis that mechanistically organizes links between systemic drivers, signaling modules, and organellar/structural lesions in podocytes, and aligns these with histologic and clinical outcomes [5,6,12]. Accordingly, this study aims to systematize and integrate evidence on podocyte injury in DN, outlining a mechanistic framework that connects hyperglycemia and comorbid drivers to intracellular hubs and barrier/adhesion failure, with implications for biomarkers and therapeutic targeting in diabetic kidney disease [13,14,15,18].
Unlike previous reviews, this work uniquely integrates temporal, methodological, and geographical trends with mechanistic insights, providing a comprehensive framework to guide both experimental and translational research.
2. Results
We identified 7769 records across four databases (Cochrane Reviews, MEDLINE/PubMed, LILACS, and Embase). After removing 5502 duplicates, 2267 records were screened at title/abstract level, of which 2021 were excluded. We sought 246 full-text reports and were unable to retrieve 32; thus, 214 reports were assessed for eligibility. Of these, 84 were excluded for predefined reasons, no podocyte injury (n = 46), no diabetes mellitus (n = 3), or no mechanistic data (n = 35), yielding 130 studies included from databases. Searches via other sources (websites, organizations, and citation chasing) located 18 additional reports; 6 could not be retrieved and 12 were assessed in full. None met the inclusion criteria (most commonly due to lack of podocyte injury), so no further studies were added. The full selection pathway is presented in Figure 1.
Methodological quality and risk of bias were appraised using the Joanna Briggs Institute (JBI) Critical Appraisal Tools. Across the 130 included studies, the mean methodological quality was 81.25% (SD 18.84; CV 23.19%), with scores ranging from 31.25% to 100.00%. Five studies achieved a perfect 100%, whereas three scored below 50% (31.25–37.50). As expected for observational designs, the most frequently unmet items concerned the use of independent control groups, within-participant controls, and the explicit identification and management of confounding variables. Despite these design-contingent limitations, the overall evidence base exhibited moderate-to-high methodological quality and no indication of bias likely to overturn the principal findings (Table 1).

Table 1.
Assessment of methodological quality and risk of bias of eligible studies (jbi).

Figure 1.
Flowchart illustrating the selection process of eligible studies. Generated using the PRISMA2020 tool [145].

Here, we summarize the annual output of studies on podocyte injury in diabetes mellitus.
Between 2002 and 2018, yearly output was intermittent and low (≤4 studies/year), totaling 23 studies across 13 reporting years. From 2019 onward, the field expanded markedly: 6 studies in 2019 (4.62%), followed by a peak of 24 in 2020 (18.46%). Subsequent years remained high: 18 in 2021 (13.85%), 9 in 2022 (6.92%), 16 in 2023 (12.31%), 19 in 2024 (14.62%), and 15 in 2025 (11.54%). Overall, 2019–2025 accounts for 107 of 130 studies (82.3%). Year-over-year changes mirror this surge: +50.00% in 2019 (vs. 2018), +300.00% in 2020 (vs. 2019), followed by contractions in 2021 (−25.00%) and 2022 (−50.00%), then renewed growth in 2023 (+77.78%) and 2024 (+18.75%), with a decline in 2025 (−21.05%). These patterns are depicted in Figure 2.
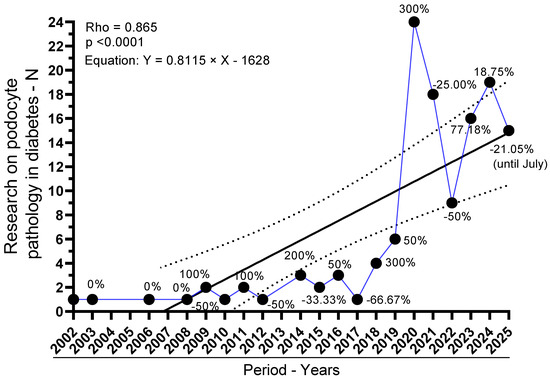
Figure 2.
Annual output of studies on podocyte injury in diabetes mellitus (2002–2025). Y-axis: number of studies per year; X-axis: publication year. Black circles mark yearly counts, linked by a blue line for visual continuity. The solid black line is the linear regression fit (Y = 0.8060 × Year − 1617; slope 0.8060, 95% CI 0.4861–1.126; R2 = 0.6089; F1,18 = 28.02; p < 0.0001). Dashed parallel lines depict the 95% confidence bands around the fitted line. A strong monotonic trend is also supported by Spearman’s p = 0.87 (95% CI, 0.6814–0.9479; p < 0.0001). Counts for 2025 reflect indexing through July only.
Trend analyses corroborate a strong temporal increase. The Spearman rank correlation between year and number of studies was ρ = 0.8652 (95% CI, 0.6773–0.9471; p < 0.0001), indicating a robust monotonic rise. Consistently, simple linear regression estimated an average increment of 0.81 studies per year (slope = 0.8115; 95% CI, 0.4581–1.165), with the model explaining 56.39% of the variance (R2 = 0.5639; F(1,18) = 23.28; p = 0.0001; residual SD = 5.084). The best-fit line was Y = 0.8115 × Year − 1628, reinforcing that publication volume has increased substantially over time. Collectively, these results indicate a pronounced and statistically significant acceleration of the field in the past decade.
Across all 130 eligible studies, human investigations were the single largest category (37/130; 28.5%), followed closely by in vitro + animal (28/130; 21.5%) and animal-only designs (27/130; 20.8%) (Figure 3A). Mixed-model designs were frequent: human + animal (10/130; 7.7%), human + in vitro + animal (10/130; 7.7%), and human + in vitro (8/130; 6.2%). In vitro-only studies were less common (7/130; 5.4%), while in silico appeared rarely (2/130; 1.5%); one report was unclassified (0.8%). Aggregating by component, any human element featured in 65/130 (50.0%), any animal element in 75/130 (57.7%), and any in vitro element in 53/130 (40.8%).
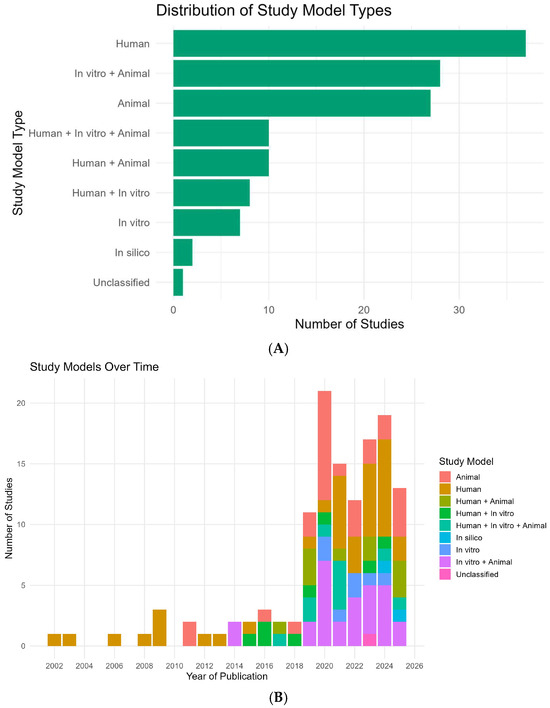
Figure 3.
Study models and their temporal distribution (2002–2025). (A) Horizontal bar chart summarizing the proportion and count of study models across all eligible records (N = 130). Bars are shown in green; labels at bar ends denote counts. Categories (n, %): Human (37, 28.5%), In vitro + Animal (28, 21.5%), Animal (27, 20.8%), Human + Animal (10, 7.7%), Human + In vitro + Animal (10, 7.7%), Human + In vitro (8, 6.2%), In vitro (7, 5.4%), In silico (2, 1.5%), Unclassified (1, 0.8%). (B) Stacked column chart showing, for each publication year, the number of studies stratified by model. The X-axis is the year (2002–2025) and the Y-axis is the number of studies. Colors encode the study models as indicated in the legend (Human, Animal, Human + Animal, Human + In vitro, Human + In vitro + Animal, In silico, In vitro, In vitro + Animal, Unclassified). The 2025 bar reflects records indexed through July only.
The temporal composition of study models (Figure 3B) shows three phases. Early phase (2002–2013): output was sparse and dominated by human-only designs (e.g., 2002, 2003, 2006, 2008, 2009, 2012, 2013 each 100% human); one exception was animal-only in 2011 (100%).
Diversification phase (2014–2018): mixed models emerge (e.g., in vitro + animal constitutes 100% in 2014; 2015–2018 introduce human + in vitro, human + animal, and human + in vitro + animal in varying proportions).
Expansion and consolidation (2019–2025): with the surge in publications (see Figure 2), portfolios broaden. In 2019, mixed designs predominate (72.7% of that year’s studies), led by human + animal (27.3%) and in vitro + animal/human + in vitro + animal (each 18.2%). In 2020, animal-only peaks (42.9%), with in vitro + animal close behind (33.3%), reflecting a shift toward mechanistic experimentation during the publication peak. The balance swings back toward human participation in 2021 (human-only 40.0%; human + in vitro + animal 26.7%) and remains mixed thereafter: 2022 distributes across in vitro + animal (33.3%), animal (25.0%), human (25.0%), in vitro (16.7%); 2023–2024 see renewed human-only leadership (35.3% and 42.1%, respectively) with persistent in vitro + animal contributions (23.5% and 26.3%). In silico analyses first appear in 2024 (5.3%) and rise modestly in 2025 (7.7%). In 2025 (data through July), animal-only (30.8%) and human + animal (23.1%) together comprise just over half of the yearly output, alongside in vitro + animal (15.4%), human-only (15.4%), and human + in vitro + animal (7.7%).
Together, these patterns indicate a clear evolution from early, single-modality human descriptions toward integrated, multi-model approaches that combine clinical relevance with mechanistic depth, a balance that has stabilized since 2019 while retaining year-to-year variability (Figure 3A,B).
The heatmap (Figure 4A) shows a clear continental gradient: Asia accounts for 60.0% of the corpus (78/130), followed by Europe 21.5% (28/130), North America 16.9% (22/130), and South America 1.5% (2/130). Within Asia, the most frequent designs were in vitro + animal (20) and human-only (19), with substantial use of animal-only (10) and mixed modalities, human + animal (8), human + in vitro + animal (9), and human + in vitro (6). In silico studies appeared exclusively in Asia (2), and the only unclassified record also originated there (1). Europe and North America emphasized human (10 and 7, respectively) and animal (9 and 8) designs; in vitro + animal was present in both (Europe 4; North America 4), whereas mixed three-arm designs were uncommon (Europe 1; North America 0). South America contributed two studies (human; human + animal).
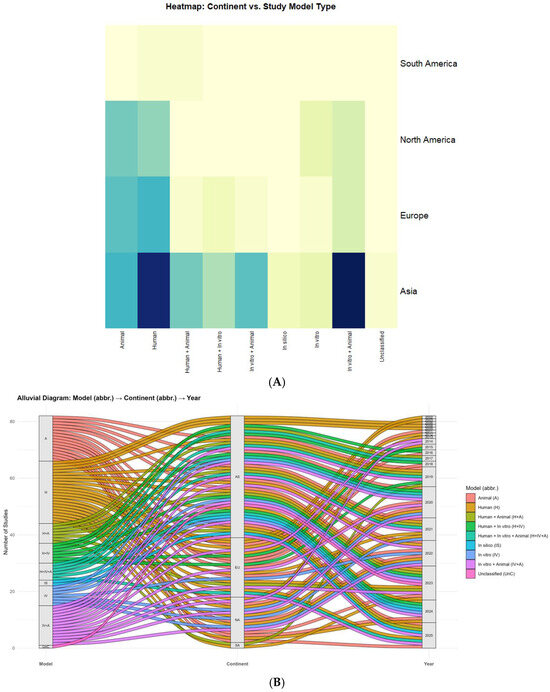
Figure 4.
Global distribution of study designs across continents and time. (A) Heatmap—Continent × study model. Matrix of absolute frequencies with rows (continents) and columns (model types); darker shades indicate a stronger association (i.e., higher frequency) between continent and model. Clustering is disabled and values are unscaled. (B) Alluvial—Model → Continent → Year. Sankey-style flows in which band width is proportional to the number of studies linking model type to continent and publication year; strata are labeled, and color encodes model category. Abbreviations: A = Animal; H = Human; H+A = Human + Animal; H+IV = Human + In vitro; IV = In vitro; IV+A = In vitro + Animal; IS = In silico; UnC = Unclassified; AF = Africa; AS = Asia; EU = Europe; NA = North America; SA = South America; OC = Oceania.
Beyond the continental view presented in Figure 4, the country-level pattern is highly concentrated. China contributes the plurality of records across 2009–2025, with visible clusters in 2019–2020 and 2024–2025; Japan is another recurrent Asian contributor (2006, 2008–2009, 2016, 2019–2022, 2023). Outside Asia, the United States appears frequently (2013, 2014, 2020–2024, 2025), often leading North American output. In Europe, Germany and Italy recur (2011–2016, 2018, 2020–2025), alongside Romania, Poland, Denmark, Finland, Switzerland, France, Croatia, the United Kingdom, and The Netherlands (Amsterdam). Turkey also appears in 2025. In South America, Brazil contributes (2019, 2023), and there is at least one cross-continental collaboration (Chile–Spain, 2023). Additional multinational efforts include China–USA, Italy–USA, Switzerland–USA, and Europe–USA, underscoring the field’s collaborative character.
Alluvial flows (Figure 4B) reveal how these patterns unfolded over time. Notable concentrations include Asia: in vitro + animal in 2020 and 2024 (each n = 5) and Asia: human in 2024 (n = 5), alongside a 2021 cluster of Asia: human + in vitro + animal (n = 4). Mechanistic surges in 2020 also involved Europe: animal (n = 4) and North America: animal (n = 3). Human-focused activity intensified in Europe 2023 (n = 3) and North America 2024 (n = 3). In silico first appears in Asia 2024–2025 (each n = 1). For 2025 (indexed through July), Asia maintains emphasis on animal (n = 3) and human + animal (n = 3). Overall, these results indicate an Asian pivot toward integrated, multi-model experimentation since 2019, with Europe/North America sustaining strong human/animal emphases and South America remaining underrepresented.
A convergent picture emerges from the eligible studies (mapped in Table 2), in which chronic hyperglycemia, hemodynamic overload, lipid toxicity, sterile inflammation, and epigenetic/post-transcriptional reprogramming drive podocyte dedifferentiation, detachment, and death, culminating in proteinuria and progressive renal decline. Mechanistic nodes and their morphologic or molecular readouts are tightly coupled, and where available, quantitative signals (e.g., inverse correlations between podocyte density and albumin excretion) reinforce causality.
Filtration barrier and adhesion failure. The slit diaphragm is an early casualty: nephrin downregulation increases glomerular permeability and tracks with albuminuria in humans; ACE inhibition partially restores nephrin and reduces albuminuria, linking mechanism to clinical phenotype [60]. Cytoskeletal and junctional scaffolds are concurrently compromised: α-actinin-4 loss correlates with podocyte dysfunction/proteinuria [49], while loss of uniform connexin-43 under hyperglycemia associates with reduced renal function, implying disrupted intercellular coupling at the slit membrane [135]. Structural and quantitative abnormalities appear early and scale inversely with albumin excretion, podocyte density falls as AER rises, implicating podocyte loss as a primary driver of albuminuria [107]; human biopsy series corroborate fewer podocytes with more proteinuria and compensatory hypertrophy [75,96]. An FSP1/Snail1/ILK/TGF-β1 program indicates EMT-like reprogramming in podocytes, mechanistically tied to GBM detachment and worse clinico-pathologic DN [121]. Adhesion dynamics are phase-dependent: α3β1-integrin is induced in early DN, facilitating foot-process effacement/detachment via TGF-β1 signaling [3], whereas late disease features reduced integrin tone with broadened processes [142]. Two amplifiers further erode the diaphragm: hyperglycemia accelerates dynein-dependent nephrin degradation (DynII1/DCTN1) [146], and anti-nephrin autoantibodies add an immune hit with ATP-depletion-related cytoskeletal injury [68]. Cell-intrinsic guardians and liabilities add texture: BASP1 co-represses WT1 to activate p53-mediated apoptosis [133], CKAP4 maintains actin–microtubule architecture (its loss precipitates effacement/detachment) [105], and SRGAP2a restrains RhoA/Cdc42 to curb motility and preserve structure under hyperglycemic/TGF-β stress [87]. A compensatory rise in podocyte ClC-5 in proteinuric patients suggests augmented albumin endocytosis within the injured barrier [26].
mTOR–stress signaling and epitranscriptomic control. Podocyte mTORC1 is a bidirectional hazard: hyperactivation dislocates slit-diaphragm proteins, induces EMT-like and ER-stress signatures, and leads to podocyte loss, mesangial expansion, and proteinuria [58]; conversely, deleting or broadly dysregulating mTORC1 yields hypertrophy, effacement, detachment, and suppressed autophagy, underscoring the need for tight rheostasis [59]. Fine-tuning layers align mechanism with lesion: miR-99a-5p constrains mTOR/EMT [118]; REDD1 lowers nephrin/podocin and heightens TRPC6-Ca2+ influx, disorganizing the cytoskeleton [102]; Rheb1 loss accelerates mitochondrial senescence via Atp5f1c acetylation independently of mTORC1 [103]. Epitranscriptomic “writers” shape vulnerability: METTL14-mediated m6A reduces Sirt1 and promotes podocyte injury [70], while METTL3 stabilizes TIMP2 (via IGF2BP2) to activate Notch-inflammation/apoptosis and, in separate work, engages an MDM2/Notch axis in dedifferentiation [57,128]. Upstream, TXNIP links hyperglycemia to oxidative stress and mTOR/EMT activation; knockdown reduces ROS and renal injury and associates with mTOR activity in human biopsies [13]. EGFR signaling elevates Rubicon and blocks autophagy through mTOR–p70S6K/RPS6, worsening podocyte injury [123]. ERK activation demonstrated in human DN podocytes further implicates VEGF/ribosome-biogenesis programs [21]. Clinically relevant counter-signaling is possible: saxagliptin suppresses p38 activity while increasing nephrin/podocin independently of glycemia [147].
Lysosomal dysfunction impairs albumin degradation, escalates cytokines, and drives glomerulosclerosis, directly connecting protein overload to scarring [23]. High-glucose mesangial signals suppress podocyte ERAD and nephrin phosphorylation, anatomically embedding the lesion in intra-glomerular crosstalk [36]. Therapeutically, HGF restores autophagy/lysosomal flux via PI3K/Akt–GSK3β–TFEB, reducing albuminuria and podocyte loss [41]. UCP2 supports macroautophagy (loss worsens proteinuria/injury) [119]. DOT1L–PLCL1 improves lipid handling and reduces lipotoxic damage [53], and BTG2 couples mTORC1 inhibition to pro-autophagic, anti-inflammatory effects [87].
Palmitate elicits an ultimately “futile” antioxidant response and oxidative ultrastructural damage [78]. Ceramides accumulate and injure mitochondria; CerS6–VDAC1 interaction triggers mtDNA leakage and cGAS–STING activation [115,140]. Insufficient FAO and mitochondrial dysfunction sustain vulnerability [148], exacerbated by PGRN deficiency with failure of the PGRN–Sirt1–PGC-1α/FoxO1 program [136]. Protective metabolic levers include β-hydroxybutyrate (GSK3β inhibition, Nrf2 activation, less senescence) [149]; SGLT2i-driven ERRα–ACOX1 activation (more FAO, less lipotoxicity, structural repair) [72]; and GM3 restoration by valproate [86]. Lipid-handling regulators add causality: CCDC92 promotes lipid deposition via ABCA1 [144], and ACSS2 epigenetically activates mTORC1 and suppresses autophagy [80]. At the calcium–mitochondria interface, TRPC6-dependent Ca2+ influx activates calpain-1/CDK5/Drp1 to drive mitochondrial fission and apoptosis [129], while Orai1-mediated SOCE activates calpain and disorganizes F-actin/nephrin [117]. Complement C3a–C3aR further disrupts podocyte bioenergetics; antagonism restores mitochondrial function/density [88]. Systemic mtDNA/OXPHOS injury and ROS parallel glomerulotubular inflammation even in normoalbuminuric DKD [104].
Podocytes upregulate CD80/B7-1 under high glucose (PI3Kα), disrupting the cytoskeleton and inducing apoptosis; CTLA4-Ig reverses this phenotype [39]. High glucose/AGEs/ROS activate NLRP3, engaging canonical and non-canonical arms that propagate podocyte dysfunction [55]; blocking caspase-1 with carnosine attenuates pyroptotic injury [150]. TRAIL–DR5 signaling triggers PANoptosis, and TRAIL/DR5 deletion reduces glomerular damage [92]. Complement disinhibition is pathogenic: DAF/CD55 loss unleashes C3 convertase, activating C3a/C3aR and an IL-1β/IL-1R1 loop that lowers nephrin and remodels actin; low DAF with C3d positivity and high urinary C3a coincide with FSGS-like lesions and proteinuria [33]. Additional nodes, HDAC4 → calcineurin apoptosis [93], ROCK-mediated mesangial fibrosis and podocyte apoptosis [84], RARRES1 → p53 apoptosis [32], RIPK3-driven NF-κB inflammation independent of necroptosis [69], and DHAP-mTORC1/ROS/NLRP3 coupling [151], further knit mechanism to lesion. In PGNMID, endothelial PV-1 overexpression links complement/IgG deposition to oxidative, inflammatory crosstalk that secondarily injures podocytes [100]. GH-TGF-β1/Notch signaling drives podocyte binucleation/mitotic catastrophe; blocking GHR/TGFBR1 prevents these cytologic lesions and DN features [82,90].
Obliterative microangiopathy (arteriolosclerosis, hyalinosis) and glomerular ischemia appear in DN and associate with collapsing glomerulopathy, tuft collapse, epithelial proliferation, VEGF overexpression, and poor outcomes/ESRD [94]. Early hyperfiltration couples to podocyte depletion and GBM thickening; endothelial stress with mesangial crosstalk activates fibrosis programs that mirror advancing histologic class [50,114]. Hyperglycemia activates the polyol/cPKC axes and podocyte loss, while DGKα/67LR maintains adhesion [31]. PECs enlarge Bowman’s capsule ECM in human DN [43] and, under severe microvascular hypoxia, associate with extracapillary hypercellularity and loss of podocyte phenotype [42]. Diabetic co-culture models confirm that HG/MGO deform GEC–podocyte transcriptomes and degrade ECM/barrier properties [34]. Uremic gut-derived metabolites and loss of retinoic-acid signaling tie systemic milieu to glomerular/endothelial dysfunction [19].
Canonical DN histology, mesangial expansion, Kimmelstiel–Wilson nodules, podocyte depletion, remains central [50]. Spatial metabolomics (MALDI-IMS/MxIF) maps lipid signatures that co-localize with podocyte loss and mesangial expansion, providing tissue-level surrogates of injury [28]. Urinary nephrin detects early podocyte injury [52]; elevated urinary podocin and intrarenal podocalyxin predict progression and track function [126]; the urinary podocin:nephrin mRNA ratio correlates with tubulointerstitial fibrosis [130]. Clinicopathologic resources ground these signals: in TRIDENT, eGFR correlates most with interstitial fibrosis and glomerular epithelial changes [85]; the nPOD-K biobank anchors histologic trajectory studies [120]. Therapeutically, pathway-directed interventions show structural dividends: HGF and DOT1L/PLCL1/BTG2 restore autophagy/lipid handling [41,53]; CTLA4-Ig, C3aR antagonism, DR5/TRAIL blockade, and inflammasome targeting blunt immune lesions [39,55,88,92]; metabolic correction with SGLT2i (reduced MAMs, AMPK activation), β-hydroxybutyrate, and VPA/GM3 improves ultrastructure [72,81,86,149]; and systemic milieu shifts after Roux-en-Y gastric bypass reverse podocyte dedifferentiation/effacement alongside reduced albuminuria [27].
Taken together, the field supports a staged lesion cascade, slit-diaphragm/adhesion failure (nephrin/integrins/connexins/trafficking), mTOR–autophagy–ER/mitochondrial disequilibrium, lipotoxicity with defective FAO and oxidative stress, immune/complement/inflammasome amplification, and epigenetic–transcriptomic reprogramming via m6A, ncRNAs, and APA, modulated by hemodynamics and endothelial/mesangial crosstalk. Each node is paired with a reproducible description at the tissue or molecular level and, in several instances, with therapeutic reversibility. For a study-by-study mapping of mechanism to lesion descriptor, see Table 2 (mechanisms and descriptions), which underpins the narrative links and citations summarized here.
Table 2.
Summary of inducing mechanisms and main characteristics of lesions associated with podocyte damage related to diabetes mellitus.
The word-cloud synthesis (Figure 5) condenses the mechanistic corpus into a high-salience vocabulary led by “podocyte” (66 mentions) and second-tier tokens such as “podocytes/injury” (20 each), “activation/via” (19 each), “signaling” (18), and a triad of “stress/inflammation/mitochondrial” (13 each). Terms that anchor the filtration barrier, “nephrin,” “foot,” “process,” “detachment,” “effacement,” “slit,” and “cytoskeleton” (≈7 each), signal a dominant barrier-failure theme, consistent with nephrin depletion and increased permeability in human DN, connexin/integrin derangements, and EMT-linked GBM disengagement [3,49,121,135,154]. Tokens denoting mTOR–ER–autophagy stress (“mTORC1/mTOR,” “autophagy,” “degradation,” “TFEB,” “rubicon”) map onto podocyte injury from mTORC1 hyperactivation and autophagy blockade, with rescue signals when lysosomal flux is restored [41,58,59,155].
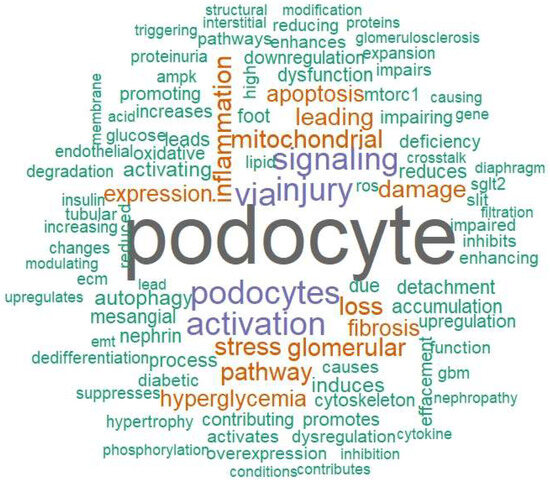
Figure 5.
Mechanistic vocabulary of podocyte injury in diabetes. Word cloud summarizing the top 100 single-word terms extracted from the mechanistic sections of all eligible studies. Font size is proportional to term frequency; color is purely esthetic (no quantitative meaning) and word placement is random. Singular/plural or closely related variants appear as separate tokens when present in the source text. The emergent lexicon maps onto the five mechanistic domains summarized in Table 2, slit-diaphragm/adhesion failure, mTOR–autophagy/ER stress, mitochondrial–lipid injury, immune/complement/inflammasome activation, and epigenetic–transcriptomic regulation—providing a compact qualitative overview of the corpus.
The prominence of mitochondrial–lipid vocabulary (“oxidative,” “ROS,” “ceramide,” “FAO,” “fission,” “TRPC6,” “calpain”) reflects lipotoxic ROS injury, defective fatty-acid oxidation, and Ca2+-coupled mitochondrial fragmentation [78,106,129,156]. Parallel immune lexemes (“inflammasome,” “pyroptosis,” “complement,” “TRAIL/DR5”) mirror CD80/B7-1 induction, C3a/C3aR-mediated mitochondrial dysfunction, NLRP3 activation, and PANoptosis in podocytes [39,55,88,92]. Epigenetic/RNA regulation is also visible (“miR-,” “METTL3/14,” “PVT1,” “LINC-,” “APA”), aligning with m6A-, lncRNA-, and microRNA-driven rewiring of stress pathways [57,61,70,137]. Finally, structural and hemodynamic tokens (“mesangial,” “GBM,” “thickening,” “proteinuria,” “hyperfiltration”) connect the lexical pattern to classic DN histology and microvascular stress [50,107,114].
Taken together, the frequency-weighted lexicon in Figure 5 independently corroborates the five mechanistic pillars summarized in Table 2, slit-diaphragm/adhesion failure; mTOR autophagy ER disequilibrium; mitochondrial lipid stress; immune/complement/inflammasome activation; and epigenetic–transcriptomic reprogramming, while situating them within the broader morphometric context of mesangial expansion, GBM remodeling, and podocyte depletion [75,96].
Figure 6A organizes the eligible studies into a directional cascade. Upstream drivers, chronic hyperglycemia, hemodynamic overload, dyslipidemia/lipotoxicity, AGEs (“metabolic memory”), microvascular stress, and sterile/viral inflammation, converge on signaling hubs, including mTORC1, ER stress/UPR and autophagy/TFEB, TXNIP, EGFR → p70S6K/RPS6, ERK/VEGF, TGF-β/EMT/Notch, ROCK, HDAC4 → calcineurin, complement C3a/C3aR, and NLRP3/pyroptosis (TRAIL–DR5), as well as epigenetic/RNA programs. These hubs impinge on organellar/structural targets, mitochondria (FAO ↓, fission ↑), lysosome/autophagic flux, ER, actin/α-actinin-4 cytoskeleton, slit diaphragm (nephrin/podocin), and adhesion complexes (Cx43, α3β1-integrin), and culminate in tissue-level readouts: foot-process effacement, podocyte loss, proteinuria, mesangial expansion, and GBM thickening/sclerosis. Representative links include mTORC1-driven mislocalization of slit proteins and autophagy suppression [58,59], TXNIP coupling hyperglycemia to oxidative and EMT programs [13], EGFR–Rubicon autophagy blockade [123], ERK/VEGF activation in human DN podocytes [21], TGF-β/Notch-dependent EMT [121], ROCK and HDAC4 → calcineurin pro-apoptotic axes [84,93], complement-driven mitochondrial dysfunction [88], inflammasome/pyroptosis and TRAIL–DR5-mediated PANoptosis [55,92], and epigenetic/RNA control that rewires these hubs [57,70,137]. At the barrier, nephrin loss, connexin-43 heterogeneity, α-actinin-4 reduction, and phase-dependent α3β1-integrin shifts map to detachment/effacement [3,49,135,142,154], with dynein-mediated nephrin degradation and anti-nephrin autoantibodies acting as amplifiers [157]. The cascade’s terminus aligns with morphometric evidence linking podocyte depletion to albuminuria and classic DN histology [50,75,96,107].
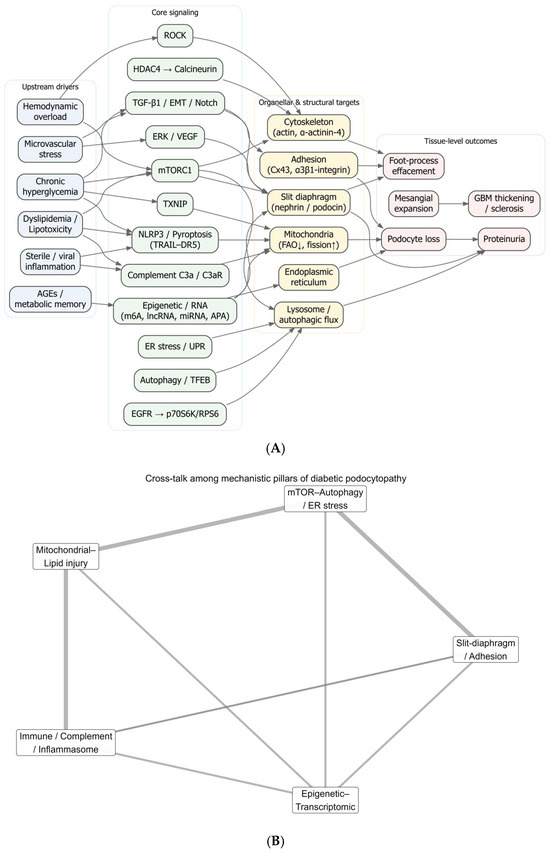
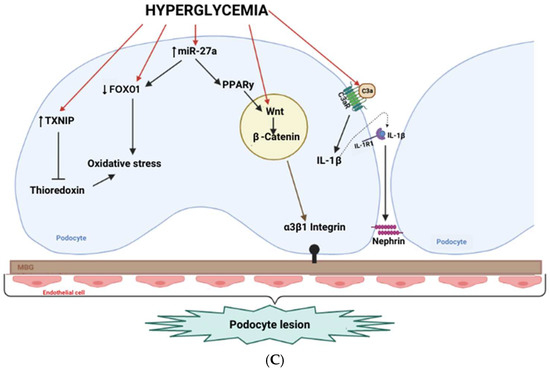
Figure 6.
Integrated architecture of diabetic podocytopathy. (A) Systems map (drivers → hubs → targets → outcomes). Directed graph summarizing hypothesized causal flow from upstream drivers (light blue: chronic hyperglycemia, hemodynamic overload, dyslipidemia/lipotoxicity, AGEs/metabolic memory, microvascular stress, sterile/viral inflammation) to core signaling hubs (light green: mTORC1, ER stress/UPR, autophagy/TFEB, TXNIP, EGFR → p70S6K/RPS6, ERK/VEGF, TGF-β/EMT/Notch, ROCK, HDAC4 → calcineurin, complement C3a/C3aR, NLRP3/pyroptosis/TRAIL-DR5, epigenetic/RNA programs) and then to organellar/structural targets (light amber: mitochondria—FAO ↓/fission ↑; lysosome/autophagic flux; ER; actin/α-actinin-4 cytoskeleton; slit diaphragm, nephrin/podocin; adhesion—Cx43 and α3β1-integrin), culminating in tissue-level outcomes (light rose: foot-process effacement, podocyte loss, proteinuria, mesangial expansion, GBM thickening/sclerosis). Arrow direction encodes putative influence; edges are drawn only where supported in the corpus (see Table 2). (B) Five-pillar interaction network. Circular network collapsing panel A into five interacting modules,) slit-diaphragm/adhesion, mTOR–autophagy/ER stress, mitochondrial lipid injury, immune/complement/inflammasome, epigenetic transcriptomic control. Edge thickness is proportional to the curated crosstalk weight (relative evidence), highlighting dense bidirectional coupling among pillars. (C) Cell-level schematic under hyperglycemia. Representative podocyte blueprint depicting hyperglycemia-induced programs (e.g., TXNIP; Wnt/β-catenin and miR-27a–PPARγ–FOXO1 axes; complement C3a/C3aR and IL-1β/IL-1R1 signaling) converging on slit-diaphragm depletion/mistrafficking (nephrin/podocin), adhesion defects (Cx43, α3β1-integrin), cytoskeletal instability, impaired autophagy/lysosome, and mitochondrial dysfunction (FAO ↓, fission ↑), which together yield effacement, detachment, podocyte loss and proteinuria. Abbreviations: AGE, advanced glycation end-product; AMPK, AMP-activated protein kinase; APA, alternative polyadenylation; C3aR, complement C3a receptor; DR5, death receptor 5; EMT, epithelial–mesenchymal transition; ER, endoplasmic reticulum; FAO, fatty-acid oxidation; GBM, glomerular basement membrane; HDAC, histone deacetylase; IL-1R1, interleukin-1 receptor type 1; lncRNA, long non-coding RNA; m6A, N6-methyladenosine; mTORC1, mechanistic target of rapamycin complex 1; NLRP3, NLR family pyrin domain containing 3; TFEB, transcription factor EB; TXNIP, thioredoxin-interacting protein; VEGF, vascular endothelial growth factor.
Figure 6B collapses the map into five interacting pillars, slit-diaphragm/adhesion, mTOR–autophagy/ER stress, mitochondrial–lipid injury, immune/complement/inflammasome, and epigenetic, transcriptomic control, revealing dense crosstalk. Examples include mTOR-driven barrier disorganization and autophagy loss [58,59]; lipotoxic ceramides and Ca2+ entry (TRPC6 or Orai1 → calpain → Drp1) driving mitochondrial fission/apoptosis [117,129,156]; complement C3a/C3aR activation and DAF loss feeding mitochondrial injury, actin remodeling, and nephrin reduction [33,88]; and writer/lncRNA/miRNA programs (METTL3/14, PVT1, miR-27a/193a, LINC01619) that gate Notch/EMT and stress responses and ultimately affect slit/adhesion components [20,57,61,70,92,158]. The network also contextualizes therapeutic reversibility observed across studies: HGF or BTG2 restoring autophagy/lysosomal flux [41,159], SGLT2 inhibition reducing MAMs and activating AMPK [81], and β-hydroxybutyrate or valproate/GM3 correcting oxidative–lipid signals [40,86].
Figure 6C depicts a representative podocyte under high glucose: TXNIP induction, Wnt/β-catenin and miR-27a–PPARγ–FOXO1 axes, and epigenetic modifiers collectively depress protective transcription and diaphragm/cytoskeletal components [13,14,20,160]. In parallel, complement C3a/C3aR signaling and an IL-1β/IL-1R1 loop amplify mitochondrial stress and actin remodeling, and DAF loss disinhibits C3 convertase [33,88]. These signals converge on slit-diaphragm depletion/mistrafficking (nephrin/podocin), adhesion failure (Cx43, α3β1-integrin), and cytoskeletal instability, precipitating effacement, detachment, podocyte loss, and proteinuria, lesions reproduced in models and observed in human biopsies [49,96,121,135,142,154].
Taken together, Figure 6 provides a results-oriented integration linking systemic drivers to intracellular hubs, organelle/barrier failure, and whole-glomerulus pathology. The panels jointly explain why diverse upstream insults can converge on a limited set of podocyte phenotypes and why targeted interventions at different nodes yield structural and functional rescue.
3. Discussion
From 7769 records, 130 studies met eligibility and collectively delineate a coherent, staged cascade of diabetic podocytopathy. Despite design-contingent limitations typical of observational work, methodological quality was moderate-to-high, and the synthesis converges on a limited set of mechanistic axes that repeatedly track with structural and clinical phenotypes. Publication activity and model diversity rose sharply after 2019, with an increasing use of mixed human/in vitro/animal designs that balance clinical relevance with mechanistic depth. Together, these features strengthen the inferential link between systemic drivers and podocyte lesions observed across models and human biospecimens.
Our integration organizes the field into five interacting “pillars”, slit diaphragm/adhesion failure; mTOR autophagy ER disequilibrium; mitochondrial lipid stress; immune/complement/inflammasome amplification; and epigenetic transcriptomic reprogramming. Human and experimental data consistently show that nephrin depletion, α-actinin-4 loss, connexin-43 heterogeneity, and phase-dependent α3β1-integrin shifts destabilize the filtration barrier and promote detachment and foot-process effacement, aligning with proteinuria and classic diabetic lesions [3,49,135,142,154].
Podocyte mTORC1 hyperactivation and proteostasis failure mislocalize slit-diaphragm proteins, induce ER-stress/EMT programs, and suppress autophagy; conversely, broad perturbation of mTOR signaling yields hypertrophy and effacement, illustrating a narrow homeostatic window for podocyte proteostasis [58,59]. Mitochondrial lipid injury is driven by palmitate-ROS toxicity, ceramide VDAC1–cGAS STING signaling, impaired FAO, and Ca2+ coupled fission via TRPC6/Orai1 calpain CDK5 Drp1, mechanistically linking metabolic stress to apoptosis [78,117,156,161,162].
Immune amplification spans CD80/B7-1 induction, C3a/C3aR-mediated bioenergetic collapse, NLRP3 pyroptosis, and TRAIL/DR5-dependent PANoptosis, with DAF loss removing complement restraint [33,39,55,88,92,150]. Epitranscriptomic and ncRNA programs, METTL3/14, PVT1/EVF-2, miR-27a/193a, and APA gate Notch/EMT and stress signaling and reshape barrier and organelle transcripts, providing a durable molecular substrate for injury [57,70,137,163].
The corpus substantiates upstream drivers, chronic hyperglycemia, hemodynamic overload, dyslipidemia and “metabolic memory,” microvascular stress, and sterile/viral inflammation, funneled through intracellular hubs that damage mitochondria, lysosomes, ER, actin architecture, slit-diaphragm components, and adhesion complexes. TXNIP links hyperglycemia to oxidative stress and EMT/mTOR activation; EGFR signaling increases Rubicon and suppresses autophagy; ERK/VEGF activation is evident in human diabetic podocytes; ROCK and HDAC4 → calcineurin promote cytoskeletal instability and apoptosis; and inflammasome/complement pathways propagate mitochondrial and actin injury [13,21,84,88,92,93,155]. Two clinically salient amplifiers, dynein-dependent nephrin degradation and anti-nephrin autoantibodies, further erode slit integrity and ATP dependent cytoskeletal homeostasis [157,164]. At the organ level, podocyte density declines inversely with albumin excretion and co-localizes with mesangial expansion and GBM remodeling, reinforcing causality between cell-level injury and whole-glomerulus pathology [50,75,96,107].
Experimental data indicate that glucocorticoids can directly preserve podocyte identity and architecture through specific pathways, e.g., dexamethasone KLF15 mediated restoration of differentiation markers and survival, maintenance of miR-30 to restrain Notch1/p53, and recovery of nephrin/synaptopodin with reduced proteinuria [165,166,167]. Angptl4 is glucocorticoid-sensitive, with sialylation conferring protection against proteinuria [168]. Notably, within our corpus and targeted check, we identified no studies that compare steroid-induced hyperglycemia versus normoglycemia on podocyte outcomes in glomerulonephritis under glucocorticoid therapy. Outside a steroid context, metabolic derangements (fasting glucose/insulin, HOMA-IR) correlate with podocyte injury in obesity-related glomerulopathy [169]. This gap merits prospective, glycaemia-stratified studies with podocyte-level endpoints.
Microangiopathy (arteriolosclerosis, hyalinosis) and ischemic remodeling compound podocyte stress and associate with collapsing patterns and adverse outcomes; early hyperfiltration couples to podocyte depletion and GBM thickening, while endothelial–mesangial crosstalk accelerates fibrosis, recapitulating ascending histologic class [50,94,114]. Mesangial-to-podocyte signals suppress ERAD and nephrin phosphorylation, anatomically embedding barrier failure within intra-glomerular signaling loops; diabetic co-culture models confirm HG/MGO-driven transcriptomic deformation and ECM degradation across GEC–podocyte units [34,36].
Conventional markers (albuminuria, eGFR) incompletely capture early podocyte injury; urinary nephrin, podocin, and podocalyxin, and the podocin:nephrin mRNA ratio track progression and tubulointerstitial fibrosis, while spatial metabolomics (MALDI-IMS/MxIF) localizes lipid signatures to podocyte loss and mesangial expansion [28,52,126,130]. Clinicopathologic resources reinforce these links: in TRIDENT, eGFR correlates most strongly with interstitial fibrosis and glomerular epithelial changes, and nPOD-K enables trajectory studies of histologic progression [85,120].
Mechanism-targeted interventions demonstrate structural dividends that validate the five-pillar architecture. HGF and BTG2 restore autophagy/lysosomal flux; DOT1L–PLCL1 improves lipid handling; β-hydroxybutyrate and valproate/GM3 mitigate oxidative–lipid stress and senescence [41,53,86,149,170]. Immune-axis interventions, CTLA4-Ig, C3aR antagonism, inflammasome blockade, DR5/TRAIL inhibition, attenuate complement/pyroptotic injury and rescue barrier components [39,55,88,92]. Systemic metabolic shifts after Roux-en-Y gastric bypass reverse podocyte dedifferentiation and effacement alongside reductions in albuminuria, emphasizing the modifiability of upstream drivers [27]. These convergences argue for “pillar-informed” combinations, e.g., SGLT2i plus an autophagy enhancer, or complement blockade alongside cytoskeletal stabilizers, tested against structural endpoints such as slit-diaphragm density, podocyte density, and EM-level ultrastructure [58,59,81,88].
Across included studies and targeted checks, gliflozins preserved podocyte architecture and reduced proteinuria in diabetic and hyperglycaemic models, with preliminary human correlates. Empagliflozin decreased foot-process width/effacement, increased podocyte number/density, and reduced albuminuria, in association with reactivation of autophagy and attenuation of oxidative stress [64,171]. Dapagliflozin suppressed podocyte epithelial–mesenchymal transition via down-regulation of IGF1R/PI3K and improved nephrin and albuminuria in STZ mice and in a small human DN cohort [172]. Canagliflozin inhibited TXNIP/NLRP3-mediated podocyte pyroptosis with improvements in albuminuria and serum creatinine [173]. Additional signals include restoration of nephrin/podocin and α-Klotho [174], protection of actin cytoskeleton and podocyte density in a nondiabetic proteinuric model [175], reduced podocyte lipotoxicity in Alport syndrome [176], and decreased urinary albumin with a related SGLT2 inhibitor [177].
Mechanistically, benefits converge on autophagy/AMPK activation and relief of mitochondria–ER stress [171,178], increased fatty-acid oxidation via ERRα–ACOX1 [72], EMT suppression [172], and restraint of inflammasome/pyroptosis [173]. Taken together, these data support SGLT2 inhibitors as a structural metabolic backbone for podocyte protection in hyperglycaemia, suitable for combination with pillar-directed modulators (e.g., autophagy, complement/inflammasome, cytoskeleton).
Strengths of this review include its large contemporary corpus, triangulation across human and experimental systems, and explicit mapping from drivers to morphologic readouts. Limitations reflect heterogeneity in models and outcome definitions, incomplete control for confounding in some observational designs, and underrepresentation of certain geographies and in silico approaches. Some promising axes, e.g., endothelial–podocyte metabolic coupling and gut-derived metabolites, remain supported by fewer studies and warrant deeper, prospective interrogation [19,34].
Priority next steps include prospective, mechanistically stratified human studies that co-measure pillar activity (e.g., complement fragments, ceramide species, mitochondrial injury markers, ncRNA and m6A signatures) with standardized morphometrics; trials of rational combinations aligned to individual pillar activation; and deeper dissection of trafficking and autoantibody amplifiers that directly govern slit-diaphragm integrity [157,179,180]. By aligning systemic drivers with intracellular hubs and organellar targets, the field is increasingly positioned to deliver precision interventions that preserve podocyte identity, adhesion, and mitochondrial fitness, thereby interrupting the progression from effacement to proteinuria and renal decline [50,58,59,88,178].
4. Materials and Methods
4.1. Ethical Aspects
This work was a secondary study and did not violate any current legislation related to ethics in research on humans and experimental models.
4.2. Type of Study and Protocol Record
This was a retrospective secondary study through a systematic review. The study was registered in the “Prospero” database under registration number CRD42020205261 and can be accessed at https://www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42020205261 (accessed on 1 August 2025). It was structured according to the recommendations of the tool Preferred Reporting Items for Systematic (PRISMA 2020) [181].
4.3. Search Strategy and Eligibility Criteria
We conducted and reported the review in accordance with PRISMA 2020 (study-selection pathway in Figure 1). Four core databases were searched from 1 January 2001 to 31 July 2025: MEDLINE (PubMed), Embase, Latin American and Caribbean Health Sciences Literature (LILACS), and the Cochrane Library (Cochrane Reviews). To broaden coverage, we also performed supplementary searches of gray literature (websites and organizations) and citation chasing (backward screening of reference lists from included studies and forward citation tracking), following principles from the University of Toronto Libraries’ Grey Literature Search Guide. Although the search window spanned 2001–2025, the earliest eligible publication identified was 2002, so included studies cover 2002–2025; counts for 2025 reflect indexing through July.
Search strategies combined controlled vocabulary and free-text terms for podocytes and diabetic kidney disease, adapted to each database. For MEDLINE, we paired Title/Abstract terms for podocytes (e.g., podocyte, podocytes, “glomerular visceral epithelial cells”) with diabetic nephropathy/kidney-disease terms (e.g., diabetic nephropathy, diabetic kidney disease, diabetic glomerulosclerosis, intracapillary glomerulosclerosis). In Embase, Emtree terms were combined with proximity operators (e.g., podocyt* OR phrases such as glomerul* NEAR/3 visceral NEAR/3 epithelial*), intersected with ‘diabetic nephropathy’ or proximity-linked diabetic kidney-disease terms. In the Cochrane Library, we used text-word/proximity formulations (e.g., podocyt* AND (diabetic NEXT nephropath* OR diabet* NEAR/3 kidney NEXT disease* OR DKD). In LILACS, DeCS and keyword combinations analogous to the above were applied. No design or model filters were imposed at the search stage to avoid missing mechanistic studies; importantly, “biopsy” was not enforced as a mandatory search term.
We included original research (human, animal, in vitro, or mixed-modality) that evaluated diabetes mellitus or a hyperglycemic milieu relevant to diabetic kidney disease; assessed podocyte injury at structural/ultrastructural or molecular levels (e.g., foot-process effacement; podocyte number/density; nephrin/podocin/synaptopodin expression; cytoskeletal or organellar injury); and provided a mechanistic context linking exposure to podocyte outcomes. We excluded non-diabetic kidney diseases; studies without podocyte outcomes or without mechanistic information; narrative reviews, editorials, and letters; conference abstracts lacking extractable data; and records for which the full text could not be retrieved.
Study selection, deduplication, and inter-rater agreement. Search results were imported into Rayyan (https://www.rayyan.ai) for automated de-duplication and blinded dual screening of titles/abstracts, followed by full-text assessment. A consolidated log was maintained in Microsoft® Excel for PRISMA accounting and manual verification of residual duplicates. Two independent reviewers (J.S.S., A.G.B.F.) screened titles/abstracts and assessed full texts against pre-specified criteria; disagreements were resolved by discussion, with a third reviewer (W.F.R.) adjudicating when required. Inter-rater agreement for screening decisions was quantified using Cohen’s kappa (κ) computed in BioEst 5.0, with κ values interpreted using conventional thresholds: <0.00 poor, 0.00–0.20 slight, 0.21–0.40 fair, 0.41–0.60 moderate, 0.61–0.80 substantial, and 0.81–1.00 almost perfect agreement. Where applicable, κ was summarized with 95% confidence intervals and two-sided p-values.
4.4. Evaluation of Study Selection and Methodological Quality
The search covered 1 January 2001 through 31 July 2025; the earliest eligible publication identified was 2002, so included studies span 2002–2025. Counts for 2025 reflect records indexed through July.
Study selection proceeded in two sequential stages: title/abstract screening and full-text review of records passing the first stage. We considered original research across human, animal, in vitro, and mixed-modality designs, provided that studies evaluated diabetes mellitus (or a hyperglycemic milieu relevant to diabetic kidney disease), reported podocyte-level outcomes (e.g., foot-process effacement; podocyte number/density; nephrin/podocin/synaptopodin; cytoskeletal/organellar injury), and offered mechanistic context linking exposure to podocyte injury. We excluded non-diabetic kidney diseases, studies without podocyte outcomes or without mechanistic information, narrative reviews/editorials/letters, conference abstracts lacking extractable data, and records for which the full text could not be retrieved.
Two independent reviewers (J.S.S. and A.G.B.F.) screened titles/abstracts and assessed full texts against pre-specified criteria; disagreements were resolved by discussion, with a third reviewer (W.F.R.) adjudicating when needed.
Methodological quality (risk of bias) was appraised using the Joanna Briggs Institute (JBI) Critical Appraisal Tools matched to study design (human observational, animal experimental, and in vitro/mixed), using the most recent versions available at the time of appraisal (available at https://jbi.global/critical-appraisal-tools; accessed on 2 August 2025). Item-level responses were summarized as absolute/relative frequencies, and overall JBI scores were expressed as percentages to permit cross-study comparison. Any differences in JBI ratings were resolved by consensus among the reviewers. Aggregate quality results are reported in the Results and Table 1.
4.5. Data Analysis and Summarization
All extracted variables were first tabulated and checked in Microsoft® Excel. The absolute and relative frequencies for categorical variables and mean, standard deviation, coefficient of variation, and 95% confidence intervals for continuous variables—were computed to summarize study characteristics and methodological quality. Item-level responses from the Joanna Briggs Institute (JBI) tools were summarized as absolute/relative frequencies, and overall JBI scores were expressed as percentages; aggregate summaries report mean, SD, and CV across studies.
Inferential analyses were limited to temporal trend testing and associations between categorical study features. Temporal trends in annual publication counts were assessed using Spearman’s rank correlation (ρ) between year and number of studies and by simple linear regression of studies per year, reporting slope with 95% CI, R2, F statistic, and two-sided p-values. Associations among study-model categories were evaluated using Pearson’s chi-square test for proportions. Normality of continuous variables was examined with the Shapiro–Wilk test to guide parametric versus non-parametric summaries. A two-sided significance level of 5% (α = 0.05) was adopted.
Trend analyses and the time-series visualization for Figure 2 were performed in GraphPad Prism, version 9.5.1 (GraphPad Software, LLC, Boston, MA, USA). All other analyses and visualizations were scripted in R within RStudio (Posit) 2025.05.0 (Build 496). For data wrangling and plotting we used ggplot2 (v3.5.2), dplyr (v1.1.4), stringr (v1.5.1), forcats (v1.0.0), and scales (v1.3.0). The alluvial diagram (Figure 4B) was generated with ggalluvial (v0.12.5); the continent-by-model heatmap (Figure 4A) used base R heatmap with palettes from RColorBrewer (v1.1-3). The word-cloud and related text features (Figure 5) were produced with tm (v0.7-16), wordcloud (v2.6), and RColorBrewer (v1.1-3); term–document matrices (DocumentTermMatrix) and frequency tables were exported to CSV, and TF–IDF summaries were computed with tidytext (v0.4.3) and visualized with ggplot2 (v3.5.2). Systems maps and schematic figures (Figure 6A) were created with DiagrammeR (v1.0.11) [Graphviz/DOT] and exported via DiagrammeRsvg (v0.1) and rsvg (v2.6.2) to PNG; the five-pillar interaction network (Figure 6B) was rendered with ggraph (v2.2.1) and igraph (v2.1.3). These schematic maps are literature-curated visual syntheses (edge weights used for layout only) and were not used for statistical inference. All figures generated in R were saved with ggsave (ggplot2 v3.5.2), and intermediate tables underlying the heatmap, alluvial diagram, word-cloud, and TF–IDF panels were exported as CSV to support reproducibility.
5. Conclusions
Diabetes mellitus (DM) remains a major driver of chronic kidney damage, with podocyte injury representing a pivotal event in the progression of diabetic nephropathy (DN). Our systematic synthesis reveals not only the mechanistic pathways underlying this process, such as redox imbalance, inflammation, apoptosis, autophagy–ER stress dysregulation, and epigenetic reprogramming, but also the temporal and methodological context in which these discoveries have emerged. Publication activity has accelerated sharply since 2019, with over 80% of all studies appearing in the past six years, underscoring the expanding global focus on podocyte biology in diabetes.
This surge has been accompanied by a clear diversification of study designs. While early work was dominated by human biopsy descriptions, more recent years have adopted integrated, multi-model approaches that combine human data, animal experimentation, and in vitro systems, thereby balancing clinical relevance with mechanistic depth. Human tissue studies remain essential for linking lesions to clinical outcomes, whereas in vitro and animal platforms provide controlled environments for dissecting molecular drivers such as mTOR, TXNIP, and NLRP3. Geographic patterns highlight Asia as the leading contributor, with increasing multinational collaborations that enhance generalizability.
Taken together, these findings indicate that the field has transitioned into a mature, multidimensional research space, where diverse methodologies converge on a reproducible cascade of podocyte injury. This evolution strengthens the evidence base for therapeutic strategies that target slit-diaphragm integrity, mitochondrial and metabolic homeostasis, immune and inflammatory pathways, and epigenetic regulators. By situating mechanistic insights within temporal, methodological, and geographical trends, our study provides a comprehensive and timely framework that can inform both experimental designs and translational efforts to mitigate podocyte injury in diabetic nephropathy. We also identify a critical clinical gap: whether steroid-induced hyperglycemia exacerbates podocyte injury in glomerulonephritis remains untested and warrants dedicated investigation.
Building on these insights, the following Future Perspectives outline key priorities to accelerate discovery and translation.
Future Perspectives
To translate convergent mechanistic insights into clinical benefit, several priorities should guide the field. Standardized, longitudinal human cohorts with harmonized phenotyping (albuminuria, eGFR), biopsy/spatial readouts, and integrated multi-omics are needed to define molecular endotypes and causal trajectories of podocyte injury.
Because our review required diabetes mellitus, studies of glomerulonephritis under glucocorticoid therapy without diabetes, and any differential effects of glucocorticoid-induced hyperglycaemia, were not captured. Prospective GN cohorts that stratify by glycaemic exposure during steroid therapy and track podocyte biomarkers (e.g., urinary nephrin/podocin transcripts, podocyte-derived extracellular vesicles) and, when feasible, ultrastructural endpoints (electron microscopy) are warranted.
Harmonization across model systems, including reporting standards for in vitro, animal, and mixed-modality designs, will improve reproducibility and facilitate cross-study synthesis. Human-relevant experimental platforms (humanized mice, organoids, co-culture microphysiological systems) should be leveraged for causal interrogation of prioritized pathways (e.g., mTOR–autophagy, complement C3a/C3aR, inflammasome, ceramide metabolism, TRPC6/Orai1-mediated Ca2+ signaling). Biomarker development should focus on minimally invasive tools (e.g., urinary nephrin/podocin transcripts, podocyte-derived extracellular vesicles) and regulatory-ready surrogate endpoints that track structural repair. Precision-intervention trials can stratify patients by molecular endotype to test pathway-directed therapies and combinations (e.g., SGLT2i backbones with immune-metabolic modulators).
Data sharing and global collaboration, particularly bridging Asian leadership with underrepresented regions, together with transparent code and repositories, will accelerate validation and generalizability. Advanced analytics, including interpretable AI/ML and causal inference frameworks, should integrate temporal, geographical, and methodological heterogeneity. Collectively, these steps can convert the field’s recent expansion into reproducible, patient-centered gains in diabetic kidney disease.
Author Contributions
W.F.R., A.L.M.d.S.M. and J.R.M. designed the study. W.F.R., C.J.F.O., J.S.S., A.G.B.F., C.B.M., R.B.M. and A.G.-N. wrote the data analysis plan. W.F.R., J.S.S., A.G.B.F., C.B.M., R.B.M., M.O.C., L.M., A.G.-N., C.J.F.O., J.R.M. and M.A.R. monitored the review process. All authors interpreted the data, J.S.S., W.F.R., A.G.B.F., C.B.M., R.B.M., M.O.C., L.M., A.G.-N., L.S.A., C.A.d.S., J.R.M. and M.A.R. assessed studies for inclusion. J.S.S., A.G.B.F., W.F.R., C.B.M., R.B.M., M.O.C., L.M. and A.G.-N. wrote the draft paper. All authors have approved the final version. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by multiple funding agencies. The Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) provided support through the “Call 012/2023—Structuring Networks for Scientific Research or Technological Development”, under the project Applications of Genomics in the Context of OneHealth, coordinated by Aristóteles Góes Neto (identifier RED-00181-23), as well as through grant APQ-02831-23. The National Council for Scientific and Technological Development (CNPq) funded this work under grant nº 406261/2023-7 and provided additional support through bench fees (Bench Fees 2). Wellington Francisco Rodrigues is a recipient of a scholarship under FAPEMIG’s Science, Technology, and Innovation Development Grant program. Camila Botelho Miguel holds a senior postdoctoral fellowship from CNPq (Call 32/2023, Process 102630/2024-0). Additional support was provided by the Coordination for the Improvement of Higher Education Personnel (CAPES) and by institutional resources from the Kidney Research Center (CePRim) at the Federal University of Triângulo Mineiro (UFTM).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original data presented in this study are fully included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors would like to express their gratitude to the research and technological innovation funding agencies, FAPEMIG and CNPq, for their invaluable support. We also extend our sincere thanks to the educational and research institutions involved in this work, including the Universidade Federal de Minas Gerais (UFMG), Universidade Federal do Triângulo Mineiro (UFTM), and the Centro Universitário de Mineiros (Unifimes), for their unwavering commitment to advancing science and fostering collaboration. We are deeply grateful to all co-authors for their essential contributions; In particular, we acknowledge the equally significant contributions of J.S.S., C.B.M., A.G.B.F., A.L.M.d.S.M., R.B.M., M.O.C. and L.M.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Dai, H.; Liu, Q.; Liu, B. Research Progress on Mechanism of Podocyte Depletion in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 2615286. [Google Scholar] [CrossRef]
- Zhang, C.; Hou, B.; Yu, S.; Chen, Q.; Zhang, N.; Li, H. HGF alleviates high glucose-induced injury in podocytes by GSK3β inhibition and autophagy restoration. Biochim. Biophys. Acta 2016, 1863, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Toyoda, M.; Kaneyama, N.; Shiraiwa, S.; Moriya, H.; Miyatake, H.; Tanaka, E.; Yamamoto, N.; Miyauchi, M.; Kimura, M.; et al. Upregulation of α3β1-Integrin in Podocytes in Early-Stage Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 9265074. [Google Scholar] [CrossRef] [PubMed]
- Parchwani, D.N.; Upadhyah, A.A. Diabetic nephropathy: Progression and pathophysiology. Int. J. Med. Sci. Public Health 2012, 1, 59–70. [Google Scholar] [CrossRef]
- Fried, L.F.; Folkerts, K.; Smela, B.; Deon Bowrin, K.; Mernagh, P.; Millier, A.; Kovesdy, C.P. Targeted literature review of the burden of illness in patients with chronic kidney disease and type 2 diabetes. Am. J. Manag. Care 2021, 27, S168–S177. [Google Scholar]
- Tereda, A. From pathophysiology to personalized care: A comprehensive review of diabetic kidney disease. J. Med. Sci. Res. 2024, 12, 246–252. [Google Scholar]
- Satirapoj, B.; Adler, S.G. Comprehensive approach to diabetic nephropathy. Kidney Res. Clin. Pract. 2014, 33, 121–131. [Google Scholar] [CrossRef]
- Romagnani, P.; Remuzzi, G. Renal progenitors in non-diabetic and diabetic nephropathies. Trends Endocrinol. Metab. 2013, 24, 13–20. [Google Scholar] [CrossRef]
- Pan, Y.; Jiang, S.; Hou, Q.; Qiu, D.; Shi, J.; Wang, L.; Chen, Z.; Zhang, M.; Duan, A.; Qin, W.; et al. Dissection of Glomerular Transcriptional Profile in Patients With Diabetic Nephropathy: SRGAP2a Protects Podocyte Structure and Function. Diabetes 2018, 67, 717–730. [Google Scholar] [CrossRef]
- Giunti, S.; Barit, D.; Cooper, M.E. Mechanisms of diabetic nephropathy: Role of hypertension. Hypertension 2006, 48, 519–526. [Google Scholar] [CrossRef]
- Shah, I.M.; Mackay, S.P.; McKay, G.A. Therapeutic strategies in the treatment of diabetic nephropathy—A translational medicine approach. Curr. Med. Chem. 2009, 16, 997–1016. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.K.; Nicholas, S.B. Pathomechanisms of diabetic kidney disease. J. Clin. Med. 2023, 12, 7349. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Qiu, D.; Shi, Y.; Wang, S.; Zhou, X.; Chen, N.; Wei, J.; Wu, M.; Wu, H.; Duan, H. Thioredoxin-interacting protein deficiency alleviates phenotypic alterations of podocytes via inhibition of mTOR activation in diabetic nephropathy. J. Cell. Physiol. 2019, 234, 16485–16502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, W.; Sun, Y.; Qiao, Y.; Zhang, L.; Zhao, Z.; Lv, S. Wnt/β-Catenin Signaling Mediated-UCH-L1 Expression in Podocytes of Diabetic Nephropathy. Int. J. Mol. Sci. 2016, 17, 1404. [Google Scholar] [CrossRef] [PubMed]
- Conserva, F.; Gesualdo, L.; Papale, M. A systems biology overview on human diabetic nephropathy: From genetic susceptibility to post-transcriptional and post-translational modifications. J. Diabetes Res. 2016, 2016, 7934504. [Google Scholar] [CrossRef]
- Mima, A. Renal protection by sodium-glucose cotransporter 2 inhibitors and its underlying mechanisms in diabetic kidney disease. J. Diabetes Its Complicat. 2018, 32, 720–725. [Google Scholar] [CrossRef]
- Jiang, H.; Shao, X.; Jia, S.; Qu, L.; Weng, C.; Shen, X.; Wang, Y.; Huang, H.; Wang, C.; Feng, S.; et al. The Mitochondria-Targeted Metabolic Tubular Injury in Diabetic Kidney Disease. Cell. Physiol. Biochem. 2019, 52, 156–171. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Reeves, W.B.; Awad, A.S. Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 2021, 17, 319–334. [Google Scholar] [CrossRef]
- Balint, L.; Socaciu, C.; Socaciu, A.I.; Vlad, A.; Gadalean, F.; Bob, F.; Milas, O.; Cretu, O.M.; Suteanu-Simulescu, A.; Glavan, M.; et al. Metabolites Potentially Derived from Gut Microbiota Associated with Podocyte, Proximal Tubule, and Renal and Cerebrovascular Endothelial Damage in Early Diabetic Kidney Disease in T2DM Patients. Metabolites 2023, 13, 893. [Google Scholar] [CrossRef]
- Zhou, Z.; Wan, J.; Hou, X.; Geng, J.; Li, X.; Bai, X. MicroRNA-27a promotes podocyte injury via PPARγ-mediated β-catenin activation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2658, Correction in Cell Death Dis. 2017, 9, 652. https://doi.org/10.1038/s41419-018-0637-3. [Google Scholar] [CrossRef]
- Yamashiro, A.; Satoh, Y.; Endo, S.; Oshima, N. Extracellular signal-regulated kinase is activated in podocytes from patients with diabetic nephropathy. Hum. Cell 2024, 37, 1553–1558. [Google Scholar] [CrossRef]
- Ivanac-Janković, R.; Ćorić, M.; Furić-Čunko, V.; Lovičić, V.; Bašić-Jukić, N.; Kes, P. BMP-7 protein expression is downregulated in human diabetic nephropathy. Acta Clin. Croat. 2015, 54, 164–168. [Google Scholar]
- Carson, J.M.; Okamura, K.; Wakashin, H.; McFann, K.; Dobrinskikh, E.; Kopp, J.B.; Blaine, J. Podocytes degrade endocytosed albumin primarily in lysosomes. PLoS ONE 2014, 9, e99771. [Google Scholar] [CrossRef] [PubMed]
- Arslan, G.; Karabulut, Y.Y.; Yeleser, İ.; Erdal, M.E.; Demir, S.; Özdemir, A.A. Correlation of hsa-mirna-342-3p and SOX 6 Expression with Diabetic Nephropathy Classification, Prognostic Histomorphological Parameters and Laboratory Findings in Diabetic Nephropathy. Ann. Diagn. Pathol. 2025, 76, 152461. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.A.; Tawhari, I.; Safar-Boueri, L.; Seif, N.; Alahmadi, A.; Gargiulo, R.; Aggarwal, V.; Usman, I.; Kisselev, S.; Gharavi, A.G.; et al. COVID-19-Associated Glomerular Disease. J. Am. Soc. Nephrol. 2021, 32, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Ceol, M.; Tiralongo, E.; Baelde, H.J.; Vianello, D.; Betto, G.; Marangelli, A.; Bonfante, L.; Valente, M.; Della Barbera, M.; D’Angelo, A.; et al. Involvement of the tubular ClC-type exchanger ClC-5 in glomeruli of human proteinuric nephropathies. PLoS ONE 2012, 7, e45605. [Google Scholar] [CrossRef]
- Canney, A.L.; Cohen, R.V.; Elliott, J.A.; Aboud, C.M.; Martin, W.P.; Docherty, N.G.; le Roux, C.W. Improvements in diabetic albuminuria and podocyte differentiation following Roux-en-Y gastric bypass surgery. Diab Vasc. Dis. Res. 2020, 17, 1479164119879039. [Google Scholar] [CrossRef]
- Esselman, A.B.; Moser, F.A.; Tideman, L.E.M.; Migas, L.G.; Djambazova, K.V.; Colley, M.E.; Pingry, E.L.; Patterson, N.H.; Farrow, M.A.; Yang, H.; et al. In situ molecular profiles of glomerular cells by integrated imaging mass spectrometry and multiplexed immunofluorescence microscopy. Kidney Int. 2025, 107, 332–337. [Google Scholar] [CrossRef]
- Denhez, B.; Rousseau, M.; Spino, C.; Dancosst, D.A.; Dumas, M.; Guay, A.; Lizotte, F.; Geraldes, P. Saturated fatty acids induce insulin resistance in podocytes through inhibition of IRS1 via activation of both IKKβ and mTORC1. Sci. Rep. 2020, 10, 21628. [Google Scholar] [CrossRef]
- Audzeyenka, I.; Rachubik, P.; Rogacka, D.; Typiak, M.; Kulesza, T.; Angielski, S.; Rychłowski, M.; Wysocka, M.; Gruba, N.; Lesner, A.; et al. Cathepsin C is a novel mediator of podocyte and renal injury induced by hyperglycemia. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118723. [Google Scholar] [CrossRef]
- Hayashi, D.; Wang, L.; Ueda, S.; Yamanoue, M.; Ashida, H.; Shirai, Y. The mechanisms of ameliorating effect of a green tea polyphenol on diabetic nephropathy based on diacylglycerol kinase α. Sci. Rep. 2020, 10, 11790. [Google Scholar] [CrossRef]
- Chen, A.; Feng, Y.; Lai, H.; Ju, W.; Li, Z.; Li, Y.; Wang, A.; Hong, Q.; Zhong, F.; Wei, C.; et al. Soluble RARRES1 induces podocyte apoptosis to promote glomerular disease progression. J. Clin. Investig. 2020, 130, 5523–5535. [Google Scholar] [CrossRef]
- Angeletti, A.; Cantarelli, C.; Petrosyan, A.; Andrighetto, S.; Budge, K.; D’Agati, V.D.; Hartzell, S.; Malvi, D.; Donadei, C.; Thurman, J.M.; et al. Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis. J. Exp. Med. 2020, 217, e20191699. [Google Scholar] [CrossRef]
- Albrecht, M.; Sticht, C.; Wagner, T.; Hettler, S.A.; De La Torre, C.; Qiu, J.; Gretz, N.; Albrecht, T.; Yard, B.; Sleeman, J.P.; et al. The crosstalk between glomerular endothelial cells and podocytes controls their responses to metabolic stimuli in diabetic nephropathy. Sci. Rep. 2023, 13, 17985. [Google Scholar] [CrossRef]
- Endlich, N.; Lange, T.; Kuhn, J.; Klemm, P.; Kotb, A.M.; Siegerist, F.; Kindt, F.; Lindenmeyer, M.T.; Cohen, C.D.; Kuss, A.W.; et al. BDNF: mRNA expression in urine cells of patients with chronic kidney disease and its role in kidney function. J. Cell. Mol. Med. 2018, 22, 5265–5277. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, D.; Kuwabara, T.; Hata, Y.; Umemoto, S.; Kanki, T.; Nishiguchi, Y.; Mizumoto, T.; Hayata, M.; Kakizoe, Y.; Izumi, Y.; et al. Suppressed ER-associated degradation by intraglomerular cross talk between mesangial cells and podocytes causes podocyte injury in diabetic kidney disease. FASEB J. 2020, 34, 15577–15590. [Google Scholar] [CrossRef]
- Hu, Y.; Ye, S.; Xing, Y.; Lv, L.; Hu, W.; Zhou, W. Saxagliptin attenuates glomerular podocyte injury by increasing the expression of renal nephrin and podocin in type 2 diabetic rats. Acta Diabetol. 2020, 57, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liao, L.; Wang, B.; Wu, Z. Identification and validation of immune and cuproptosis-related genes for diabetic nephropathy by WGCNA and machine learning. Front. Immunol. 2024, 15, 1332279. [Google Scholar] [CrossRef]
- Fiorina, P.; Vergani, A.; Bassi, R.; Niewczas, M.A.; Altintas, M.M.; Pezzolesi, M.G.; D’Addio, F.; Chin, M.; Tezza, S.; Ben Nasr, M.; et al. Role of podocyte B7-1 in diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1415–1429. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, B.; Gong, A.Y.; Malhotra, D.K.; Gupta, R.; Dworkin, L.D.; Gong, R. The ketone body β-hydroxybutyrate mitigates the senescence response of glomerular podocytes to diabetic insults. Kidney Int. 2021, 100, 1037–1053, Correction in Kidney Int. 2022, 101, 1301–1302. https://doi.org/10.1016/j.kint.2022.04.002. [Google Scholar] [CrossRef]
- Hou, B.; Li, Y.; Li, X.; Zhang, C.; Zhao, Z.; Chen, Q.; Zhang, N.; Li, H. HGF protected against diabetic nephropathy via autophagy-lysosome pathway in podocyte by modulating PI3K/Akt-GSK3β-TFEB axis. Cell. Signal. 2020, 75, 109744. [Google Scholar] [CrossRef]
- Han, W.; Zheng, Q.; Zhang, Z.; Wang, X.; Gao, L.; Niu, D.; Li, R.; Wang, C. Association of the podocyte phenotype with extracapillary hypercellularity in patients with diabetic kidney disease. J. Nephrol. 2024, 37, 2209–2222. [Google Scholar] [CrossRef]
- Holderied, A.; Romoli, S.; Eberhard, J.; Konrad, L.A.; Devarapu, S.K.; Marschner, J.A.; Müller, S.; Anders, H.J. Glomerular parietal epithelial cell activation induces collagen secretion and thickening of Bowman’s capsule in diabetes. Lab. Investig. 2015, 95, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Gujarati, N.A.; Frimpong, B.O.; Zaidi, M.; Bronstein, R.; Revelo, M.P.; Haley, J.D.; Kravets, I.; Guo, Y.; Mallipattu, S.K. Podocyte-specific KLF6 primes proximal tubule CaMK1D signaling to attenuate diabetic kidney disease. Nat. Commun. 2024, 15, 8038. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.; Li, J.; Asadi, M.; Basgen, J.M.; Zhu, B.; Yi, Z.; Jiang, S.; Doke, T.; El Shamy, O.; Patel, N.; et al. DACH1 protects podocytes from experimental diabetic injury and modulates PTIP-H3K4Me3 activity. J. Clin. Investig. 2021, 131, 141279. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Cui, H.; Ding, J. Smad3 signalling affects high glucose-induced podocyte injury via regulation of the cytoskeletal protein transgelin. Nephrology 2020, 25, 659–666. [Google Scholar] [CrossRef]
- Fu, Y.; Sun, Y.; Wang, M.; Hou, Y.; Huang, W.; Zhou, D.; Wang, Z.; Yang, S.; Tang, W.; Zhen, J.; et al. Elevation of JAML Promotes Diabetic Kidney Disease by Modulating Podocyte Lipid Metabolism. Cell Metab. 2020, 32, 1052–1062.e1058. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Z.; Hu, H.; Yang, K.; Zhu, Z.; Yang, Q.; Liang, W. LRH-1 activation alleviates diabetes-induced podocyte injury by promoting GLS2-mediated glutaminolysis. Cell Prolif. 2023, 56, e13479. [Google Scholar] [CrossRef]
- Kimura, M.; Toyoda, M.; Kato, M.; Kobayashi, K.; Abe, M.; Kobayashi, T.; Miyauchi, M.; Yamamoto, N.; Umezono, T.; Suzuki, D. Expression of alpha-actinin-4 in human diabetic nephropathy. Intern. Med. 2008, 47, 1099–1106. [Google Scholar] [CrossRef]
- Lei, Q.; Hou, X.; Liu, X.; Liang, D.; Fan, Y.; Xu, F.; Liang, S.; Yang, J.; Xie, G.; Liu, Z.; et al. Artificial intelligence assists identification and pathologic classification of glomerular lesions in patients with diabetic nephropathy. J. Transl. Med. 2024, 22, 397. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Q.; Fan, X.; Zhen, J.; Wang, C.; Chen, H.; Liu, Y.; Zhou, P.; Zhang, T.; Huang, T.; et al. Long noncoding RNA ENST00000436340 promotes podocyte injury in diabetic kidney disease by facilitating the association of PTBP1 with RAB3B. Cell Death Dis. 2023, 14, 130. [Google Scholar] [CrossRef]
- Kondapi, K.; Kumar, N.L.; Moorthy, S.; Silambanan, S. A Study of Association of Urinary Nephrin with Albuminuria in Patients with Diabetic Nephropathy. Indian J. Nephrol. 2021, 31, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ye, S.; Kong, J.; Zhou, Q.; Wang, Z.; Zhang, Y.; Yan, H.; Wang, Y.; Li, T.; Xie, Y.; et al. DOT1L protects against podocyte injury in diabetic kidney disease through phospholipase C-like 1. Cell Commun. Signal. 2024, 22, 519. [Google Scholar] [CrossRef] [PubMed]
- Kondapi, K.; Silambanan, S.; Moorthy, S.; Kumar, N.L. A Study of the Risk Factors and Urinary Podocin as an Early Prognostic Indicator of Renal Injury in Diabetic Nephropathy. J. Assoc. Physicians India 2021, 69, 11–12. [Google Scholar] [PubMed]
- Shahzad, K.; Fatima, S.; Khawaja, H.; Elwakiel, A.; Gadi, I.; Ambreen, S.; Zimmermann, S.; Mertens, P.R.; Biemann, R.; Isermann, B. Podocyte-specific Nlrp3 inflammasome activation promotes diabetic kidney disease. Kidney Int. 2022, 102, 766–779. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Hasegawa, K.; Yasuda, I.; Muraoka, H.; Umino, H.; Tokuyama, H.; Hashiguchi, A.; Wakino, S.; Itoh, H. Diabetic condition induces hypertrophy and vacuolization in glomerular parietal epithelial cells. Sci. Rep. 2021, 11, 1515. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, X.; Hu, X.; Gao, L.; Zeng, H.; Wang, X.; Huang, Y.; Zhu, W.; Wang, J.; Wen, J.; et al. METTL3-mediated m(6)A modification of TIMP2 mRNA promotes podocyte injury in diabetic nephropathy. Mol. Ther. 2022, 30, 1721–1740. [Google Scholar] [CrossRef]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Rüegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef]
- Gödel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mór, A.; Lindenmeyer, M.T.; Rastaldi, M.P.; Hartleben, G.; et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef]
- Langham, R.G.; Kelly, D.J.; Cox, A.J.; Thomson, N.M.; Holthöfer, H.; Zaoui, P.; Pinel, N.; Cordonnier, D.J.; Gilbert, R.E. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: Effects of angiotensin converting enzyme inhibition. Diabetologia 2002, 45, 1572–1576. [Google Scholar] [CrossRef]
- Bai, X.; Geng, J.; Li, X.; Wan, J.; Liu, J.; Zhou, Z.; Liu, X. Long Noncoding RNA LINC01619 Regulates MicroRNA-27a/Forkhead Box Protein O1 and Endoplasmic Reticulum Stress-Mediated Podocyte Injury in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 29, 355–376. [Google Scholar] [CrossRef]
- Wang, L.; Li, H. MiR-770-5p facilitates podocyte apoptosis and inflammation in diabetic nephropathy by targeting TIMP3. Biosci. Rep. 2020, 40, BSR20193653. [Google Scholar] [CrossRef]
- Lai, H.; Chen, A.; Cai, H.; Fu, J.; Salem, F.; Li, Y.; He, J.C.; Schlondorff, D.; Lee, K. Podocyte and endothelial-specific elimination of BAMBI identifies differential transforming growth factor-β pathways contributing to diabetic glomerulopathy. Kidney Int. 2020, 98, 601–614. [Google Scholar] [CrossRef]
- Hudkins, K.L.; Li, X.; Holland, A.L.; Swaminathan, S.; Alpers, C.E. Regression of diabetic nephropathy by treatment with empagliflozin in BTBR ob/ob mice. Nephrol. Dial. Transplant. 2022, 37, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Kostic, S.; Hauke, T.; Ghahramani, N.; Filipovic, N.; Vukojevic, K. Expression pattern of apoptosis-inducing factor in the kidneys of streptozotocin-induced diabetic rats. Acta Histochem. 2020, 122, 151655. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, M.; Wolf, G. AGE-Induced Suppression of EZH2 Mediates Injury of Podocytes by Reducing H3K27me3. Am. J. Nephrol. 2020, 51, 676–692. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Jiang, L.; Li, Y.Y.; Huang, Y.B.; Hu, X.R.; Zhu, W.; Wang, X.; Wu, Y.G.; Meng, X.M.; Qi, X.M. Wogonin protects glomerular podocytes by targeting Bcl-2-mediated autophagy and apoptosis in diabetic kidney disease. Acta Pharmacol. Sin. 2022, 43, 96–110. [Google Scholar] [CrossRef]
- Li, X.; Zhang, P.; Jiang, S.; Shang, S.; Zhang, J.; Liu, J.; Li, C.; Gao, Y.; Zhang, H.; Li, W. Utilizing Podocyte Foot Process Morphology for the Identification of Diabetic Nephropathy with or without Minimal Change Disease: Establishment of an Artificial Intelligence-Assisted Diagnostic Model. Diabetes Metab. Syndr. Obes. 2025, 18, 2141–2153. [Google Scholar] [CrossRef]
- Li, L.; Li, J.; Li, R.; Zhao, X.; Chen, Y.; Cai, Y.; Yang, Y.; Wang, W.; Zheng, S.; Zhang, L.; et al. Podocyte RIPK3 Deletion Improves Diabetic Kidney Disease by Attenuating NF-κB p65 Driven Inflammation. Adv. Sci. 2025, 12, e03325. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, H.; Song, N.; Liang, Y.; Zhu, J.; Chen, J.; Ning, Y.; Hu, J.; Fang, Y.; Teng, J.; et al. METTL14 aggravates podocyte injury and glomerulopathy progression through N(6)-methyladenosine-dependent downregulating of Sirt1. Cell Death Dis. 2021, 12, 881. [Google Scholar] [CrossRef]
- Liang, X.; Wang, P.; Chen, B.; Ge, Y.; Gong, A.Y.; Flickinger, B.; Malhotra, D.K.; Wang, L.J.; Dworkin, L.D.; Liu, Z.; et al. Glycogen synthase kinase 3β hyperactivity in urinary exfoliated cells predicts progression of diabetic kidney disease. Kidney Int. 2020, 97, 175–192. [Google Scholar] [CrossRef]
- Hu, H.; Wang, J.; Peng, Z.; Fan, Y.; Yang, Q.; Hu, J. Dapagliflozin attenuates diabetes-induced podocyte lipotoxicity via ERRα-Mediated lipid metabolism. Free Radic. Biol. Med. 2025, 234, 178–191. [Google Scholar] [CrossRef]
- Lv, Z.; Hu, J.; Su, H.; Yu, Q.; Lang, Y.; Yang, M.; Fan, X.; Liu, Y.; Liu, B.; Zhao, Y.; et al. TRAIL induces podocyte PANoptosis via death receptor 5 in diabetic kidney disease. Kidney Int. 2025, 107, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chen, P.P.; Zhang, J.X.; Li, X.Q.; Wang, G.H.; Yuan, B.Y.; Huang, S.J.; Liu, X.Q.; Jiang, T.T.; Wang, M.Y.; et al. GPR43 deficiency protects against podocyte insulin resistance in diabetic nephropathy through the restoration of AMPKα activity. Theranostics 2021, 11, 4728–4742. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, M.; Toyoda, M.; Kobayashi, K.; Abe, M.; Kobayashi, T.; Kato, M.; Yamamoto, N.; Kimura, M.; Umezono, T.; Suzuki, D. Hypertrophy and loss of podocytes in diabetic nephropathy. Intern. Med. 2009, 48, 1615–1620. [Google Scholar] [CrossRef][Green Version]
- Martins, A.; Bernardes, A.B.; Ferreira, V.A.; Wanderley, D.C.; Araújo, S.A.; do Carmo Neto, J.R.; da Silva, C.A.; Lira, R.C.P.; Araújo, L.S.; Dos Reis, M.A.; et al. In situ assessment of Mindin as a biomarker of podocyte lesions in diabetic nephropathy. PLoS ONE 2023, 18, e0284789. [Google Scholar] [CrossRef] [PubMed]
- Lizotte, F.; Rousseau, M.; Denhez, B.; Lévesque, D.; Guay, A.; Liu, H.; Moreau, J.; Higgins, S.; Sabbagh, R.; Susztak, K.; et al. Deletion of protein tyrosine phosphatase SHP-1 restores SUMOylation of podocin and reverses the progression of diabetic kidney disease. Kidney Int. 2023, 104, 787–802, Erratum in Kidney Int. 2023, 104, 1228. https://doi.org/10.1016/j.kint.2023.10.007. [Google Scholar] [CrossRef]
- Lee, E.; Lee, H.S. Peroxidase expression is decreased by palmitate in cultured podocytes but increased in podocytes of advanced diabetic nephropathy. J. Cell. Physiol. 2018, 233, 9060–9069. [Google Scholar] [CrossRef]
- Löwen, J.; Gröne, E.F.; Groß-Weißmann, M.L.; Bestvater, F.; Gröne, H.J.; Kriz, W. Pathomorphological sequence of nephron loss in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2021, 321, F600–F616, Correction in Am. J. Physiol. Ren. Physiol. 2022, 322, F245–F377. [Google Scholar] [CrossRef]
- Lu, J.; Li, X.Q.; Chen, P.P.; Zhang, J.X.; Liu, L.; Wang, G.H.; Liu, X.Q.; Jiang, T.T.; Wang, M.Y.; Liu, W.T.; et al. Activation of acetyl-CoA synthetase 2 mediates kidney injury in diabetic nephropathy. JCI Insight 2023, 8, 165817. [Google Scholar] [CrossRef]
- Li, X.; Li, Q.; Jiang, X.; Song, S.; Zou, W.; Yang, Q.; Liu, S.; Chen, S.; Wang, C. Inhibition of SGLT2 protects podocytes in diabetic kidney disease by rebalancing mitochondria-associated endoplasmic reticulum membranes. Cell Commun. Signal. 2024, 22, 534. [Google Scholar] [CrossRef]
- Nishad, R.; Mukhi, D.; Singh, A.K.; Motrapu, M.; Chintala, K.; Tammineni, P.; Pasupulati, A.K. Growth hormone induces mitotic catastrophe of glomerular podocytes and contributes to proteinuria. Cell Death Dis. 2021, 12, 342. [Google Scholar] [CrossRef]
- Petrica, L.; Hogea, E.; Gadalean, F.; Vlad, A.; Vlad, M.; Dumitrascu, V.; Velciov, S.; Gluhovschi, C.; Bob, F.; Ursoniu, S.; et al. Long noncoding RNAs may impact podocytes and proximal tubule function through modulating miRNAs expression in Early Diabetic Kidney Disease of Type 2 Diabetes Mellitus patients. Int. J. Med. Sci. 2021, 18, 2093–2101. [Google Scholar] [CrossRef]
- Matoba, K.; Sekiguchi, K.; Nagai, Y.; Takeda, Y.; Takahashi, H.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Renal ROCK Activation and Its Pharmacological Inhibition in Patients with Diabetes. Front. Pharmacol. 2021, 12, 738121. [Google Scholar] [CrossRef]
- Palmer, M.B.; Abedini, A.; Jackson, C.; Blady, S.; Chatterjee, S.; Sullivan, K.M.; Townsend, R.R.; Brodbeck, J.; Almaani, S.; Srivastava, A.; et al. The Role of Glomerular Epithelial Injury in Kidney Function Decline in Patients with Diabetic Kidney Disease in the TRIDENT Cohort. Kidney Int. Rep. 2021, 6, 1066–1080. [Google Scholar] [CrossRef] [PubMed]
- Naito, S.; Nakayama, K.; Kawashima, N. Enhanced Levels of Glycosphingolipid GM3 Delay the Progression of Diabetic Nephropathy. Int. J. Mol. Sci. 2023, 24, 11355. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Teng, Y.; Wang, R.; Chen, D.; Chen, H. Deciphering the molecular nexus of BTG2 in periodontitis and diabetic kidney disease. BMC Med. Genom. 2024, 17, 152. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Corna, D.; Locatelli, M.; Cassis, P.; Carminati, C.E.; Bolognini, S.; Zoja, C.; Remuzzi, G.; Benigni, A.; et al. C3a receptor blockade protects podocytes from injury in diabetic nephropathy. JCI Insight 2020, 5, 131849. [Google Scholar] [CrossRef]
- Khurana, I.; Kaipananickal, H.; Maxwell, S.; Birkelund, S.; Syreeni, A.; Forsblom, C.; Okabe, J.; Ziemann, M.; Kaspi, A.; Rafehi, H.; et al. Reduced methylation correlates with diabetic nephropathy risk in type 1 diabetes. J. Clin. Investig. 2023, 133, 160959. [Google Scholar] [CrossRef]
- Mukhi, D.; Kolligundla, L.P.; Maruvada, S.; Nishad, R.; Pasupulati, A.K. Growth hormone induces transforming growth factor-β1 in podocytes: Implications in podocytopathy and proteinuria. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119391. [Google Scholar] [CrossRef]
- Li, C.; Ng, J.K.; Chan, G.C.; Fung, W.W.; Chow, K.M.; Szeto, C.C. Preservation of Urinary Podocyte Markers in Diabetic Kidney Disease by Sodium-Glucose Cotransporter 2 Inhibitor Therapy. Kidney Dis. 2025, 11, 218–225. [Google Scholar] [CrossRef]
- Lv, Z.; Wang, Z.; Hu, J.; Su, H.; Liu, B.; Lang, Y.; Yu, Q.; Liu, Y.; Fan, X.; Yang, M.; et al. LncRNA PVT1 induces mitochondrial dysfunction of podocytes via TRIM56 in diabetic kidney disease. Cell Death Dis. 2024, 15, 697. [Google Scholar] [CrossRef]
- Shi, W.; Huang, Y.; Zhao, X.; Xie, Z.; Dong, W.; Li, R.; Chen, Y.; Li, Z.; Wang, W.; Ye, Z.; et al. Histone deacetylase 4 mediates high glucose-induced podocyte apoptosis via upregulation of calcineurin. Biochem. Biophys. Res. Commun. 2020, 533, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, S.P.; Reddi, A.S.; Chandran, C.B.; Chevalier, J.M.; Okechukwu, C.N.; Seshan, S.V. Collapsing glomerulopathy superimposed on diabetic nephropathy: Insights into etiology of an under-recognized, severe pattern of glomerular injury. Nephrol. Dial. Transplant. 2014, 29, 392–399. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, L.; Zeng, H.; Gao, L.; Guo, S.; Chen, C.; Zhang, M.; Ma, L.; Li, Y.; Qi, X.; et al. Circ-0000953 deficiency exacerbates podocyte injury and autophagy disorder by targeting Mir665-3p-Atg4b in diabetic nephropathy. Autophagy 2024, 20, 1072–1097. [Google Scholar] [CrossRef]
- Su, J.; Li, S.J.; Chen, Z.H.; Zeng, C.H.; Zhou, H.; Li, L.S.; Liu, Z.H. Evaluation of podocyte lesion in patients with diabetic nephropathy: Wilms’ tumor-1 protein used as a podocyte marker. Diabetes Res. Clin. Pract. 2010, 87, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Motrapu, M.; Świderska, M.K.; Mesas, I.; Marschner, J.A.; Lei, Y.; Martinez Valenzuela, L.; Fu, J.; Lee, K.; Angelotti, M.L.; Antonelli, G.; et al. Drug Testing for Residual Progression of Diabetic Kidney Disease in Mice Beyond Therapy with Metformin, Ramipril, and Empagliflozin. J. Am. Soc. Nephrol. 2020, 31, 1729–1745. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, S.; Ramanand, A.; Stark, A.; Varghese, V.; Chalmers, D.; Au-Yeung, N.; Kanduri, S.R.; Lukitsch, I.; Poloni, J.A.T.; Keitel, E.; et al. Urinary Vacuolar Casts Are a Unique Type of Casts in Advanced Proteinuric Glomerulopathies. Kidney360 2024, 5, 216–227. [Google Scholar] [CrossRef]
- Minakawa, A.; Fukuda, A.; Sato, Y.; Kikuchi, M.; Kitamura, K.; Wiggins, R.C.; Fujimoto, S. Podocyte hypertrophic stress and detachment precedes hyperglycemia or albuminuria in a rat model of obesity and type2 diabetes-associated nephropathy. Sci. Rep. 2019, 9, 18485. [Google Scholar] [CrossRef]
- Sawada, A.; Kawanishi, K.; Igarashi, Y.; Taneda, S.; Hattori, M.; Ishida, H.; Tanabe, K.; Koike, J.; Honda, K.; Nagashima, Y.; et al. Overexpression of Plasmalemmal Vesicle-Associated Protein-1 Reflects Glomerular Endothelial Injury in Cases of Proliferative Glomerulonephritis with Monoclonal IgG Deposits. Kidney Int. Rep. 2023, 8, 151–163. [Google Scholar] [CrossRef]
- Sharma, K.R.; Heckler, K.; Stoll, S.J.; Hillebrands, J.L.; Kynast, K.; Herpel, E.; Porubsky, S.; Elger, M.; Hadaschik, B.; Bieback, K.; et al. ELMO1 protects renal structure and ultrafiltration in kidney development and under diabetic conditions. Sci. Rep. 2016, 6, 37172. [Google Scholar] [CrossRef]
- Sunilkumar, S.; Yerlikaya, E.I.; VanCleave, A.; Subrahmanian, S.M.; Toro, A.L.; Kimball, S.R.; Dennis, M.D. REDD1-dependent GSK3β signaling in podocytes promotes canonical NF-κB activation in diabetic nephropathy. J. Biol. Chem. 2025, 301, 108244. [Google Scholar] [CrossRef]
- Lu, Q.; Hu, X.; Hou, Q.; Yu, L.; Cao, K.; Ding, D.; Lu, Y.; Dai, C. Rheb1 deficiency elicits mitochondrial dysfunction and accelerates podocyte senescence through promoting Atp5f1c acetylation. Cell Signal. 2024, 124, 111451. [Google Scholar] [CrossRef]
- Petrica, L.; Vlad, A.; Gadalean, F.; Muntean, D.M.; Vlad, D.; Dumitrascu, V.; Bob, F.; Milas, O.; Suteanu-Simulescu, A.; Glavan, M.; et al. Mitochondrial DNA Changes in Blood and Urine Display a Specific Signature in Relation to Inflammation in Normoalbuminuric Diabetic Kidney Disease in Type 2 Diabetes Mellitus Patients. Int. J. Mol. Sci. 2023, 24, 9803. [Google Scholar] [CrossRef]
- Boi, R.; Lassén, E.; Johansson, A.; Liu, P.; Chaudhari, A.; Tati, R.; Müller-Deile, J.; Schiffer, M.; Ebefors, K.; Nyström, J. Cytoskeleton-associated protein 4 affects podocyte cytoskeleton dynamics in diabetic kidney disease. JCI Insight 2025, 10, 181298. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, M.; Zhao, Y.; Gong, J.; Su, H.; Yuan, F.; Fang, K.; Yuan, X.; Yu, X.; Dong, H.; et al. Berberine protects against diabetic kidney disease via promoting PGC-1α-regulated mitochondrial energy homeostasis. Br. J. Pharmacol. 2020, 177, 3646–3661. [Google Scholar] [CrossRef] [PubMed]
- Dalla Vestra, M.; Masiero, A.; Roiter, A.M.; Saller, A.; Crepaldi, G.; Fioretto, P. Is podocyte injury relevant in diabetic nephropathy? Studies in patients with type 2 diabetes. Diabetes 2003, 52, 1031–1035. [Google Scholar] [CrossRef]
- Tian, X.; Inoue, K.; Zhang, Y.; Wang, Y.; Sperati, C.J.; Pedigo, C.E.; Zhao, T.; Yan, M.; Groener, M.; Moledina, D.G.; et al. Inhibiting calpain 1 and 2 in cyclin G associated kinase-knockout mice mitigates podocyte injury. JCI Insight 2020, 5, 142740. [Google Scholar] [CrossRef]
- Veron, D.; Aggarwal, P.K.; Li, Q.; Moeckel, G.; Kashgarian, M.; Tufro, A. Podocyte VEGF-A Knockdown Induces Diffuse Glomerulosclerosis in Diabetic and in eNOS Knockout Mice. Front. Pharmacol. 2021, 12, 788886. [Google Scholar] [CrossRef] [PubMed]
- Su, P.P.; Liu, D.W.; Zhou, S.J.; Chen, H.; Wu, X.M.; Liu, Z.S. Down-regulation of Risa improves podocyte injury by enhancing autophagy in diabetic nephropathy. Mil. Med. Res. 2022, 9, 23. [Google Scholar] [CrossRef]
- Suarez, R.; Villarreal, C.; Nahuelpán, Y.; Jara, C.; Oyarzún, C.; Alarcón, S.; Díaz-Encarnación, M.M.; Guillén-Gómez, E.; Quezada, C.; San Martín, R. Defective insulin-stimulated equilibrative nucleoside transporter-2 activity and altered subcellular transporter distribution drive the loss of adenosine homeostasis in diabetic kidney disease progression. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 166890. [Google Scholar] [CrossRef]
- Song, S.; Shi, C.; Bian, Y.; Yang, Z.; Mu, L.; Wu, H.; Duan, H.; Shi, Y. Sestrin2 remedies podocyte injury via orchestrating TSP-1/TGF-β1/Smad3 axis in diabetic kidney disease. Cell Death Dis. 2022, 13, 663. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Weidner, J.; Allamargot, C.; Piper, R.C.; Misurac, J.; Nester, C. Dynein-Mediated Trafficking: A New Mechanism of Diabetic Podocytopathy. Kidney360 2023, 4, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, V.T.N.; Nair, V.; Melsom, T.; Looker, H.C.; Mariani, L.H.; Fermin, D.; Eichinger, F.; Menon, R.; Subramanian, L.; Ladd, P.; et al. Molecular programs associated with glomerular hyperfiltration in early diabetic kidney disease. Kidney Int. 2022, 102, 1345–1358. [Google Scholar] [CrossRef]
- Woo, C.Y.; Baek, J.Y.; Kim, A.R.; Hong, C.H.; Yoon, J.E.; Kim, H.S.; Yoo, H.J.; Park, T.S.; Kc, R.; Lee, K.U.; et al. Inhibition of Ceramide Accumulation in Podocytes by Myriocin Prevents Diabetic Nephropathy. Diabetes Metab. J. 2020, 44, 581–591. [Google Scholar] [CrossRef]
- Sun, S.; Yang, S.; Cheng, Y.; Fang, T.; Qu, J.; Tian, L.; Zhang, M.; Wu, S.; Sun, B.; Chen, L. Jinlida granules alleviate podocyte apoptosis and mitochondrial dysfunction via the AMPK/PGC-1α pathway in diabetic nephropathy. Int. J. Mol. Med. 2025, 55, 26. [Google Scholar] [CrossRef]
- Tao, Y.; Chaudhari, S.; Shotorbani, P.Y.; Ding, Y.; Chen, Z.; Kasetti, R.; Zode, G.; Ma, R. Enhanced Orai1-mediated store-operated Ca2+ channel/calpain signaling contributes to high glucose-induced podocyte injury. J. Biol. Chem. 2022, 298, 101990. [Google Scholar] [CrossRef]
- Uil, M.; Hau, C.M.; Ahdi, M.; Mills, J.D.; Kers, J.; Saleem, M.A.; Florquin, S.; Gerdes, V.E.A.; Nieuwland, R.; Roelofs, J. Cellular origin and microRNA profiles of circulating extracellular vesicles in different stages of diabetic nephropathy. Clin. Kidney J. 2021, 14, 358–365. [Google Scholar] [CrossRef]
- Yang, Q.; Yang, S.; Liang, Y.; Sun, Q.; Fang, Y.; Jiang, L.; Wen, P.; Yang, J. UCP2 deficiency impairs podocyte autophagy in diabetic nephropathy. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166705. [Google Scholar] [CrossRef]
- Ward, H.H.; Anquetil, F.; Das, V.; Gibson, C.B.; Dovmark, T.H.; Kusmartseva, I.; Yang, M.; Beery, M.; Atkinson, M.A.; Zeng, X.; et al. Network for Pancreatic Organ donors with Diabetes-Kidney: A Heterogenous Donor Cohort for the Investigation of Diabetic Kidney Disease Pathogenesis and Progression. Kidney360 2025, 6, 15–26. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Iwano, M.; Suzuki, D.; Nakatani, K.; Kimura, K.; Harada, K.; Kubo, A.; Akai, Y.; Toyoda, M.; Kanauchi, M.; et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am. J. Kidney Dis. 2009, 54, 653–664. [Google Scholar] [CrossRef]
- Yao, T.; Zha, D.; Hu, C.; Wu, X. Circ_0000285 promotes podocyte injury through sponging miR-654-3p and activating MAPK6 in diabetic nephropathy. Gene 2020, 747, 144661. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pan, Y.; Cao, S.; Sasaki, K.; Wang, Y.; Niu, A.; Fan, X.; Wang, S.; Zhang, M.Z.; Harris, R.C. Podocyte EGFR Inhibits Autophagy Through Upregulation of Rubicon in Type 2 Diabetic Nephropathy. Diabetes 2021, 70, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Ng, J.K.; Fung, W.W.; Chan, G.C.; Chow, K.M.; Szeto, C.C. Intrarenal and Urinary Glycogen Synthase Kinase-3 Beta Levels in Diabetic and Nondiabetic Chronic Kidney Disease. Kidney Blood Press. Res. 2023, 48, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Zhai, R.; Xie, L.; Zheng, Z.; Jian, G.; Chen, T.; Su, J.; Gao, C.; Wang, N.; Yang, X.; et al. Xuesaitong Protects Podocytes from Apoptosis in Diabetic Rats through Modulating PTEN-PDK1-Akt-mTOR Pathway. J. Diabetes Res. 2020, 2020, 9309768. [Google Scholar] [CrossRef]
- Zeng, L.; Fung, W.W.; Chan, G.C.; Ng, J.K.; Chow, K.M.; Szeto, C.C. Urinary and Kidney Podocalyxin and Podocin Levels in Diabetic Kidney Disease: A Kidney Biopsy Study. Kidney Med. 2023, 5, 100569. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Y.; He, X.; Yang, L.; Wang, J.; Xia, S.; Liu, D.; Liu, S.; Liu, W.; Duan, H. Cdk5-Mediated Phosphorylation of Sirt1 Contributes to Podocyte Mitochondrial Dysfunction in Diabetic Nephropathy. Antioxid. Redox Signal. 2021, 34, 171–190. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Z.; Yang, Y.; Han, Z.; Pan, Q.; Chen, Y.; Yu, H.; Shen, S.; Xu, L. The Impact of METTL3 on MDM2 Promotes Podocytes Injury During Diabetic Kidney Disease. J. Cell. Mol. Med. 2025, 29, e70627. [Google Scholar] [CrossRef]
- Yu, H.; Chen, Y.; Ma, H.; Wang, Z.; Zhang, R.; Jiao, J. TRPC6 mediates high glucose-induced mitochondrial fission through activation of CDK5 in cultured human podocytes. Front. Physiol. 2022, 13, 984760. [Google Scholar] [CrossRef]
- Li, C.; Szeto, C.C. Urinary podocyte markers in diabetic kidney disease. Kidney Res. Clin. Pract. 2024, 43, 274–286. [Google Scholar] [CrossRef]
- Wang, S.; Ai, H.; Liu, L.; Zhang, X.; Gao, F.; Zheng, L.; Yi, J.; Sun, L.; Yu, C.; Zhao, H.; et al. Micro-RNA-27a/b negatively regulates hepatic gluconeogenesis by targeting FOXO1. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E911–E924. [Google Scholar] [CrossRef]
- Xu, D.; Tong, Z.; Yang, P.; Chen, Q.; Wang, S.; Zhao, W.; Han, L.; Yin, Y.; Xu, R.; Zhang, M.; et al. G protein-coupled receptor 107 deficiency promotes development of diabetic nephropathy. Mol. Biomed. 2025, 6, 10. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, C.; Ye, Q.; Tong, L.; Jiang, H.; Zhu, X.; Huang, L.; Lin, W.; Fu, H.; Wang, J.; et al. Podocyte apoptosis in diabetic nephropathy by BASP1 activation of the p53 pathway via WT1. Acta Physiol. 2021, 232, e13634. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, J.; Dai, S.; Gao, P.; Qi, Y.; Zhao, X.; Liu, J.; Wang, Y.; Gao, Y. miRNA-193a-mediated WT1 suppression triggers podocyte injury through activation of the EZH2/β-catenin/NLRP3 pathway in children with diabetic nephropathy. Exp. Cell Res. 2024, 442, 114238. [Google Scholar] [CrossRef] [PubMed]
- Sawai, K.; Mukoyama, M.; Mori, K.; Yokoi, H.; Koshikawa, M.; Yoshioka, T.; Takeda, R.; Sugawara, A.; Kuwahara, T.; Saleem, M.A.; et al. Redistribution of connexin43 expression in glomerular podocytes predicts poor renal prognosis in patients with type 2 diabetes and overt nephropathy. Nephrol. Dial. Transplant. 2006, 21, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef]
- Zhao, T.; Zhan, D.; Qu, S.; Jiang, S.; Gan, W.; Qin, W.; Zheng, C.; Cheng, F.; Lu, Y.; Liu, M.; et al. Transcriptomics-proteomics Integration reveals alternative polyadenylation driving inflammation-related protein translation in patients with diabetic nephropathy. J. Transl. Med. 2023, 21, 86. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, H.; Yan, Y.; Li, Y.; Lei, M.; Liu, Y.; Yang, L.; Zhou, S.; Pan, S.; Liu, Z.; et al. LncRNA evf-2 Exacerbates Podocyte Injury in Diabetic Nephropathy by Inducing Cell Cycle Re-entry and Inflammation Through Distinct Mechanisms Triggered by hnRNPU. Adv. Sci. 2024, 11, e2406532. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, H.; Luo, Q.; Yang, K.; Zou, Z.; Shi, M.; Liang, W. Dihydroxyacetone phosphate accumulation leads to podocyte pyroptosis in diabetic kidney disease. J. Cell. Mol. Med. 2024, 28, e18073. [Google Scholar] [CrossRef]
- Zhu, Z.; Cao, Y.; Jian, Y.; Hu, H.; Yang, Q.; Hao, Y.; Jiang, H.; Luo, Z.; Yang, X.; Li, W.; et al. CerS6 links ceramide metabolism to innate immune responses in diabetic kidney disease. Nat. Commun. 2025, 16, 1528. [Google Scholar] [CrossRef]
- Zhang, C.; Ren, W.; Lu, X.; Feng, L.; Li, J.; Zhu, B. The compound XueShuanTong promotes podocyte mitochondrial autophagy via the AMPK/mTOR pathway to alleviate diabetic nephropathy injury. Mitochondrion 2025, 83, 102024. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, S.; Huang, X.Y.; Chang, D.Y.; Wang, H.; Nie, L.; Xiong, Z.Y.; Chen, M.; Zhao, M.H.; Wang, S.X. Association of podocyte ultrastructural changes with proteinuria and pathological classification in type 2 diabetic nephropathy. Diabetes Metab. 2024, 50, 101547. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, S.; Sun, X.; Lou, Y.; Bao, J.; Yu, J. Hyperoside ameliorates diabetic nephropathy induced by STZ via targeting the miR-499-5p/APC axis. J. Pharmacol. Sci. 2021, 146, 10–20. [Google Scholar] [CrossRef]
- Zuo, F.; Wang, Y.; Xu, X.; Ding, R.; Tang, W.; Sun, Y.; Wang, X.; Zhang, Y.; Wu, J.; Xie, Y.; et al. CCDC92 deficiency ameliorates podocyte lipotoxicity in diabetic kidney disease. Metabolism 2024, 150, 155724. [Google Scholar] [CrossRef] [PubMed]
- Haddaway, N.R.; Page, M.J.; Pritchard, C.C.; McGuinness, L.A. PRISMA2020: An R package and Shiny app for producing PRISMA 2020-compliant flow diagrams, with interactivity for optimised digital transparency and Open Synthesis. Campbell Syst. Rev. 2022, 18, e1230. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, X.; Wang, S.; Chen, D.; Shu, J.; Chong, N.; Wang, Q.; Xu, Y. Dapagliflozin improves podocytes injury in diabetic nephropathy via regulating cholesterol balance through KLF5 targeting the ABCA1 signalling pathway. Diabetol. Metab. Syndr. 2024, 16, 38. [Google Scholar] [CrossRef]
- Hu, J.; Yang, Q.; Chen, Z.; Liang, W.; Feng, J.; Ding, G. Small GTPase Arf6 regulates diabetes-induced cholesterol accumulation in podocytes. J. Cell. Physiol. 2019, 234, 23559–23570. [Google Scholar] [CrossRef]
- Qin, Z.; Hoh, C.K.; Olson, E.S.; Jahromi, A.H.; Hall, D.J.; Barback, C.V.; You, Y.H.; Yanagita, M.; Sharma, K.; Vera, D.R. Molecular Imaging of the Glomerulus via Mesangial Cell Uptake of Radiolabeled Tilmanocept. J. Nucl. Med. 2019, 60, 1325–1332. [Google Scholar] [CrossRef]
- Fang, R.; Cao, X.; Zhu, Y.; Chen, Q. Hsa_circ_0037128 aggravates high glucose-induced podocytes injury in diabetic nephropathy through mediating miR-31-5p/KLF9. Autoimmunity 2022, 55, 254–263. [Google Scholar] [CrossRef]
- Zhu, W.; Li, Y.Y.; Zeng, H.X.; Liu, X.Q.; Sun, Y.T.; Jiang, L.; Xia, L.L.; Wu, Y.G. Carnosine alleviates podocyte injury in diabetic nephropathy by targeting caspase-1-mediated pyroptosis. Int. Immunopharmacol. 2021, 101, 108236. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Yang, J.; Liu, G. Melatonin Protects Against Diabetic Kidney Disease via the SIRT1/NLRP3 Signalling Pathway. Nephrology 2025, 30, e70073. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, Y.; Wang, T.; Wang, X.; He, J.; Xu, J.; Wu, B.; Li, Y. Ginsenoside Rg1 Alleviates Podocyte EMT Passage by Regulating AKT/GSK3 β/β-Catenin Pathway by Restoring Autophagic Activity. Evid. Based Complement. Altern. Med. 2020, 2020, 1903627. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Q.; Xia, C.; Zheng, H.; Jiang, W.; Wang, Y.; Sun, W. Qing-Re-Xiao-Zheng-(Yi-Qi) formula attenuates the renal podocyte ferroptosis in diabetic kidney disease through AMPK pathway. J. Ethnopharmacol. 2025, 351, 120157. [Google Scholar] [CrossRef]
- Kelly, D.J.; Aaltonen, P.; Cox, A.J.; Rumble, J.R.; Langham, R.; Panagiotopoulos, S.; Jerums, G.; Holthöfer, H.; Gilbert, R.E. Expression of the slit-diaphragm protein, nephrin, in experimental diabetic nephropathy: Differing effects of anti-proteinuric therapies. Nephrol. Dial. Transplant. 2002, 17, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Guan, X.M.; Wang, R.Y.; Xie, Y.S.; Zhou, H.; Ni, W.J.; Tang, L.Q. Berberine mitigates high glucose-induced podocyte apoptosis by modulating autophagy via the mTOR/P70S6K/4EBP1 pathway. Life Sci. 2020, 243, 117277. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.Y.; Kc, R.; Kim, M.; Kim, H.S.; Baek, J.Y.; Koh, E.H. Autophagic flux defect in diabetic kidney disease results in megamitochondria formation in podocytes. Biochem. Biophys. Res. Commun. 2020, 521, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, H.; Yan, J.; Wang, X.; Xu, M.; Wang, M.; Fan, B.; Liu, J.; Lin, N.; Li, L.; et al. Loss of CLDN5 in podocytes deregulates WIF1 to activate WNT signaling and contributes to kidney disease. Nat. Commun. 2022, 13, 1600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Tu, W.; Long, Y.N.; Li, P.F.; He, K.Y.; Wu, J. Notch signaling in diabetic kidney disease: Recent progress. Front. Endocrinol. 2025, 16, 1537769. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, L.; Wei, X.; Chen, M.; Zhang, X.; Mo, R.; Huang, R.; Liang, T.; Xu, X. Puerarin alleviates diabetic nephropathy by inhibiting Caspase-1-mediated pyroptosis. J. Pharm. Pharmacol. 2024, 76, 213–223. [Google Scholar] [CrossRef]
- Bai, X.; Hou, X.; Tian, J.; Geng, J.; Li, X. CDK5 promotes renal tubulointerstitial fibrosis in diabetic nephropathy via ERK1/2/PPARγ pathway. Oncotarget 2016, 7, 36510–36528. [Google Scholar] [CrossRef]
- Qin, X.; Zhao, Y.; Gong, J.; Huang, W.; Su, H.; Yuan, F.; Fang, K.; Wang, D.; Li, J.; Zou, X.; et al. Berberine Protects Glomerular Podocytes via Inhibiting Drp1-Mediated Mitochondrial Fission and Dysfunction. Theranostics 2019, 9, 1698–1713. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jiang, N.; Li, J.; Li, X.; He, S. Upregulation of BRD7 protects podocytes against high glucose-induced apoptosis by enhancing Nrf2 in a GSK-3β-dependent manner. Tissue Cell 2022, 76, 101813. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dai, S.; Yang, J.; Ma, J.; Wang, P.; Zhao, X.; Liu, J.; Xiao, A.; Song, Y.; Gao, L. MiR-33a Overexpression Exacerbates Diabetic Nephropathy Through Sirt6-dependent Notch Signaling. Iran. J. Kidney Dis. 2024, 18, 168–178. [Google Scholar] [PubMed]
- Li, Z.; Tan, D.; Lin, J.; Zhang, T.; Liu, B.; Zhao, B.; Liang, D.; Li, L.; Wei, X.; Lv, Z.; et al. Podocyte TLR4 deletion alleviates diabetic kidney disease through prohibiting PKCδ/SHP-1-dependent ER stress and relieving podocyte damage and inflammation. J. Adv. Res. 2025, in press. [Google Scholar] [CrossRef]
- Mallipattu, S.K.; Guo, Y.; Revelo, M.P.; Roa-Peña, L.; Miller, T.; Ling, J.; Shankland, S.J.; Bialkowska, A.B.; Ly, V.; Estrada, C.; et al. Krüppel-Like Factor 15 Mediates Glucocorticoid-Induced Restoration of Podocyte Differentiation Markers. J. Am. Soc. Nephrol. 2017, 28, 166–184. [Google Scholar] [CrossRef]
- Wu, J.; Zheng, C.; Fan, Y.; Zeng, C.; Chen, Z.; Qin, W.; Zhang, C.; Zhang, W.; Wang, X.; Zhu, X.; et al. Downregulation of microRNA-30 facilitates podocyte injury and is prevented by glucocorticoids. J. Am. Soc. Nephrol. 2014, 25, 92–104. [Google Scholar] [CrossRef]
- Agrawal, S.; Chanley, M.A.; Westbrook, D.; Nie, X.; Kitao, T.; Guess, A.J.; Benndorf, R.; Hidalgo, G.; Smoyer, W.E. Pioglitazone Enhances the Beneficial Effects of Glucocorticoids in Experimental Nephrotic Syndrome. Sci. Rep. 2016, 6, 24392. [Google Scholar] [CrossRef]
- Clement, L.C.; Avila-Casado, C.; Macé, C.; Soria, E.; Bakker, W.W.; Kersten, S.; Chugh, S.S. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat. Med. 2011, 17, 117–122. [Google Scholar] [CrossRef]
- Chen, H.M.; Liu, Z.H.; Zeng, C.H.; Li, S.J.; Wang, Q.W.; Li, L.S. Podocyte lesions in patients with obesity-related glomerulopathy. Am. J. Kidney Dis. 2006, 48, 772–779. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, M.; Zhu, W.; Zhang, Y.; Liu, P.; Li, P. Morroniside attenuates podocytes lipid deposition in diabetic nephropathy: A network pharmacology, molecular docking and experimental validation study. Int. Immunopharmacol. 2024, 138, 112560. [Google Scholar] [CrossRef]
- Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.S.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 2987. [Google Scholar] [CrossRef]
- Guo, R.; Wang, P.; Zheng, X.; Cui, W.; Shang, J.; Zhao, Z. SGLT2 inhibitors suppress epithelial-mesenchymal transition in podocytes under diabetic conditions via downregulating the IGF1R/PI3K pathway. Front. Pharmacol. 2022, 13, 897167, Correction in Front. Pharmacol. 2022, 13, 1074294. https://doi.org/10.3389/fphar.2022.1074294. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Chen, Y.; Cheng, Y.; Wang, Y.; Xu, N.; Wang, H.; Wang, L.; Chi, Y.; Ye, X.; et al. Canagliflozin Attenuates Podocyte Inflammatory Injury through Suppressing the TXNIP/NLRP3 Signaling Pathway in Diabetic Kidney Disease Mice. Inflammation 2025. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Ertracht, O.; Nakhoul, F. #993 The molecular effects of SGLT2i (Empagliflozin) on α-Klotho and Nephrin/Podocin proteins in type II diabetic mice model. Nephrol. Dial. Transplant. 2024, 39, 1057. [Google Scholar] [CrossRef]
- Cassis, P.; Locatelli, M.; Cerullo, D.; Corna, D.; Buelli, S.; Zanchi, C.; Villa, S.; Morigi, M.; Remuzzi, G.; Benigni, A.; et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight 2018, 3, 98720. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Molina, J.; Kim, J.J.; Mallela, S.K.; Ahmad, A.; Varona Santos, J.; Al-Ali, H.; Mitrofanova, A.; Sharma, K.; Fontanesi, F.; et al. Empagliflozin reduces podocyte lipotoxicity in experimental Alport syndrome. elife 2023, 12, 83353. [Google Scholar] [CrossRef]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy: SGLT2 protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Yang, Y.; Li, F.; Chen, Q.; Zhao, Z.; Zhang, N.; Li, H. Hepatocyte growth factor attenuates high glucose-disturbed mitochondrial dynamics in podocytes by decreasing ARF6-dependent DRP1 translocation. Biochim. Biophys. Acta Mol. Cell Res. 2024, 1871, 119623. [Google Scholar] [CrossRef]
- Li, H.; Li, Q.; Fan, Z.; Shen, Y.; Li, J.; Zhang, F. Identification of podocyte molecular markers in diabetic kidney disease via single-cell RNA sequencing and machine learning. PLoS ONE 2025, 20, e0328352. [Google Scholar] [CrossRef]
- Jiang, Z.; Qian, L.; Yang, R.; Wu, Y.; Guo, Y.; Chen, T. LncRNA TCF7 contributes to high glucose-induced damage in human podocytes by up-regulating SEMA3A via sponging miR-16-5p. J. Diabetes Investig. 2023, 14, 193–204. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.; Bossuyt, P.; Boutron, I.; Hoffmann, T.; Mulrow, C.; Shamseer, L.; Tetzlaff, J.; Moher, D. Updating guidance for reporting systematic reviews: Development of the PRISMA 2020 statement. J. Clin. Epidemiol. 2020, 134, 104–112. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).