Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results


Abstract
1. Introduction
2. J-Domain Proteins
2.1. DNAJB6
2.1.1. Structure of DNAJB6b
2.1.2. Functions of DNAJB6
Cochaperone Function
Antiaggregation and Cytoprotection
Cytoskeletal Maintenance
DNAJB6a in ER Stress Protection
Signal Transduction and Gene Regulation
2.1.3. DNAJB6 Mutations in Muscle Disease
2.1.4. Clinical and Pathological Features
2.1.5. Pathomechanistic Effects of DNAJB6 Mutations
Altered Antiaggregation Function
DNAJB6 Turnover
Dominant Toxicity
GSK3β Signaling
2.2. DNAJB2
2.2.1. DNAJB2 Expression
2.2.2. Functions of DNAJB2
2.2.3. DNAJB2 Mutations in Neuromuscular Disease
2.2.4. Clinical Features of DNAJB2-Related Neuropathies
2.2.5. Pathomechanisms of DNAJB2 Mutations
3. Small Heat Shock Proteins
3.1. Structure and Function of sHSPs
3.2. αB-Crystallin (HSPB5)
3.2.1. Neuromuscular Diseases Due to CRYAB Mutations
3.2.2. Pathomechanisms of CRYAB Mutations
Animal Models
Structural Effects
Chaperone Activity and Client Interactions
Aggregation and Amyloid Formation
Mitochondria and Redox Status
Conclusions
Recessive αB-Crystallinopathy
3.3. HSPB1
3.3.1. HSPB1 in Neuromuscular Disease
3.3.2. Pathomechanisms of HSPB1 Mutations
Properties of Mutant Proteins
Downstream Pathomechanisms
3.4. HSPB3
3.4.1. Functions of HSPB3
3.4.2. HSPB3 in Neuromuscular Disease
3.5. HSPB8
3.5.1. HSPB8 in Neuromuscular Disease
3.5.2. Animal Models
3.5.3. Pathomechanisms of HSPB8 Mutations
Chaperone Activity and Autophagy
Aggregation and Cytotoxicity
Mitochondria and Oxidative Stress
RNA Metabolism
HSPB8-Related Myopathy
4. BAG3
4.1. Structure and Functions of BAG3
4.1.1. Regulation of BAG3 Expression
4.1.2. Regulation of Expression by BAG3
4.1.3. BAG3 Proteostasis and Transport
4.1.4. BAG3 in Autophagy
4.1.5. Stress Granules and Defective Ribosomal Products
4.1.6. BAG3/BAG1 Ratio and Aging
4.1.7. Other Functions of BAG3 in Muscle Cells
4.2. BAG3 in Neuromuscular Disease
4.2.1. BAG3 Animal Models
BAG3-Deficient Models
BAG3 Mutation Models
4.2.2. Pathomechanisms
4.3. New Results on the Effects of BAG3 p.P209L on DNAJB6
5. Conclusions
6. Materials and Methods
6.1. Filter Trap Assay
6.2. DNAJB6 Turnover Assay
6.3. Microscopy and Image Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACD | α-crystallin domain |
| ALS | amyotrophic lateral sclerosis |
| AxM | axial myopathy |
| CASA | chaperone-assisted selective autophagy |
| CMA | chaperone-mediated autophagy |
| CMT | Charcot–Marie–Tooth disease |
| CNS | central nervous system |
| CTD | C-terminal domain |
| DCM | dilated cardiomyopathy |
| dHMN | distal hereditary motor neuropathy |
| DM | distal myopathy |
| DRIP | defective ribosomal product |
| ERAD | endoplasmic-reticulum-associated degradation |
| FRAP | fluorescence recovery after photobleaching |
| FTA | filter trap assay |
| G/F region | glycine/phenylalanine-rich domain |
| HCM | hypertrophic cardiomyopathy |
| HPD motif | histidine–proline–aspartate -motif |
| IF | intermediate filament |
| IPV | Ile-Pro-Val motif |
| JD | J domain |
| JDP | J-domain protein (Hsp40) |
| KI | knock-in |
| KO | knockout |
| LGMD | limb-girdle muscular dystrophy |
| MFM | myofibrillar myopathy |
| MSP | multisystem proteinopathy |
| MT | microtubule |
| MTOC | microtubule organizing center |
| N2B-us | N2B unique sequence (in titin) |
| NEF | nucleotide exchange factor |
| NF | neurofilament |
| NRC | neonatal rat cardiomyocytes |
| NTD | N-terminal domain |
| polyQ | polyglutamine |
| PTP | permeability transition pore |
| RCM | restrictive cardiomyopathy |
| ROS | reactive oxygen species |
| SG | stress granule |
| sHSP | small heat shock protein |
| SMN | survival of motor neuron |
| S/T region | serine/threonine-rich region |
| UIM | ubiquitin interaction motif |
| UPS | ubiquitin-proteasome system |
References
- Dekker, S.L.; Kampinga, H.H.; Bergink, S. DNAJs: More than Substrate Delivery to HSPA. Front. Mol. Biosci. 2015, 2, 35. [Google Scholar] [CrossRef]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.Y.; Larsen, B.; Choi, H.; Berger, B.; Gingras, A.C.; Lindquist, S. A Quantitative Chaperone Interaction Network Reveals the Architecture of Cellular Protein Homeostasis Pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Freilich, R.; Arhar, T.; Abrams, J.L.; Gestwicki, J.E. Protein-Protein Interactions in the Molecular Chaperone Network. Acc. Chem. Res. 2018, 51, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Alberti, S.; Benesch, J.L.P.; Boelens, W.; Buchner, J.; Carver, J.A.; Cecconi, C.; Ecroyd, H.; Gusev, N.; Hightower, L.E.; et al. Small Heat Shock Proteins: Multifaceted Proteins with Important Implications for Life. Cell Stress Chaperones 2019, 24, 295–308. [Google Scholar] [CrossRef]
- Shiber, A.; Ravid, T. Chaperoning Proteins for Destruction: Diverse Roles of Hsp70 Chaperones and their Co-Chaperones in Targeting Misfolded Proteins to the Proteasome. Biomolecules 2014, 4, 704–724. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperones in Autophagy. Pharmacol. Res. 2012, 66, 484–493. [Google Scholar] [CrossRef][Green Version]
- Dubińska-Magiera, M.; Jabłońska, J.; Saczko, J.; Kulbacka, J.; Jagla, T.; Daczewska, M. Contribution of Small Heat Shock Proteins to Muscle Development and Function. FEBS Lett. 2014, 588, 517–530. [Google Scholar] [CrossRef]
- Bell, R.A.; Al-Khalaf, M.; Megeney, L.A. The Beneficial Role of Proteolysis in Skeletal Muscle Growth and Stress Adaptation. Skelet. Muscle 2016, 6. [Google Scholar] [CrossRef]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2020 Version of the Gene Table of Neuromuscular Disorders (Nuclear Genome). Neuromuscul. Disord. 2019, 29, 980–1018. [Google Scholar] [CrossRef]
- Cheetham, M.E.; Jackson, A.P.; Anderton, B.H. Regulation of 70-kDa Heat-Shock-Protein ATPase Activity and Substrate Binding by Human DnaJ-Like Proteins, HSJ1a and HSJ1b. Eur. J. Biochem. 1994, 226, 99–107. [Google Scholar] [CrossRef]
- Chuang, J.Z.; Zhou, H.; Zhu, M.; Li, S.H.; Li, X.J.; Sung, C.H. Characterization of a Brain-Enriched Chaperone, MRJ, that Inhibits Huntingtin Aggregation and Toxicity Independently. J. Biol. Chem. 2002, 277, 19831–19838. [Google Scholar] [CrossRef] [PubMed]
- Doong, H.; Rizzo, K.; Fang, S.; Kulpa, V.; Weissman, A.M.; Kohn, E.C. CAIR-1/BAG-3 Abrogates Heat Shock Protein-70 Chaperone Complex-Mediated Protein Degradation: Accumulation of Poly-Ubiquitinated Hsp90 Client Proteins. J. Biol. Chem. 2003, 278, 28490–28500. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the Human Interactome Defines Protein Communities and Disease Networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Sarparanta, J.; Jonson, P.H.; Golzio, C.; Sandell, S.; Luque, H.; Screen, M.; McDonald, K.; Stajich, J.M.; Mahjneh, I.; Vihola, A.; et al. Mutations Affecting the Cytoplasmic Functions of the Co-Chaperone DNAJB6 Cause Limb-Girdle Muscular Dystrophy. Nat. Genet. 2012, 44, 450–455. [Google Scholar] [CrossRef]
- Carra, S.; Seguin, S.J.; Lambert, H.; Landry, J. HspB8 Chaperone Activity Toward Poly(Q)-Containing Proteins Depends on its Association with Bag3, a Stimulator of Macroautophagy. J. Biol. Chem. 2008, 283, 1437–1444. [Google Scholar] [CrossRef]
- Rauch, J.N.; Tse, E.; Freilich, R.; Mok, S.A.; Makley, L.N.; Southworth, D.R.; Gestwicki, J.E. BAG3 is a Modular, Scaffolding Protein that Physically Links Heat Shock Protein 70 (Hsp70) to the Small Heat Shock Proteins. J. Mol. Biol. 2017, 429, 128–141. [Google Scholar] [CrossRef]
- Aquilina, J.A.; Shrestha, S.; Morris, A.M.; Ecroyd, H. Structural and Functional Aspects of Hetero-Oligomers Formed by the Small Heat Shock Proteins αB-Crystallin and HSP27. J. Biol. Chem. 2013, 288, 13602–13609. [Google Scholar] [CrossRef]
- Morelli, F.F.; Mediani, L.; Heldens, L.; Bertacchini, J.; Bigi, I.; Carrà, A.D.; Vinet, J.; Carra, S. An Interaction Study in Mammalian Cells Demonstrates Weak Binding of HSPB2 to BAG3, which is Regulated by HSPB3 and Abrogated by HSPB8. Cell Stress Chaperones 2017, 22, 531–540. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Suzuki, A.; Kishikawa, M.; Akutsu, R.; Hirose, T.; Waye, M.M.; Tsui, S.K.; Yoshida, S.; Ohno, S. Muscle Develops a Specific Form of Small Heat Shock Protein Complex Composed of MKBP/HSPB2 and HSPB3 during Myogenic Differentiation. J. Biol. Chem. 2000, 275, 1095–1104. [Google Scholar] [CrossRef]
- Fontaine, J.M.; Sun, X.; Hoppe, A.D.; Simon, S.; Vicart, P.; Welsh, M.J.; Benndorf, R. Abnormal Small Heat Shock Protein Interactions Involving Neuropathy-Associated HSP22 (HSPB8) Mutants. FASEB J. 2006, 20, 2168–2170. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 Chaperone Machinery: J Proteins as Drivers of Functional Specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Andreasson, C.; Barducci, A.; Cheetham, M.E.; Cyr, D.; Emanuelsson, C.; Genevaux, P.; Gestwicki, J.E.; Goloubinoff, P.; Huerta-Cepas, J.; et al. Function, Evolution, and Structure of J-Domain Proteins. Cell Stress Chaperones 2019, 24, 7–15. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the Nomenclature of the Human Heat Shock Proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Craig, E.A. The Glycine-Phenylalanine-Rich Region Determines the Specificity of the Yeast Hsp40 Sis1. Mol. Cell Biol. 1999, 19, 7751–7758. [Google Scholar] [CrossRef] [PubMed]
- Wall, D.; Zylicz, M.; Georgopoulos, C. The Conserved G/F Motif of the DnaJ Chaperone is Necessary for the Activation of the Substrate Binding Properties of the DnaK Chaperone. J. Biol. Chem. 1995, 270, 2139–2144. [Google Scholar] [CrossRef] [PubMed]
- Perales-Calvo, J.; Muga, A.; Moro, F. Role of DnaJ G/F-Rich Domain in Conformational Recognition and Binding of Protein Substrates. J. Biol. Chem. 2010, 285, 34231–34239. [Google Scholar] [CrossRef]
- Cajo, G.C.; Horne, B.E.; Kelley, W.L.; Schwager, F.; Georgopoulos, C.; Genevaux, P. The Role of the DIF Motif of the DnaJ (Hsp40) Co-Chaperone in the Regulation of the DnaK (Hsp70) Chaperone Cycle. J. Biol. Chem. 2006, 281, 12436–12444. [Google Scholar] [CrossRef]
- Karamanos, T.K.; Tugarinov, V.; Clore, G.M. Unraveling the Structure and Dynamics of the Human DNAJB6b Chaperone by NMR Reveals Insights into Hsp40-Mediated Proteostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 21529–21538. [Google Scholar] [CrossRef]
- Craig, E.A.; Marszalek, J. How do J-Proteins Get Hsp70 to do so Many Different Things? Trends Biochem. Sci. 2017, 42, 355–368. [Google Scholar] [CrossRef]
- Hageman, J.; Rujano, M.A.; van Waarde, M.A.; Kakkar, V.; Dirks, R.P.; Govorukhina, N.; Oosterveld-Hut, H.M.; Lubsen, N.H.; Kampinga, H.H. A DNAJB Chaperone Subfamily with HDAC-Dependent Activities Suppresses Toxic Protein Aggregation. Mol. Cell 2010, 37, 355–369. [Google Scholar] [CrossRef]
- Kakkar, V.; Kuiper, E.F.; Pandey, A.; Braakman, I.; Kampinga, H.H. Versatile Members of the DNAJ Family show Hsp70 Dependent Anti-Aggregation Activity on RING1 Mutant Parkin C289G. Sci. Rep. 2016, 6, 34830. [Google Scholar] [CrossRef] [PubMed]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 Co-Chaperone Complex Unlocks Metazoan Protein Disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Nillegoda, N.B.; Stank, A.; Malinverni, D.; Alberts, N.; Szlachcic, A.; Barducci, A.; De Los Rios, P.; Wade, R.C.; Bukau, B. Evolution of an Intricate J-Protein Network Driving Protein Disaggregation in Eukaryotes. Elife 2017, 6. [Google Scholar] [CrossRef]
- Engert, J.C.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a Spastic Ataxia Common in Northeastern Québec, is Caused by Mutations in a New Gene Encoding an 11.5-Kb ORF. Nat. Genet. 2000, 24, 120–125. [Google Scholar] [CrossRef]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The Ataxia Protein Sacsin is a Functional Co-Chaperone that Protects Against Polyglutamine-Expanded Ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef]
- Anderson, J.F.; Siller, E.; Barral, J.M. The Neurodegenerative-Disease-Related Protein Sacsin is a Molecular Chaperone. J. Mol. Biol. 2011, 411, 870–880. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell. Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef]
- Farhan, S.M.K.; Howrigan, D.P.; Abbott, L.E.; Klim, J.R.; Topp, S.D.; Byrnes, A.E.; Churchhouse, C.; Phatnani, H.; Smith, B.N.; Rampersaud, E.; et al. Exome Sequencing in Amyotrophic Lateral Sclerosis Implicates a Novel Gene, DNAJC7, Encoding a Heat-Shock Protein. Nat. Neurosci. 2019, 22, 1966–1974. [Google Scholar] [CrossRef]
- Seki, N.; Hattori, A.; Hayashi, A.; Kozuma, S.; Miyajima, N.; Saito, T. Cloning, Tissue Expression, and Chromosomal Assignment of Human MRJ Gene for a Member of the DNAJ Protein Family. J. Hum. Genet. 1999, 44, 185–189. [Google Scholar] [CrossRef]
- Hanai, R.; Mashima, K. Characterization of Two Isoforms of a Human DnaJ Homologue, HSJ2. Mol. Biol. Rep. 2003, 30, 149–153. [Google Scholar] [CrossRef]
- Ding, Y.; Long, P.A.; Bos, J.M.; Shih, Y.H.; Ma, X.; Sundsbak, R.S.; Chen, J.; Jiang, Y.; Zhao, L.; Hu, X.; et al. A Modifier Screen Identifies DNAJB6 as a Cardiomyopathy Susceptibility Gene. JCI Insight 2016, 1, e88797. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Fillmore, R.A.; Metge, B.J.; Rajesh, M.; Xi, Y.; King, J.; Ju, J.; Pannell, L.; Shevde, L.A.; Samant, R.S. Large Isoform of MRJ (DNAJB6) Reduces Malignant Activity of Breast Cancer. Breast Cancer Res. 2008, 10, R22. [Google Scholar] [CrossRef] [PubMed]
- Izawa, I.; Nishizawa, M.; Ohtakara, K.; Ohtsuka, K.; Inada, H.; Inagaki, M. Identification of Mrj, a DnaJ/Hsp40 Family Protein, as a Keratin 8/18 Filament Regulatory Protein. J. Biol. Chem. 2000, 275, 34521–34527. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.D.; Geary-Joo, C.; Hughes, M.; Cross, J.C. The Mrj Co-Chaperone Mediates Keratin Turnover and Prevents the Formation of Toxic Inclusion Bodies in Trophoblast Cells of the Placenta. Development 2007, 134, 1809–1817. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dai, Y.S.; Xu, J.; Molkentin, J.D. The DnaJ-Related Factor Mrj Interacts with Nuclear Factor of Activated T Cells c3 and Mediates Transcriptional Repression through Class II Histone Deacetylase Recruitment. Mol. Cell Biol. 2005, 25, 9936–9948. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andrews, J.F.; Sykora, L.J.; Barik Letostak, T.; Menezes, M.E.; Mitra, A.; Barik, S.; Shevde, L.A.; Samant, R.S. Cellular Stress Stimulates Nuclear Localization Signal (NLS) Independent Nuclear Transport of MRJ. Exp. Cell Res. 2012, 318, 1086–1093. [Google Scholar] [CrossRef][Green Version]
- Månsson, C.; Kakkar, V.; Monsellier, E.; Sourigues, Y.; Härmark, J.; Kampinga, H.H.; Melki, R.; Emanuelsson, C. DNAJB6 is a Peptide-Binding Chaperone which can Suppress Amyloid Fibrillation of Polyglutamine Peptides at Substoichiometric Molar Ratios. Cell Stress Chaperones 2014, 19, 227–239. [Google Scholar] [CrossRef]
- Söderberg, C.A.G.; Månsson, C.; Bernfur, K.; Rutsdottir, G.; Härmark, J.; Rajan, S.; Al-Karadaghi, S.; Rasmussen, M.; Höjrup, P.; Hebert, H.; et al. Structural Modelling of the DNAJB6 Oligomeric Chaperone shows a Peptide-Binding Cleft Lined with Conserved S/T-Residues at the Dimer Interface. Sci. Rep. 2018, 8, 5199. [Google Scholar] [CrossRef]
- Bengoechea, R.; Pittman, S.K.; Tuck, E.P.; True, H.L.; Weihl, C.C. Myofibrillar Disruption and RNA-Binding Protein Aggregation in a Mouse Model of Limb-Girdle Muscular Dystrophy 1D. Hum. Mol. Genet. 2015, 24, 6588–6602. [Google Scholar] [CrossRef]
- Månsson, C.; Arosio, P.; Hussein, R.; Kampinga, H.H.; Hashem, R.M.; Boelens, W.C.; Dobson, C.M.; Knowles, T.P.; Linse, S.; Emanuelsson, C. Interaction of the Molecular Chaperone DNAJB6 with Growing Amyloid-Beta 42 (Aβ42) Aggregates Leads to Sub-Stoichiometric Inhibition of Amyloid Formation. J. Biol. Chem. 2014, 289, 31066–31076. [Google Scholar] [CrossRef]
- Hageman, J.; van Waarde-Verhagen, M.; Zylicz, A.; Walerych, D.; Kampinga, H.H. The Diverse Members of the Mammalian HSP70 Machine show Distinct Chaperone-Like Activities. Biochem. J. 2011, 435, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, N.; Arkan, S.; Hansen, C. The Molecular Chaperone DNAJB6, but Not DNAJB1, Suppresses the Seeded Aggregation of Alpha-Synuclein in Cells. Int. J. Mol. Sci. 2019, 20, 4495. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, V.; Månsson, C.; de Mattos, E.P.; Bergink, S.; van der Zwaag, M.; van Waarde, M.A.; Kloosterhuis, N.J.; Melki, R.; van Cruchten, R.T.; Al-Karadaghi, S.; et al. The S/T-Rich Motif in the DNAJB6 Chaperone Delays Polyglutamine Aggregation and the Onset of Disease in a Mouse Model. Mol. Cell 2016, 62, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Fayazi, Z.; Ghosh, S.; Marion, S.; Bao, X.; Shero, M.; Kazemi-Esfarjani, P. A Drosophila Ortholog of the Human MRJ Modulates Polyglutamine Toxicity and Aggregation. Neurobiol. Dis. 2006, 24, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Gillis, J.; Schipper-Krom, S.; Juenemann, K.; Gruber, A.; Coolen, S.; van den Nieuwendijk, R.; van Veen, H.; Overkleeft, H.; Goedhart, J.; Kampinga, H.H.; et al. The DNAJB6 and DNAJB8 Protein Chaperones Prevent Intracellular Aggregation of Polyglutamine Peptides. J. Biol. Chem. 2013, 288, 17225–17237. [Google Scholar] [CrossRef] [PubMed]
- Månsson, C.; van Cruchten, R.T.P.; Weininger, U.; Yang, X.; Cukalevski, R.; Arosio, P.; Dobson, C.M.; Knowles, T.; Akke, M.; Linse, S.; et al. Conserved S/T Residues of the Human Chaperone DNAJB6 are Required for Effective Inhibition of Aβ42 Amyloid Fibril Formation. Biochemistry 2018, 57, 4891–4902. [Google Scholar] [CrossRef] [PubMed]
- Reidy, M.; Sharma, R.; Roberts, B.L.; Masison, D.C. Human J-Protein DnaJB6b Cures a Subset of Saccharomyces Cerevisiae Prions and Selectively Blocks Assembly of Structurally Related Amyloids. J. Biol. Chem. 2016, 291, 4035–4047. [Google Scholar] [CrossRef]
- Udan-Johns, M.; Bengoechea, R.; Bell, S.; Shao, J.; Diamond, M.I.; True, H.L.; Weihl, C.C.; Baloh, R.H. Prion-Like Nuclear Aggregation of TDP-43 during Heat Shock is Regulated by HSP40/70 Chaperones. Hum. Mol. Genet. 2014, 23, 157–170. [Google Scholar] [CrossRef]
- Li, S.; Zhang, P.; Freibaum, B.D.; Kim, N.C.; Kolaitis, R.M.; Molliex, A.; Kanagaraj, A.P.; Yabe, I.; Tanino, M.; Tanaka, S.; et al. Genetic Interaction of hnRNPA2B1 and DNAJB6 in a Drosophila Model of Multisystem Proteinopathy. Hum. Mol. Genet. 2016, 25, 936–950. [Google Scholar] [CrossRef]
- Rose, J.M.; Novoselov, S.S.; Robinson, P.A.; Cheetham, M.E. Molecular Chaperone-Mediated Rescue of Mitophagy by a Parkin RING1 Domain Mutant. Hum. Mol. Genet. 2011, 20, 16–27. [Google Scholar] [CrossRef]
- Aprile, F.A.; Källstig, E.; Limorenko, G.; Vendruscolo, M.; Ron, D.; Hansen, C. The Molecular Chaperones DNAJB6 and Hsp70 Cooperate to Suppress α-Synuclein Aggregation. Sci. Rep. 2017, 7, 9039. [Google Scholar] [CrossRef] [PubMed]
- Bason, M.; Meister-Broekema, M.; Alberts, N.; Dijkers, P.; Bergink, S.; Sibon, O.C.M.; Kampinga, H.H. Astrocytic Expression of the Chaperone DNAJB6 Results in Non-Cell Autonomous Protection in Huntington’s Disease. Neurobiol. Dis. 2019, 124, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Kline, N.L.; Masison, D.C. Human DnaJB6 Anti-Amyloid Chaperone Protects Yeast from Polyglutamine Toxicity Separately from Spatial Segregation of Aggregates. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef]
- Li, Z.F.; Wu, X.; Jiang, Y.; Liu, J.; Wu, C.; Inagaki, M.; Izawa, I.; Mizisin, A.P.; Engvall, E.; Shelton, G.D. Non-Pathogenic Protein Aggregates in Skeletal Muscle in MLF1 Transgenic Mice. J. Neurol. Sci. 2008, 264, 77–86. [Google Scholar] [CrossRef]
- Dyer, J.O.; Dutta, A.; Gogol, M.; Weake, V.M.; Dialynas, G.; Wu, X.; Seidel, C.; Zhang, Y.; Florens, L.; Washburn, M.P.; et al. Myeloid Leukemia Factor Acts in a Chaperone Complex to Regulate Transcription Factor Stability and Gene Expression. J. Mol. Biol. 2017, 429, 2093–2107. [Google Scholar] [CrossRef]
- Kim, W.Y.; Fayazi, Z.; Bao, X.; Higgins, D.; Kazemi-Esfarjani, P. Evidence for Sequestration of Polyglutamine Inclusions by Drosophila Myeloid Leukemia Factor. Mol. Cell. Neurosci. 2005, 29, 536–544. [Google Scholar] [CrossRef]
- Banerjee, M.; Datta, M.; Bhattacharyya, N.P. Modulation of Mutant Huntingtin Aggregates and Toxicity by Human Myeloid Leukemia Factors. Int. J. Biochem. Cell Biol. 2017, 82, 1–9. [Google Scholar] [CrossRef]
- Miller, M.; Chen, A.; Gobert, V.; Auge, B.; Beau, M.; Burlet-Schiltz, O.; Haenlin, M.; Waltzer, L. Control of RUNX-Induced Repression of Notch Signaling by MLF and its Partner DnaJ-1 during Drosophila Hematopoiesis. PLoS Genet. 2017, 13, e1006932. [Google Scholar] [CrossRef]
- Hnia, K.; Ramspacher, C.; Vermot, J.; Laporte, J. Desmin in Muscle and Associated Diseases: Beyond the Structural Function. Cell Tissue Res. 2015, 360, 591–608. [Google Scholar] [CrossRef]
- Stone, M.R.; O’Neill, A.; Lovering, R.M.; Strong, J.; Resneck, W.G.; Reed, P.W.; Toivola, D.M.; Ursitti, J.A.; Omary, M.B.; Bloch, R.J. Absence of Keratin 19 in Mice Causes Skeletal Myopathy with Mitochondrial and Sarcolemmal Reorganization. J. Cell. Sci. 2007, 120, 3999–4008. [Google Scholar] [CrossRef]
- Muriel, J.M.; O’Neill, A.; Kerr, J.P.; Kleinhans-Welte, E.; Lovering, R.M.; Bloch, R.J. Keratin 18 is an Integral Part of the Intermediate Filament Network in Murine Skeletal Muscle. Am. J. Physiol. Cell. Physiol. 2020, 318, C215–C224. [Google Scholar] [CrossRef] [PubMed]
- Claeys, K.G.; Fardeau, M. Myofibrillar Myopathies. Handb. Clin. Neurol. 2013, 113, 1337–1342. [Google Scholar] [PubMed]
- Sandell, S.; Huovinen, S.; Palmio, J.; Raheem, O.; Lindfors, M.; Zhao, F.; Haapasalo, H.; Udd, B. Diagnostically Important Muscle Pathology in DNAJB6 Mutated LGMD1D. Acta Neuropathol. Commun. 2016, 4, 9. [Google Scholar] [CrossRef]
- Kedia, N.; Arhzaouy, K.; Pittman, S.K.; Sun, Y.; Batchelor, M.; Weihl, C.C.; Bieschke, J. Desmin Forms Toxic, Seeding-Competent Amyloid Aggregates that Persist in Muscle Fibers. Proc. Natl. Acad. Sci. USA 2019, 116, 16835–16840. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Salvans, M.; Cavazza, T.; Espadas, G.; Sabido, E.; Vernos, I. Proteomic Profiling of Microtubule Self-Organization in M-Phase. Mol. Cell. Proteomics 2018, 17, 1991–2004. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Salvans, M.; Scrofani, J.; Modol, A.; Vernos, I. DnaJB6 is a RanGTP-Regulated Protein Required for Microtubule Organization during Mitosis. J. Cell. Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.R.; Mehta, A.; Moore, B.P.; Phadke, P.A.; Meehan, W.J.; Accavitti, M.A.; Shevde, L.A.; Hopper, J.E.; Xie, Y.; Welch, D.R.; et al. Breast Cancer Metastasis Suppressor 1 (BRMS1) is Stabilized by the Hsp90 Chaperone. Biochem. Biophys. Res. Commun. 2006, 348, 1429–1435. [Google Scholar] [CrossRef]
- Pan, Z.; Sikandar, S.; Witherspoon, M.; Dizon, D.; Nguyen, T.; Benirschke, K.; Wiley, C.; Vrana, P.; Lipkin, S.M. Impaired Placental Trophoblast Lineage Differentiation in Alkbh1(-/-) Mice. Dev. Dyn. 2008, 237, 316–327. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, Z.; Cao, Y.; Zhang, S.; Li, H.; Huang, Y.; Ding, Y.Q.; Liu, X. The Hsp40 Family Chaperone Protein DnaJB6 Enhances Schlafen1 Nuclear Localization which is Critical for Promotion of Cell-Cycle Arrest in T-Cells. Biochem. J. 2008, 413, 239–250. [Google Scholar] [CrossRef]
- Mitra, A.; Menezes, M.E.; Shevde, L.A.; Samant, R.S. DNAJB6 Induces Degradation of β-Catenin and Causes Partial Reversal of Mesenchymal Phenotype. J. Biol. Chem. 2010, 285, 24686–24694. [Google Scholar] [CrossRef]
- Mitra, A.; Menezes, M.E.; Pannell, L.K.; Mulekar, M.S.; Honkanen, R.E.; Shevde, L.A.; Samant, R.S. DNAJB6 Chaperones PP2A Mediated Dephosphorylation of GSK3β to Downregulate β-Catenin Transcription Target, Osteopontin. Oncogene 2012, 31, 4472–4483. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.D.; Mattar, P.; Schuurmans, C.; Cross, J.C. Neural Stem Cell Self-Renewal Requires the Mrj Co-Chaperone. Dev. Dyn. 2009, 238, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Shevde, L.A.; Samant, R.S. Emerging Roles and Underlying Molecular Mechanisms of DNAJB6 in Cancer. Oncotarget 2016, 7, 53984–53996. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.R.; Bengoechea, R.; Pittman, S.K.; Chou, T.F.; True, H.L.; Weihl, C.C. Lithium Chloride Corrects Weakness and Myopathology in a Preclinical Model of LGMD1D. Neurol. Genet. 2019, 5, e318. [Google Scholar] [CrossRef]
- van der Velden, J.L.; Schols, A.M.; Willems, J.; Kelders, M.C.; Langen, R.C. Glycogen Synthase Kinase 3β Suppresses Myogenic Differentiation through Negative Regulation of NFATc3. J. Biol. Chem. 2008, 283, 358–366. [Google Scholar] [CrossRef]
- Harms, M.B.; Sommerville, R.B.; Allred, P.; Bell, S.; Ma, D.; Cooper, P.; Lopate, G.; Pestronk, A.; Weihl, C.C.; Baloh, R.H. Exome Sequencing Reveals DNAJB6 Mutations in Dominantly-Inherited Myopathy. Ann. Neurol. 2012, 71, 407–416. [Google Scholar] [CrossRef]
- Straub, V.; Murphy, A.; Udd, B.; LGMD Workshop Study Group. 229th ENMC International Workshop: Limb Girdle Muscular Dystrophies—Nomenclature and Reformed Classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef]
- Ruggieri, A.; Brancati, F.; Zanotti, S.; Maggi, L.; Pasanisi, M.B.; Saredi, S.; Terracciano, C.; Antozzi, C.; Apice, M.R.D.; Sangiuolo, F.; et al. Complete Loss of the DNAJB6 G/F Domain and Novel Missense Mutations Cause Distal-Onset DNAJB6 Myopathy. Acta Neuropathol. Commun. 2015, 3, 44. [Google Scholar] [CrossRef]
- Jonson, P.H.; Palmio, J.; Johari, M.; Penttilä, S.; Evilä, A.; Nelson, I.; Bonne, G.; Wiart, N.; Meyer, V.; Boland, A.; et al. Novel Mutations in DNAJB6 Cause LGMD1D and Distal Myopathy in French Families. Eur. J. Neurol. 2018, 25, 790–794. [Google Scholar] [CrossRef]
- Palmio, J.; Jonson, P.H.; Inoue, M.; Sarparanta, J.; Bengoechea, R.; Savarese, M.; Vihola, A.; Jokela, M.; Nakagawa, M.; Noguchi, S.; et al. Mutations in the J Domain of DNAJB6 Cause Dominant Distal Myopathy. Neuromuscul. Disord. 2020. [Google Scholar] [CrossRef]
- Carvalho, A.A.S.; Lacene, E.; Brochier, G.; Labasse, C.; Madelaine, A.; Silva, V.G.D.; Corazzini, R.; Papadopoulos, K.; Behin, A.; Laforet, P.; et al. Genetic Mutations and Demographic, Clinical, and Morphological Aspects of Myofibrillar Myopathy in a French Cohort. Genet. Test. Mol. Biomark. 2018, 22, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Nallamilli, B.R.R.; Chakravorty, S.; Kesari, A.; Tanner, A.; Ankala, A.; Schneider, T.; da Silva, C.; Beadling, R.; Alexander, J.J.; Askree, S.H.; et al. Genetic Landscape and Novel Disease Mechanisms from a Large LGMD Cohort of 4656 Patients. Ann. Clin. Transl. Neurol. 2018, 5, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Couthouis, J.; Raphael, A.R.; Siskind, C.; Findlay, A.R.; Buenrostro, J.D.; Greenleaf, W.J.; Vogel, H.; Day, J.W.; Flanigan, K.M.; Gitler, A.D. Exome Sequencing Identifies a DNAJB6 Mutation in a Family with Dominantly-Inherited Limb-Girdle Muscular Dystrophy. Neuromuscul. Disord. 2014, 24, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Cedeno, G.; Winder, T.; Milone, M. DNAJB6 Myopathy: A Vacuolar Myopathy with Childhood Onset. Muscle Nerve 2014, 49, 607–610. [Google Scholar] [CrossRef]
- Palmio, J.; Jonson, P.H.; Evilä, A.; Auranen, M.; Straub, V.; Bushby, K.; Sarkozy, A.; Kiuru-Enari, S.; Sandell, S.; Pihko, H.; et al. Novel Mutations in DNAJB6 Gene Cause a very Severe Early-Onset Limb-Girdle Muscular Dystrophy 1D Disease. Neuromuscul. Disord. 2015, 25, 835–842. [Google Scholar] [CrossRef]
- Zima, J.; Eaton, A.; Pál, E.; Till, Á.; Ito, Y.A.; Warman-Chardon, J.; Hartley, T.; Cagnone, G.; Melegh, B.I.; Care4Rare Canada; et al. Intrafamilial Variability of Limb-Girdle Muscular Dystrophy, LGMD1D Type. Eur. J. Med. Genet. 2019, 63, 103655. [Google Scholar] [CrossRef]
- Nam, T.S.; Li, W.; Heo, S.H.; Lee, K.H.; Cho, A.; Shin, J.H.; Kim, Y.O.; Chae, J.H.; Kim, D.S.; Kim, M.K.; et al. A Novel Mutation in DNAJB6, p.(Phe91Leu), in Childhood-Onset LGMD1D with a Severe Phenotype. Neuromuscul. Disord. 2015, 25, 843–851. [Google Scholar] [CrossRef]
- Sato, T.; Hayashi, Y.K.; Oya, Y.; Kondo, T.; Sugie, K.; Kaneda, D.; Houzen, H.; Yabe, I.; Sasaki, H.; Noguchi, S.; et al. DNAJB6 Myopathy in an Asian Cohort and cytoplasmic/nuclear Inclusions. Neuromuscul. Disord. 2013, 23, 269–276. [Google Scholar] [CrossRef]
- Kojima, Y.; Noto, Y.I.; Takewaki, D.; Tokuda, N.; Shiga, K.; Hamano, A.; Mizuta, I.; Muranishi, M.; Kasai, T.; Nakagawa, M.; et al. Characteristic Posterior-Dominant Lower Limb Muscle Involvement in Limb-Girdle Muscular Dystrophy due to a DNAJB6 Phe93Leu Mutation. Intern. Med. 2017, 56, 2347–2351. [Google Scholar] [CrossRef][Green Version]
- Monies, D.; Alhindi, H.N.; Almuhaizea, M.A.; Abouelhoda, M.; Alazami, A.M.; Goljan, E.; Alyounes, B.; Jaroudi, D.; AlIssa, A.; Alabdulrahman, K.; et al. A First-Line Diagnostic Assay for Limb-Girdle Muscular Dystrophy and Other Myopathies. Hum. Genom. 2016, 10, 32. [Google Scholar] [CrossRef]
- Bohlega, S.A.; Alfawaz, S.; Abou-Al Shaar, H.; Al-Hindi, H.N.; Murad, H.N.; Bohlega, M.S.; Meyer, B.F.; Monies, D. LGMD1D Myopathy with Cytoplasmic and Nuclear Inclusions in a Saudi Family due to DNAJB6 Mutation. Acta Myol. 2018, 37, 221–226. [Google Scholar] [PubMed]
- Tsai, P.C.; Tsai, Y.S.; Soong, B.W.; Huang, Y.H.; Wu, H.T.; Chen, Y.H.; Lin, K.P.; Liao, Y.C.; Lee, Y.C. A Novel DNAJB6 Mutation Causes Dominantly Inherited Distal-Onset Myopathy and Compromises DNAJB6 Function. Clin. Genet. 2017, 92, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Park, H.J.; Lee, J.H.; Hong, J.; Ahn, S.W.; Choi, Y.C. Two Korean Families with Limb-Girdle Muscular Dystrophy Type 1D Associated with DNAJB6 Mutations. Yonsei Med. J. 2018, 59, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Yabe, I.; Tanino, M.; Yaguchi, H.; Takiyama, A.; Cai, H.; Kanno, H.; Takahashi, I.; Hayashi, Y.K.; Watanabe, M.; Takahashi, H.; et al. Pathology of Frontotemporal Dementia with Limb Girdle Muscular Dystrophy Caused by a DNAJB6 Mutation. Clin. Neurol. Neurosurg. 2014, 127, 10–12. [Google Scholar] [CrossRef]
- Stein, K.C.; Bengoechea, R.; Harms, M.B.; Weihl, C.C.; True, H.L. Myopathy-Causing Mutations in an HSP40 Chaperone Disrupt Processing of Specific Client Conformers. J. Biol. Chem. 2014, 289, 21120–21130. [Google Scholar] [CrossRef]
- Meister-Broekema, M.; Freilich, R.; Jagadeesan, C.; Rauch, J.N.; Bengoechea, R.; Motley, W.W.; Kuiper, E.F.E.; Minoia, M.; Furtado, G.V.; van Waarde, M.A.W.H.; et al. Myopathy Associated BAG3 Mutations Lead to Protein Aggregation by Stalling Hsp70 Networks. Nat. Commun. 2018, 9, 5342. [Google Scholar] [CrossRef]
- Aweida, D.; Rudesky, I.; Volodin, A.; Shimko, E.; Cohen, S. GSK3-Beta Promotes Calpain-1-Mediated Desmin Filament Depolymerization and Myofibril Loss in Atrophy. J. Cell Biol. 2018, 217, 3698–3714. [Google Scholar] [CrossRef]
- Homma, S.; Iwasaki, M.; Shelton, G.D.; Engvall, E.; Reed, J.C.; Takayama, S. BAG3 Deficiency Results in Fulminant Myopathy and Early Lethality. Am. J. Pathol. 2006, 169, 761–773. [Google Scholar] [CrossRef]
- Cheetham, M.E.; Brion, J.P.; Anderton, B.H. Human Homologues of the Bacterial Heat-Shock Protein DnaJ are Preferentially Expressed in Neurons. Biochem. J. 1992, 284 Pt 2, 469–476. [Google Scholar] [CrossRef]
- Chapple, J.P.; Cheetham, M.E. The Chaperone Environment at the Cytoplasmic Face of the Endoplasmic Reticulum can Modulate Rhodopsin Processing and Inclusion Formation. J. Biol. Chem. 2003, 278, 19087–19094. [Google Scholar] [CrossRef]
- Westhoff, B.; Chapple, J.P.; van der Spuy, J.; Höhfeld, J.; Cheetham, M.E. HSJ1 is a Neuronal Shuttling Factor for the Sorting of Chaperone Clients to the Proteasome. Curr. Biol. 2005, 15, 1058–1064. [Google Scholar] [CrossRef]
- Blumen, S.C.; Astord, S.; Robin, V.; Vignaud, L.; Toumi, N.; Cieslik, A.; Achiron, A.; Carasso, R.L.; Gurevich, M.; Braverman, I.; et al. A Rare Recessive Distal Hereditary Motor Neuropathy with HSJ1 Chaperone Mutation. Ann. Neurol. 2012, 71, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Gess, B.; Auer-Grumbach, M.; Schirmacher, A.; Strom, T.; Zitzelsberger, M.; Rudnik-Schöneborn, S.; Röhr, D.; Halfter, H.; Young, P.; Senderek, J. HSJ1-Related Hereditary Neuropathies: Novel Mutations and Extended Clinical Spectrum. Neurology 2014, 83, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Borrell-Pagès, M.; Canals, J.M.; Cordelières, F.P.; Parker, J.A.; Pineda, J.R.; Grange, G.; Bryson, E.A.; Guillermier, M.; Hirsch, E.; Hantraye, P.; et al. Cystamine and Cysteamine Increase Brain Levels of BDNF in Huntington Disease Via HSJ1b and Transglutaminase. J. Clin. Investig. 2006, 116, 1410–1424. [Google Scholar] [CrossRef] [PubMed]
- Claeys, K.G.; Sozanska, M.; Martin, J.J.; Lacene, E.; Vignaud, L.; Stockholm, D.; Laforêt, P.; Eymard, B.; Kichler, A.; Scherman, D.; et al. DNAJB2 Expression in Normal and Diseased Human and Mouse Skeletal Muscle. Am. J. Pathol. 2010, 176, 2901–2910. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Safdar, A.; Parise, G.; Melov, S.; Fu, M.; MacNeil, L.; Kaczor, J.; Payne, E.T.; Tarnopolsky, M.A. Gene Expression Profiling in Human Skeletal Muscle during Recovery from Eccentric Exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1901–R1910. [Google Scholar] [CrossRef]
- Howarth, J.L.; Kelly, S.; Keasey, M.P.; Glover, C.; Lee, Y.B.; Mitrophanous, K.; Chapple, J.P.; Gallo, J.M.; Cheetham, M.E.; Uney, J.B. Hsp40 Molecules that Target to the Ubiquitin-Proteasome System Decrease Inclusion Formation in Models of Polyglutamine Disease. Mol. Ther. 2007, 15, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, D.; Marin, O.; Arrigoni, G.; Franchin, C.; Vilardell, J.; Sandre, M.; Li, W.; Parfitt, D.A.; Pinna, L.A.; Cheetham, M.E.; et al. Protein Kinase CK2 Modulates HSJ1 Function through Phosphorylation of the UIM2 Domain. Hum. Mol. Genet. 2017, 26, 611–623. [Google Scholar] [CrossRef]
- Schnaider, T.; Soti, C.; Cheetham, M.E.; Miyata, Y.; Yahara, I.; Csermely, P. Interaction of the Human DnaJ Homologue, HSJ1b with the 90 kDa Heat Shock Protein, Hsp90. Life Sci. 2000, 67, 1455–1465. [Google Scholar] [CrossRef]
- Labbadia, J.; Novoselov, S.S.; Bett, J.S.; Weiss, A.; Paganetti, P.; Bates, G.P.; Cheetham, M.E. Suppression of Protein Aggregation by Chaperone Modification of High Molecular Weight Complexes. Brain 2012, 135, 1180–1196. [Google Scholar] [CrossRef]
- Novoselov, S.S.; Mustill, W.J.; Gray, A.L.; Dick, J.R.; Kanuga, N.; Kalmar, B.; Greensmith, L.; Cheetham, M.E. Molecular Chaperone Mediated Late-Stage Neuroprotection in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e73944. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Mitchell, J.C.; Novoselov, S.; Miller, J.; Nishimura, A.L.; Scotter, E.L.; Vance, C.A.; Cheetham, M.E.; Shaw, C.E. The Heat Shock Response Plays an Important Role in TDP-43 Clearance: Evidence for Dysfunction in Amyotrophic Lateral Sclerosis. Brain 2016, 139, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.C.; Zhou, C.J.; Zhou, Z.R.; Zhang, Y.H.; Zheng, X.M.; Song, A.X.; Hu, H.Y. Co-Chaperone HSJ1a Dually Regulates the Proteasomal Degradation of Ataxin-3. PLoS ONE 2011, 6, e19763. [Google Scholar] [CrossRef] [PubMed]
- Meimaridou, E.; Gooljar, S.B.; Ramnarace, N.; Anthonypillai, L.; Clark, A.J.; Chapple, J.P. The Cytosolic Chaperone Hsc70 Promotes Traffic to the Cell Surface of Intracellular Retained Melanocortin-4 Receptor Mutants. Mol. Endocrinol. 2011, 25, 1650–1660. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Barriere, H.; Bagdany, M.; Rabeh, W.M.; Du, K.; Hohfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral Protein Quality Control Removes Unfolded CFTR from the Plasma Membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef]
- Cheetham, M.E.; Anderton, B.H.; Jackson, A.P. Inhibition of hsc70-Catalysed Clathrin Uncoating by HSJ1 Proteins. Biochem. J. 1996, 319 Pt 1, 103–108. [Google Scholar] [CrossRef]
- Lupo, V.; García-García, F.; Sancho, P.; Tello, C.; García-Romero, M.; Villarreal, L.; Alberti, A.; Sivera, R.; Dopazo, J.; Pascual-Pascual, S.I.; et al. Assessment of Targeted Next-Generation Sequencing as a Tool for the Diagnosis of Charcot-Marie-Tooth Disease and Hereditary Motor Neuropathy. J. Mol. Diagn. 2016, 18, 225–234. [Google Scholar] [CrossRef]
- Frasquet, M.; Chumillas, M.J.; Vílchez, J.J.; Márquez-Infante, C.; Palau, F.; Vázquez-Costa, J.F.; Lupo, V.; Espinós, C.; Sevilla, T. Phenotype and Natural History of Inherited Neuropathies Caused by HSJ1 c.352+1G>A Mutation. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1265–1268. [Google Scholar] [CrossRef]
- Sanchez, E.; Darvish, H.; Mesias, R.; Taghavi, S.; Firouzabadi, S.G.; Walker, R.H.; Tafakhori, A.; Paisán-Ruiz, C. Identification of a Large DNAJB2 Deletion in a Family with Spinal Muscular Atrophy and Parkinsonism. Hum. Mutat. 2016, 37, 1180–1189. [Google Scholar] [CrossRef]
- Teive, H.; Kok, F.; Raskin, S.; Arruda, W. Distal hereditary motor neuropathy with HSJ1 chaperone mutation, presenting with peripheral motor neuropathy, associated to parkinsonism, and cerebellar ataxia. Case report. Parkinsonism Relat. Disord. 2016, 22, e154–e192. [Google Scholar] [CrossRef]
- Frasquet, M.; Vázquez-Costa, J.F.; Sevilla, T. The Role of DNAJB2 in Amyotrophic Lateral Sclerosis. Brain 2016, 139, e57. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-Wide Rare Variant Analysis Identifies TUBA4A Mutations Associated with Familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Kappé, G.; Franck, E.; Verschuure, P.; Boelens, W.C.; Leunissen, J.A.; de Jong, W.W. The Human Genome Encodes 10 α-Crystallin-Related Small Heat Shock Proteins: HspB1-10. Cell Stress Chaperones 2003, 8, 53–61. [Google Scholar] [CrossRef]
- Kappé, G.; Boelens, W.C.; de Jong, W.W. Why Proteins without an α-Crystallin Domain should Not be Included in the Human Small Heat Shock Protein Family HSPB. Cell Stress Chaperones 2010, 15, 457–461. [Google Scholar] [CrossRef]
- Kriehuber, T.; Rattei, T.; Weinmaier, T.; Bepperling, A.; Haslbeck, M.; Buchner, J. Independent Evolution of the Core Domain and its Flanking Sequences in Small Heat Shock Proteins. FASEB J. 2010, 24, 3633–3642. [Google Scholar] [CrossRef]
- Sudnitsyna, M.V.; Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. The Role of Intrinsically Disordered Regions in the Structure and Functioning of Small Heat Shock Proteins. Curr. Protein Pept. Sci. 2012, 13, 76–85. [Google Scholar] [CrossRef]
- Haslbeck, M.; Weinkauf, S.; Buchner, J. Small Heat Shock Proteins: Simplicity Meets Complexity. J. Biol. Chem. 2019, 294, 2121–2132. [Google Scholar] [CrossRef]
- Clouser, A.F.; Baughman, H.E.; Basanta, B.; Guttman, M.; Nath, A.; Klevit, R.E. Interplay of Disordered and Ordered Regions of a Human Small Heat Shock Protein Yields an Ensemble of ‘Quasi-Ordered’ States. Elife 2019, 8. [Google Scholar] [CrossRef]
- Jehle, S.; Vollmar, B.S.; Bardiaux, B.; Dove, K.K.; Rajagopal, P.; Gonen, T.; Oschkinat, H.; Klevit, R.E. N-Terminal Domain of αB-Crystallin Provides a Conformational Switch for Multimerization and Structural Heterogeneity. Proc. Natl. Acad. Sci. USA 2011, 108, 6409–6414. [Google Scholar] [CrossRef]
- Delbecq, S.P.; Jehle, S.; Klevit, R. Binding Determinants of the Small Heat Shock Protein, αB-Crystallin: Recognition of the ’IxI’ Motif. EMBO J. 2012, 31, 4587–4594. [Google Scholar] [CrossRef]
- Delbecq, S.P.; Rosenbaum, J.C.; Klevit, R.E. A Mechanism of Subunit Recruitment in Human Small Heat Shock Protein Oligomers. Biochemistry 2015, 54, 4276–4284. [Google Scholar] [CrossRef] [PubMed]
- Bova, M.P.; McHaourab, H.S.; Han, Y.; Fung, B.K. Subunit Exchange of Small Heat Shock Proteins. Analysis of Oligomer Formation of αA-Crystallin and Hsp27 by Fluorescence Resonance Energy Transfer and Site-Directed Truncations. J. Biol. Chem. 2000, 275, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Aquilina, J.A.; Benesch, J.L.; Bateman, O.A.; Slingsby, C.; Robinson, C.V. Polydispersity of a Mammalian Chaperone: Mass Spectrometry Reveals the Population of Oligomers in αB-Crystallin. Proc. Natl. Acad. Sci. USA 2003, 100, 10611–10616. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.T.; Bortolus, M.; Koteiche, H.A.; Mchaourab, H.S. Sequence, Structure, and Dynamic Determinants of Hsp27 (HspB1) Equilibrium Dissociation are Encoded by the N-Terminal Domain. Biochemistry 2012, 51, 1257–1268. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Brunsting, J.F.; Stege, G.J.; Konings, A.W.; Landry, J. Cells Overexpressing Hsp27 show Accelerated Recovery from Heat-Induced Nuclear Protein Aggregation. Biochem. Biophys. Res. Commun. 1994, 204, 1170–1177. [Google Scholar] [CrossRef]
- Mogk, A.; Schlieker, C.; Friedrich, K.L.; Schonfeld, H.J.; Vierling, E.; Bukau, B. Refolding of Substrates Bound to Small Hsps Relies on a Disaggregation Reaction Mediated most Efficiently by ClpB/DnaK. J. Biol. Chem. 2003, 278, 31033–31042. [Google Scholar] [CrossRef]
- Cashikar, A.G.; Duennwald, M.; Lindquist, S.L. A Chaperone Pathway in Protein Disaggregation. Hsp26 Alters the Nature of Protein Aggregates to Facilitate Reactivation by Hsp104. J. Biol. Chem. 2005, 280, 23869–23875. [Google Scholar] [CrossRef]
- Ungelenk, S.; Moayed, F.; Ho, C.T.; Grousl, T.; Scharf, A.; Mashaghi, A.; Tans, S.; Mayer, M.P.; Mogk, A.; Bukau, B. Small Heat Shock Proteins Sequester Misfolding Proteins in Near-Native Conformation for Cellular Protection and Efficient Refolding. Nat. Commun. 2016, 7, 13673. [Google Scholar] [CrossRef]
- Specht, S.; Miller, S.B.; Mogk, A.; Bukau, B. Hsp42 is Required for Sequestration of Protein Aggregates into Deposition Sites in Saccharomyces Cerevisiae. J. Cell Biol. 2011, 195, 617–629. [Google Scholar] [CrossRef]
- Escusa-Toret, S.; Vonk, W.I.; Frydman, J. Spatial Sequestration of Misfolded Proteins by a Dynamic Chaperone Pathway Enhances Cellular Fitness during Stress. Nat. Cell Biol. 2013, 15, 1231–1243. [Google Scholar] [CrossRef]
- Grousl, T.; Ungelenk, S.; Miller, S.; Ho, C.T.; Khokhrina, M.; Mayer, M.P.; Bukau, B.; Mogk, A. A Prion-Like Domain in Hsp42 Drives Chaperone-Facilitated Aggregation of Misfolded Proteins. J. Cell Biol. 2018, 217, 1269–1285. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.T.; Grousl, T.; Shatz, O.; Jawed, A.; Ruger-Herreros, C.; Semmelink, M.; Zahn, R.; Richter, K.; Bukau, B.; Mogk, A. Cellular Sequestrases Maintain Basal Hsp70 Capacity Ensuring Balanced Proteostasis. Nat. Commun. 2019, 10, 4851. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread Proteome Remodeling and Aggregation in Aging C. Elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Ehrnsperger, M.; Graber, S.; Gaestel, M.; Buchner, J. Binding of Non-Native Protein to Hsp25 during Heat Shock Creates a Reservoir of Folding Intermediates for Reactivation. EMBO J. 1997, 16, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.J.; Vierling, E. A Small Heat Shock Protein Cooperates with Heat Shock Protein 70 Systems to Reactivate a Heat-Denatured Protein. Plant Physiol. 2000, 122, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Bukau, B. Role of sHsps in Organizing Cytosolic Protein Aggregation and Disaggregation. Cell Stress Chaperones 2017, 22, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Żwirowski, S.; Kłosowska, A.; Obuchowski, I.; Nillegoda, N.B.; Piróg, A.; Ziętkiewicz, S.; Bukau, B.; Mogk, A.; Liberek, K. Hsp70 Displaces Small Heat Shock Proteins from Aggregates to Initiate Protein Refolding. EMBO J. 2017, 36, 783–796. [Google Scholar] [CrossRef]
- Takayama, S.; Xie, Z.; Reed, J.C. An Evolutionarily Conserved Family of Hsp70/Hsc70 Molecular Chaperone Regulators. J. Biol. Chem. 1999, 274, 781–786. [Google Scholar] [CrossRef]
- Fuchs, M.; Poirier, D.J.; Seguin, S.J.; Lambert, H.; Carra, S.; Charette, S.J.; Landry, J. Identification of the Key Structural Motifs Involved in HspB8/HspB6-Bag3 Interaction. Biochem. J. 2009, 425, 245–255. [Google Scholar] [CrossRef]
- Basha, E.; Friedrich, K.L.; Vierling, E. The N-Terminal Arm of Small Heat Shock Proteins is Important for both Chaperone Activity and Substrate Specificity. J. Biol. Chem. 2006, 281, 39943–39952. [Google Scholar] [CrossRef]
- Ecroyd, H.; Meehan, S.; Horwitz, J.; Aquilina, J.A.; Benesch, J.L.; Robinson, C.V.; Macphee, C.E.; Carver, J.A. Mimicking Phosphorylation of αB-Crystallin Affects its Chaperone Activity. Biochem. J. 2007, 401, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Jaya, N.; Garcia, V.; Vierling, E. Substrate Binding Site Flexibility of the Small Heat Shock Protein Molecular Chaperones. Proc. Natl. Acad. Sci. USA 2009, 106, 15604–15609. [Google Scholar] [CrossRef] [PubMed]
- Delbecq, S.P.; Klevit, R.E. One Size does Not Fit all: The Oligomeric States of αB Crystallin. FEBS Lett. 2013, 587, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Mainz, A.; Peschek, J.; Stavropoulou, M.; Back, K.C.; Bardiaux, B.; Asami, S.; Prade, E.; Peters, C.; Weinkauf, S.; Buchner, J.; et al. The Chaperone αB-Crystallin Uses Different Interfaces to Capture an Amorphous and an Amyloid Client. Nat. Struct. Mol. Biol. 2015, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, C.; Li, Y.; Zhao, C.; Li, T.; Li, D.; Zhang, S.; Liu, C. Mechanistic Insights into the Switch of αB-Crystallin Chaperone Activity and Self-Multimerization. J. Biol. Chem. 2018, 293, 14880–14890. [Google Scholar] [CrossRef] [PubMed]
- Freilich, R.; Betegon, M.; Tse, E.; Mok, S.A.; Julien, O.; Agard, D.A.; Southworth, D.R.; Takeuchi, K.; Gestwicki, J.E. Competing Protein-Protein Interactions Regulate Binding of Hsp27 to its Client Protein Tau. Nat. Commun. 2018, 9, 4563. [Google Scholar] [CrossRef] [PubMed]
- Delbecq, S.P.; Klevit, R.E. HSPB5 Engages Multiple States of a Destabilized Client to Enhance Chaperone Activity in a Stress-Dependent Manner. J. Biol. Chem. 2019, 294, 3261–3270. [Google Scholar] [CrossRef]
- Benesch, J.L.; Ayoub, M.; Robinson, C.V.; Aquilina, J.A. Small Heat Shock Protein Activity is Regulated by Variable Oligomeric Substructure. J. Biol. Chem. 2008, 283, 28513–28517. [Google Scholar] [CrossRef]
- Shemetov, A.A.; Seit-Nebi, A.S.; Bukach, O.V.; Gusev, N.B. Phosphorylation by Cyclic AMP-Dependent Protein Kinase Inhibits Chaperone-Like Activity of Human HSP22 in Vitro. Biochemistry (Mosc) 2008, 73, 200–208. [Google Scholar] [CrossRef]
- Almeida-Souza, L.; Goethals, S.; de Winter, V.; Dierick, I.; Gallardo, R.; Van Durme, J.; Irobi, J.; Gettemans, J.; Rousseau, F.; Schymkowitz, J.; et al. Increased Monomerization of Mutant HSPB1 Leads to Protein Hyperactivity in Charcot-Marie-Tooth Neuropathy. J. Biol. Chem. 2010, 285, 12778–12786. [Google Scholar] [CrossRef]
- Alderson, T.R.; Roche, J.; Gastall, H.Y.; Dias, D.M.; Pritisanac, I.; Ying, J.; Bax, A.; Benesch, J.L.P.; Baldwin, A.J. Local Unfolding of the HSP27 Monomer Regulates Chaperone Activity. Nat. Commun. 2019, 10, 1068. [Google Scholar] [CrossRef] [PubMed]
- Rogalla, T.; Ehrnsperger, M.; Préville, X.; Kotlyarov, A.; Lutsch, G.; Ducasse, C.; Paul, C.; Wieske, M.; Arrigo, A.P.; Buchner, J.; et al. Regulation of Hsp27 Oligomerization, Chaperone Function, and Protective Activity Against Oxidative stress/tumor Necrosis Factor Alpha by Phosphorylation. J. Biol. Chem. 1999, 274, 18947–18956. [Google Scholar] [CrossRef] [PubMed]
- Giese, K.C.; Vierling, E. Changes in Oligomerization are Essential for the Chaperone Activity of a Small Heat Shock Protein in Vivo and in Vitro. J. Biol. Chem. 2002, 277, 46310–46318. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, M.J.; Bernock, L.J.; Landry, J.; Spitz, D.R.; Weber, L.A.; Hickey, E.; Freeman, M.L.; Corry, P.M. Stress Protection by a Fluorescent Hsp27 Chimera that is Independent of Nuclear Translocation Or Multimeric Dissociation. Cell Stress Chaperones 2002, 7, 281–296. [Google Scholar] [CrossRef]
- Franzmann, T.M.; Wühr, M.; Richter, K.; Walter, S.; Buchner, J. The Activation Mechanism of Hsp26 does Not Require Dissociation of the Oligomer. J. Mol. Biol. 2005, 350, 1083–1093. [Google Scholar] [CrossRef]
- Shashidharamurthy, R.; Koteiche, H.A.; Dong, J.; McHaourab, H.S. Mechanism of Chaperone Function in Small Heat Shock Proteins: Dissociation of the HSP27 Oligomer is Required for Recognition and Binding of Destabilized T4 Lysozyme. J. Biol. Chem. 2005, 280, 5281–5289. [Google Scholar] [CrossRef]
- Peschek, J.; Braun, N.; Rohrberg, J.; Back, K.C.; Kriehuber, T.; Kastenmüller, A.; Weinkauf, S.; Buchner, J. Regulated Structural Transitions Unleash the Chaperone Activity of αB-Crystallin. Proc. Natl. Acad. Sci. USA 2013, 110, E3780–E3789. [Google Scholar] [CrossRef]
- Santhanagopalan, I.; Degiacomi, M.T.; Shepherd, D.A.; Hochberg, G.K.A.; Benesch, J.L.P.; Vierling, E. It Takes a Dimer to Tango: Oligomeric Small Heat Shock Proteins Dissociate to Capture Substrate. J. Biol. Chem. 2018, 293, 19511–19521. [Google Scholar] [CrossRef]
- Rajagopal, P.; Tse, E.; Borst, A.J.; Delbecq, S.P.; Shi, L.; Southworth, D.R.; Klevit, R.E. A Conserved Histidine Modulates HSPB5 Structure to Trigger Chaperone Activity in Response to Stress-Related Acidosis. Elife 2015, 4. [Google Scholar] [CrossRef]
- Mymrikov, E.V.; Riedl, M.; Peters, C.; Weinkauf, S.; Haslbeck, M.; Buchner, J. Regulation of Small Heat Shock Proteins by Hetero-Oligomer Formation. J. Biol. Chem. 2019, 295, 158–169. [Google Scholar] [CrossRef]
- Lambert, H.; Charette, S.J.; Bernier, A.F.; Guimond, A.; Landry, J. HSP27 Multimerization Mediated by Phosphorylation-Sensitive Intermolecular Interactions at the Amino Terminus. J. Biol. Chem. 1999, 274, 9378–9385. [Google Scholar] [CrossRef] [PubMed]
- Koteiche, H.A.; McHaourab, H.S. Mechanism of Chaperone Function in Small Heat-Shock Proteins. Phosphorylation-Induced Activation of Two-Mode Binding in αB-Crystallin. J. Biol. Chem. 2003, 278, 10361–10367. [Google Scholar] [CrossRef] [PubMed]
- Shemetov, A.A.; Gusev, N.B. Biochemical Characterization of Small Heat Shock Protein HspB8 (Hsp22)-Bag3 Interaction. Arch. Biochem. Biophys. 2011, 513, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Collier, M.P.; Alderson, T.R.; de Villiers, C.P.; Nicholls, D.; Gastall, H.Y.; Allison, T.M.; Degiacomi, M.T.; Jiang, H.; Mlynek, G.; Fürst, D.O.; et al. HspB1 Phosphorylation Regulates its Intramolecular Dynamics and Mechanosensitive Molecular Chaperone Interaction with Filamin C. Sci. Adv. 2019, 5, eaav8421. [Google Scholar] [CrossRef] [PubMed]
- Ehrnsperger, M.; Lilie, H.; Gaestel, M.; Buchner, J. The Dynamics of Hsp25 Quaternary Structure. Structure and Function of Different Oligomeric Species. J. Biol. Chem. 1999, 274, 14867–14874. [Google Scholar] [CrossRef] [PubMed]
- Haslbeck, M.; Walke, S.; Stromer, T.; Ehrnsperger, M.; White, H.E.; Chen, S.; Saibil, H.R.; Buchner, J. Hsp26: A Temperature-Regulated Chaperone. EMBO J. 1999, 18, 6744–6751. [Google Scholar] [CrossRef]
- Clouser, A.F.; Klevit, R.E. pH-Dependent Structural Modulation is Conserved in the Human Small Heat Shock Protein HSBP1. Cell Stress Chaperones 2017, 22, 569–575. [Google Scholar] [CrossRef]
- Mainz, A.; Bardiaux, B.; Kuppler, F.; Multhaup, G.; Felli, I.C.; Pierattelli, R.; Reif, B. Structural and Mechanistic Implications of Metal Binding in the Small Heat-Shock Protein αB-Crystallin. J. Biol. Chem. 2012, 287, 1128–1138. [Google Scholar] [CrossRef]
- Kim, M.V.; Kasakov, A.S.; Seit-Nebi, A.S.; Marston, S.B.; Gusev, N.B. Structure and Properties of K141E Mutant of Small Heat Shock Protein HSP22 (HspB8, H11) that is Expressed in Human Neuromuscular Disorders. Arch. Biochem. Biophys. 2006, 454, 32–41. [Google Scholar] [CrossRef]
- Mymrikov, E.V.; Daake, M.; Richter, B.; Haslbeck, M.; Buchner, J. The Chaperone Activity and Substrate Spectrum of Human Small Heat Shock Proteins. J. Biol. Chem. 2017, 292, 672–684. [Google Scholar] [CrossRef]
- Boelens, W.C. Cell Biological Roles of αB-Crystallin. Prog. Biophys. Mol. Biol. 2014, 115, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Dimauro, I.; Antonioni, A.; Mercatelli, N.; Caporossi, D. The Role of αB-Crystallin in Skeletal and Cardiac Muscle Tissues. Cell Stress Chaperones 2018, 23, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.P. Mammalian HspB1 (Hsp27) is a Molecular Sensor Linked to the Physiology and Environment of the Cell. Cell Stress Chaperones 2017, 22, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Ruger-Herreros, C.; Bukau, B. Cellular Functions and Mechanisms of Action of Small Heat Shock Proteins. Annu. Rev. Microbiol. 2019, 73, 89–110. [Google Scholar] [CrossRef]
- Inaguma, Y.; Hasegawa, K.; Goto, S.; Ito, H.; Kato, K. Induction of the Synthesis of hsp27 and αB Crystallin in Tissues of Heat-Stressed Rats and its Suppression by Ethanol Or an α1-Adrenergic Antagonist. J. Biochem. 1995, 117, 1238–1243. [Google Scholar] [CrossRef]
- Neufer, P.D.; Benjamin, I.J. Differential Expression of αB-Crystallin and Hsp27 in Skeletal Muscle during Continuous Contractile Activity. Relationship to Myogenic Regulatory Factors. J. Biol. Chem. 1996, 271, 24089–24095. [Google Scholar] [CrossRef]
- Golenhofen, N.; Perng, M.D.; Quinlan, R.A.; Drenckhahn, D. Comparison of the Small Heat Shock Proteins αB-Crystallin, MKBP, HSP25, HSP20, and cvHSP in Heart and Skeletal Muscle. Histochem. Cell Biol. 2004, 122, 415–425. [Google Scholar] [CrossRef]
- Larkins, N.T.; Murphy, R.M.; Lamb, G.D. Absolute Amounts and Diffusibility of HSP72, HSP25, and αB-Crystallin in Fast- and Slow-Twitch Skeletal Muscle Fibers of Rat. Am. J. Physiol. Cell. Physiol. 2012, 302, C228–C239. [Google Scholar] [CrossRef]
- Atomi, Y.; Yamada, S.; Strohman, R.; Nonomura, Y. αB-Crystallin in Skeletal Muscle: Purification and Localization. J. Biochem. 1991, 110, 812–822. [Google Scholar] [CrossRef]
- Golenhofen, N.; Ness, W.; Koob, R.; Htun, P.; Schaper, W.; Drenckhahn, D. Ischemia-Induced Phosphorylation and Translocation of Stress Protein αB-Crystallin to Z Lines of Myocardium. Am. J. Physiol. 1998, 274, H1457–H1464. [Google Scholar] [CrossRef]
- Golenhofen, N.; Arbeiter, A.; Koob, R.; Drenckhahn, D. Ischemia-Induced Association of the Stress Protein αB-Crystallin with I-Band Portion of Cardiac Titin. J. Mol. Cell. Cardiol. 2002, 34, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Bullard, B.; Ferguson, C.; Minajeva, A.; Leake, M.C.; Gautel, M.; Labeit, D.; Ding, L.; Labeit, S.; Horwitz, J.; Leonard, K.R.; et al. Association of the Chaperone αB-Crystallin with Titin in Heart Muscle. J. Biol. Chem. 2004, 279, 7917–7924. [Google Scholar] [CrossRef] [PubMed]
- Koh, T.J.; Escobedo, J. Cytoskeletal Disruption and Small Heat Shock Protein Translocation Immediately after Lengthening Contractions. Am. J. Physiol. Cell. Physiol. 2004, 286, C713–C722. [Google Scholar] [CrossRef] [PubMed]
- Kötter, S.; Unger, A.; Hamdani, N.; Lang, P.; Vorgerd, M.; Nagel-Steger, L.; Linke, W.A. Human Myocytes are Protected from Titin Aggregation-Induced Stiffening by Small Heat Shock Proteins. J. Cell Biol. 2014, 204, 187–202. [Google Scholar] [CrossRef]
- Bennardini, F.; Wrzosek, A.; Chiesi, M. αB-Crystallin in Cardiac Tissue. Association with Actin and Desmin Filaments. Circ. Res. 1992, 71, 288–294. [Google Scholar] [CrossRef]
- Nicholl, I.D.; Quinlan, R.A. Chaperone Activity of α-Crystallins Modulates Intermediate Filament Assembly. EMBO J. 1994, 13, 945–953. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Valdez, M.M.; Clark, J.I. αB-Crystallin Selectively Targets Intermediate Filament Proteins during Thermal Stress. Investig. Ophthalmol. Vis. Sci. 1999, 40, 951–958. [Google Scholar]
- Perng, M.D.; Cairns, L.; van den IJssel, P.; Prescott, A.; Hutcheson, A.M.; Quinlan, R.A. Intermediate Filament Interactions can be Altered by HSP27 and αB-Crystallin. J. Cell. Sci. 1999, 112 Pt 13, 2099–2112. [Google Scholar]
- Perng, M.D.; Wen, S.F.; van den IJssel, P.; Prescott, A.R.; Quinlan, R.A. Desmin Aggregate Formation by R120G αB-Crystallin is Caused by Altered Filament Interactions and is Dependent upon Network Status in Cells. Mol. Biol. Cell 2004, 15, 2335–2346. [Google Scholar] [CrossRef]
- Elliott, J.L.; Der Perng, M.; Prescott, A.R.; Jansen, K.A.; Koenderink, G.H.; Quinlan, R.A. The Specificity of the Interaction between αB-Crystallin and Desmin Filaments and its Impact on Filament Aggregation and Cell Viability. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120375. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Houck, S.A.; Clark, J.I. Interactive Sequences in the Stress Protein and Molecular Chaperone Human αB Crystallin Recognize and Modulate the Assembly of Filaments. Int. J. Biochem. Cell Biol. 2007, 39, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, I.; Jabłońska, J.; Zmojdzian, M.; Taghli-Lamallem, O.; Renaud, Y.; Junion, G.; Daczewska, M.; Huelsmann, S.; Jagla, K.; Jagla, T. Drosophila Small Heat Shock Protein CryAB Ensures Structural Integrity of Developing Muscles, and Proper Muscle and Heart Performance. Development 2015, 142, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Tang, S.; Xu, J.; Sun, J.; Zhang, X.; Li, Y.; Bao, E. CRYAB Protects Cardiomyocytes Against Heat Stress by Preventing Caspase-Mediated Apoptosis and Reducing F-Actin Aggregation. Cell Stress Chaperones 2019, 24, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Atomi, Y. Chaperone Activity of αB-Crystallin Suppresses Tubulin Aggregation through Complex Formation. Cell Struct. Funct. 1997, 22, 539–544. [Google Scholar] [CrossRef]
- Xi, J.H.; Bai, F.; McGaha, R.; Andley, U.P. Alpha-Crystallin Expression Affects Microtubule Assembly and Prevents their Aggregation. FASEB J. 2006, 20, 846–857. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Houck, S.A.; Clark, J.I. Interactive Domains in the Molecular Chaperone Human αB Crystallin Modulate Microtubule Assembly and Disassembly. PLoS ONE 2007, 2, e498. [Google Scholar] [CrossRef]
- Houck, S.A.; Clark, J.I. Dynamic Subunit Exchange and the Regulation of Microtubule Assembly by the Stress Response Protein Human αB Crystallin. PLoS ONE 2010, 5, e11795. [Google Scholar] [CrossRef]
- Zhu, Y.; Bogomolovas, J.; Labeit, S.; Granzier, H. Single Molecule Force Spectroscopy of the Cardiac Titin N2B Element: Effects of the Molecular Chaperone αB-Crystallin with Disease-Causing Mutations. J. Biol. Chem. 2009, 284, 13914–13923. [Google Scholar] [CrossRef]
- Kamradt, M.C.; Chen, F.; Sam, S.; Cryns, V.L. The Small Heat Shock Protein αB-Crystallin Negatively Regulates Apoptosis during Myogenic Differentiation by Inhibiting Caspase-3 Activation. J. Biol. Chem. 2002, 277, 38731–38736. [Google Scholar] [CrossRef]
- Acunzo, J.; Katsogiannou, M.; Rocchi, P. Small Heat Shock Proteins HSP27 (HspB1), αB-Crystallin (HspB5) and HSP22 (HspB8) as Regulators of Cell Death. Int. J. Biochem. Cell Biol. 2012, 44, 1622–1631. [Google Scholar] [CrossRef]
- Ganguly, S.; Mitra, A.; Sarkar, S. Role of α-Crystallin B in Regulation of Stress Induced Cardiomyocyte Apoptosis. Cardiovasc. Hematol. Agents Med. Chem. 2014, 12, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prevost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tome, F.; Dupret, J.M.; et al. A Missense Mutation in the αB-Crystallin Chaperone Gene Causes a Desmin-Related Myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Engel, A.G. Myofibrillar Myopathy Caused by Novel Dominant Negative αB-Crystallin Mutations. Ann. Neurol. 2003, 54, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Reilich, P.; Schoser, B.; Schramm, N.; Krause, S.; Schessl, J.; Kress, W.; Müller-Höcker, J.; Walter, M.C.; Lochmüller, H. The p.G154S Mutation of the Alpha-B Crystallin Gene (CRYAB) Causes Late-Onset Distal Myopathy. Neuromuscul. Disord. 2010, 20, 255–259. [Google Scholar] [CrossRef]
- Sacconi, S.; Féasson, L.; Antoine, J.C.; Pécheux, C.; Bernard, R.; Cobo, A.M.; Casarin, A.; Salviati, L.; Desnuelle, C.; Urtizberea, A. A Novel CRYAB Mutation Resulting in Multisystemic Disease. Neuromuscul. Disord. 2012, 22, 66–72. [Google Scholar] [CrossRef]
- Fichna, J.P.; Potulska-Chromik, A.; Miszta, P.; Redowicz, M.J.; Kaminska, A.M.; Zekanowski, C.; Filipek, S. A Novel Dominant D109A CRYAB Mutation in a Family with Myofibrillar Myopathy Affects αB-Crystallin Structure. BBA Clin. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschroder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The Novel αB-Crystallin (CRYAB) Mutation p.D109G Causes Restrictive Cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef]
- Inagaki, N.; Hayashi, T.; Arimura, T.; Koga, Y.; Takahashi, M.; Shibata, H.; Teraoka, K.; Chikamori, T.; Yamashina, A.; Kimura, A. ΑB-Crystallin Mutation in Dilated Cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 342, 379–386. [Google Scholar] [CrossRef]
- Pilotto, A.; Marziliano, N.; Pasotti, M.; Grasso, M.; Costante, A.M.; Arbustini, E. αB-Crystallin Mutation in Dilated Cardiomyopathies: Low Prevalence in a Consecutive Series of 200 Unrelated Probands. Biochem. Biophys. Res. Commun. 2006, 346, 1115–1117. [Google Scholar] [CrossRef]
- Claeys, K.G.; Fardeau, M.; Schröder, R.; Suominen, T.; Tolksdorf, K.; Behin, A.; Dubourg, O.; Eymard, B.; Maisonobe, T.; Stojkovic, T.; et al. Electron Microscopy in Myofibrillar Myopathies Reveals Clues to the Mutated Gene. Neuromuscul. Disord. 2008, 18, 656–666. [Google Scholar] [CrossRef]
- Schröder, R.; Schoser, B. Myofibrillar Myopathies: A Clinical and Myopathological Guide. Brain Pathol. 2009, 19, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Del Bigio, M.R.; Chudley, A.E.; Sarnat, H.B.; Campbell, C.; Goobie, S.; Chodirker, B.N.; Selcen, D. Infantile Muscular Dystrophy in Canadian Aboriginals is an αB-Crystallinopathy. Ann. Neurol. 2011, 69, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Forrest, K.M.; Al-Sarraj, S.; Sewry, C.; Buk, S.; Tan, S.V.; Pitt, M.; Durward, A.; McDougall, M.; Irving, M.; Hanna, M.G.; et al. Infantile Onset Myofibrillar Myopathy due to Recessive CRYAB Mutations. Neuromuscul. Disord. 2011, 21, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Luo, D.; Tian, T.; Li, N.; He, X.; Rao, C.; Zhong, B.; Lu, X. A Novel Homozygous Initiation Codon Variant Associated with Infantile Alpha-Bcrystallinopathy in a Chinese Family. Mol. Genet. Genom. Med. 2019, 7, e825. [Google Scholar] [CrossRef]
- Wang, X.; Osinska, H.; Klevitsky, R.; Gerdes, A.M.; Nieman, M.; Lorenz, J.; Hewett, T.; Robbins, J. Expression of R120G-αB-Crystallin Causes Aberrant Desmin and αB-Crystallin Aggregation and Cardiomyopathy in Mice. Circ. Res. 2001, 89, 84–91. [Google Scholar] [CrossRef]
- Sanbe, A.; Osinska, H.; Villa, C.; Gulick, J.; Klevitsky, R.; Glabe, C.G.; Kayed, R.; Robbins, J. Reversal of Amyloid-Induced Heart Disease in Desmin-Related Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2005, 102, 13592–13597. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human αB-Crystallin Mutation Causes Oxido-Reductive Stress and Protein Aggregation Cardiomyopathy in Mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef]
- Tannous, P.; Zhu, H.; Johnstone, J.L.; Shelton, J.M.; Rajasekaran, N.S.; Benjamin, I.J.; Nguyen, L.; Gerard, R.D.; Levine, B.; Rothermel, B.A.; et al. Autophagy is an Adaptive Response in Desmin-Related Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 9745–9750. [Google Scholar] [CrossRef]
- Maloyan, A.; Osinska, H.; Lammerding, J.; Lee, R.T.; Cingolani, O.H.; Kass, D.A.; Lorenz, J.N.; Robbins, J. Biochemical and Mechanical Dysfunction in a Mouse Model of Desmin-Related Myopathy. Circ. Res. 2009, 104, 1021–1028. [Google Scholar] [CrossRef]
- Wang, X.; Klevitsky, R.; Huang, W.; Glasford, J.; Li, F.; Robbins, J. αB-Crystallin Modulates Protein Aggregation of Abnormal Desmin. Circ. Res. 2003, 93, 998–1005. [Google Scholar] [CrossRef]
- Maloyan, A.; Sanbe, A.; Osinska, H.; Westfall, M.; Robinson, D.; Imahashi, K.; Murphy, E.; Robbins, J. Mitochondrial Dysfunction and Apoptosis Underlie the Pathogenic Process in α-B-Crystallin Desmin-Related Cardiomyopathy. Circulation 2005, 112, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Mani, K.; Liu, H.; Kovacs, A.; Murphy, J.T.; Foroughi, L.; French, B.A.; Weinheimer, C.J.; Kraja, A.; Benjamin, I.J.; et al. Transcription Factor EB Activation Rescues Advanced αB-Crystallin Mutation-Induced Cardiomyopathy by Normalizing Desmin Localization. J. Am. Heart Assoc. 2019, 8, e010866. [Google Scholar] [CrossRef] [PubMed]
- Andley, U.P.; Hamilton, P.D.; Ravi, N.; Weihl, C.C. A Knock-in Mouse Model for the R120G Mutation of αB-Crystallin Recapitulates Human Hereditary Myopathy and Cataracts. PLoS ONE 2011, 6, e17671. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.P.; Garland, D.L.; Green, D.E.; Tamm, E.R.; Giblin, F.J.; Wawrousek, E.F. αB-Crystallin in Lens Development and Muscle Integrity: A Gene Knockout Approach. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2924–2934. [Google Scholar]
- Morrison, L.E.; Whittaker, R.J.; Klepper, R.E.; Wawrousek, E.F.; Glembotski, C.C. Roles for αB-Crystallin and HSPB2 in Protecting the Myocardium from Ischemia-Reperfusion-Induced Damage in a KO Mouse Model. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H847–H855. [Google Scholar] [CrossRef]
- Golenhofen, N.; Redel, A.; Wawrousek, E.F.; Drenckhahn, D. Ischemia-Induced Increase of Stiffness of αB-crystallin/HSPB2-Deficient Myocardium. Pflugers Arch. 2006, 451, 518–525. [Google Scholar] [CrossRef]
- Neppl, R.L.; Kataoka, M.; Wang, D.Z. Crystallin-αB Regulates Skeletal Muscle Homeostasis Via Modulation of argonaute2 Activity. J. Biol. Chem. 2014, 289, 17240–17248. [Google Scholar] [CrossRef]
- Kadono, T.; Zhang, X.Q.; Srinivasan, S.; Ishida, H.; Barry, W.H.; Benjamin, I.J. CRYAB and HSPB2 Deficiency Increases Myocyte Mitochondrial Permeability Transition and Mitochondrial Calcium Uptake. J. Mol. Cell. Cardiol. 2006, 40, 783–789. [Google Scholar] [CrossRef]
- Benjamin, I.J.; Guo, Y.; Srinivasan, S.; Boudina, S.; Taylor, R.P.; Rajasekaran, N.S.; Gottlieb, R.; Wawrousek, E.F.; Abel, E.D.; Bolli, R. CRYAB and HSPB2 Deficiency Alters Cardiac Metabolism and Paradoxically Confers Protection Against Myocardial Ischemia in Aging Mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3201–H3209. [Google Scholar] [CrossRef]
- Bova, M.P.; Yaron, O.; Huang, Q.; Ding, L.; Haley, D.A.; Stewart, P.L.; Horwitz, J. Mutation R120G in αB-Crystallin, which is Linked to a Desmin-Related Myopathy, Results in an Irregular Structure and Defective Chaperone-Like Function. Proc. Natl. Acad. Sci. USA 1999, 96, 6137–6142. [Google Scholar] [CrossRef]
- Kumar, L.V.; Ramakrishna, T.; Rao, C.M. Structural and Functional Consequences of the Mutation of a Conserved Arginine Residue in αA and αB Crystallins. J. Biol. Chem. 1999, 274, 24137–24141. [Google Scholar] [CrossRef] [PubMed]
- Treweek, T.M.; Rekas, A.; Lindner, R.A.; Walker, M.J.; Aquilina, J.A.; Robinson, C.V.; Horwitz, J.; Perng, M.D.; Quinlan, R.A.; Carver, J.A. R120G αB-Crystallin Promotes the Unfolding of Reduced Alpha-Lactalbumin and is Inherently Unstable. FEBS J. 2005, 272, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Michiel, M.; Skouri-Panet, F.; Lechaire, J.P.; Vicart, P.; Tardieu, A. Residue R120 is Essential for the Quaternary Structure and Functional Integrity of Human αB-Crystallin. Biochemistry 2007, 46, 9605–9614. [Google Scholar] [CrossRef] [PubMed]
- Bagnéris, C.; Bateman, O.A.; Naylor, C.E.; Cronin, N.; Boelens, W.C.; Keep, N.H.; Slingsby, C. Crystal Structures of α-Crystallin Domain Dimers of αB-Crystallin and Hsp20. J. Mol. Biol. 2009, 392, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.R.; Naylor, C.E.; Bagnéris, C.; Keep, N.H.; Slingsby, C. Crystal Structure of R120G Disease Mutant of Human αB-Crystallin Domain Dimer shows Closure of a Groove. J. Mol. Biol. 2011, 408, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Michiel, M.; Skouri-Panet, F.; Duprat, E.; Simon, S.; Férard, C.; Tardieu, A.; Finet, S. Abnormal Assemblies and Subunit Exchange of αB-Crystallin R120 Mutants could be Associated with Destabilization of the Dimeric Substructure. Biochemistry 2009, 48, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Dimitrova, V.; Gibert, B.; Virot, S.; Mounier, N.; Nivon, M.; Kretz-Remy, C.; Corset, V.; Mehlen, P.; Arrigo, A.P. Analysis of the Dominant Effects Mediated by Wild Type Or R120G Mutant of alphaB-Crystallin (HspB5) Towards Hsp27 (HspB1). PLoS ONE 2013, 8, e70545. [Google Scholar] [CrossRef]
- Hayes, V.H.; Devlin, G.; Quinlan, R.A. Truncation of αB-Crystallin by the Myopathy-Causing Q151X Mutation significantly Destabilizes the Protein Leading to Aggregate Formation in Transfected Cells. J. Biol. Chem. 2008, 283, 10500–10512. [Google Scholar] [CrossRef]
- Simon, S.; Fontaine, J.M.; Martin, J.L.; Sun, X.; Hoppe, A.D.; Welsh, M.J.; Benndorf, R.; Vicart, P. Myopathy-Associated αB-Crystallin Mutants: Abnormal Phosphorylation, Intracellular Location, and Interactions with Other Small Heat Shock Proteins. J. Biol. Chem. 2007, 282, 34276–34287. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Luo, L.; Wu, M.; Zeng, R.; Cheng, G.; Hu, B.; Liu, B.; Liang, J.J.; Shang, F. A Novel αB-Crystallin Mutation Associated with Autosomal Dominant Congenital Lamellar Cataract. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1069–1075. [Google Scholar] [CrossRef]
- Gerasimovich, E.S.; Strelkov, S.V.; Gusev, N.B. Some Properties of Three αB-Crystallin Mutants Carrying Point Substitutions in the C-Terminal Domain and Associated with Congenital Diseases. Biochimie 2017, 142, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Chávez Zobel, A.T.; Loranger, A.; Marceau, N.; Thériault, J.R.; Lambert, H.; Landry, J. Distinct Chaperone Mechanisms can Delay the Formation of Aggresomes by the Myopathy-Causing R120G αB-Crystallin Mutant. Hum. Mol. Genet. 2003, 12, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Kamei, K.; Iwamoto, I.; Inaguma, Y.; Tsuzuki, M.; Kishikawa, M.; Shimada, A.; Hosokawa, M.; Kato, K. Hsp27 Suppresses the Formation of Inclusion Bodies Induced by Expression of R120G αB-Crystallin, a Cause of Desmin-Related Myopathy. Cell Mol. Life Sci. 2003, 60, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Hanson, M.J.; Liu, B.F.; Chylack, L.T.; Liang, J.J. Protein-Protein Interactions between Lens Vimentin and αB-Crystallin using FRET Acceptor Photobleaching. Mol. Vis. 2008, 14, 1282–1287. [Google Scholar]
- Chen, F.; Chang, R.; Trivedi, M.; Capetanaki, Y.; Cryns, V.L. Caspase Proteolysis of Desmin Produces a Dominant-Negative Inhibitor of Intermediate Filaments and Promotes Apoptosis. J. Biol. Chem. 2003, 278, 6848–6853. [Google Scholar] [CrossRef]
- den Engelsman, J.; Bennink, E.J.; Doerwald, L.; Onnekink, C.; Wunderink, L.; Andley, U.P.; Kato, K.; de Jong, W.W.; Boelens, W.C. Mimicking Phosphorylation of the Small Heat-Shock Protein αB-Crystallin Recruits the F-Box Protein FBX4 to Nuclear SC35 Speckles. Eur. J. Biochem. 2004, 271, 4195–4203. [Google Scholar] [CrossRef]
- den Engelsman, J.; Gerrits, D.; de Jong, W.W.; Robbins, J.; Kato, K.; Boelens, W.C. Nuclear Import of αB-Crystallin is Phosphorylation-Dependent and Hampered by Hyperphosphorylation of the Myopathy-Related Mutant R120G. J. Biol. Chem. 2005, 280, 37139–37148. [Google Scholar] [CrossRef]
- van den IJssel, P.; Wheelock, R.; Prescott, A.; Russell, P.; Quinlan, R.A. Nuclear Speckle Localisation of the Small Heat Shock Protein αB-Crystallin and its Inhibition by the R120G Cardiomyopathy-Linked Mutation. Exp. Cell Res. 2003, 287, 249–261. [Google Scholar] [CrossRef]
- den Engelsman, J.; van de Schootbrugge, C.; Yong, J.; Pruijn, G.J.; Boelens, W.C. Pseudophosphorylated αB-Crystallin is a Nuclear Chaperone Imported into the Nucleus with Help of the SMN Complex. PLoS ONE 2013, 8, e73489. [Google Scholar] [CrossRef]
- Adhikari, A.S.; Sridhar Rao, K.; Rangaraj, N.; Parnaik, V.K.; Mohan Rao, C. Heat Stress-Induced Localization of Small Heat Shock Proteins in Mouse Myoblasts: Intranuclear Lamin A/C Speckles as Target for alphaB-Crystallin and Hsp25. Exp. Cell Res. 2004, 299, 393–403. [Google Scholar] [CrossRef]
- Sanbe, A.; Osinska, H.; Saffitz, J.E.; Glabe, C.G.; Kayed, R.; Maloyan, A.; Robbins, J. Desmin-Related Cardiomyopathy in Transgenic Mice: A Cardiac Amyloidosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10132–10136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rajasekaran, N.S.; Orosz, A.; Xiao, X.; Rechsteiner, M.; Benjamin, I.J. Selective Degradation of Aggregate-Prone CryAB Mutants by HSPB1 is Mediated by Ubiquitin-Proteasome Pathways. J. Mol. Cell. Cardiol. 2010, 49, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Min, X.; Li, C.; Benjamin, I.J.; Qian, B.; Zhang, X.; Ding, Z.; Gao, X.; Yao, Y.; Ma, Y.; et al. Involvement of Reductive Stress in the Cardiomyopathy in Transgenic Mice with Cardiac-Specific Overexpression of Heat Shock Protein 27. Hypertension 2010, 55, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Mitzelfelt, K.A.; Limphong, P.; Choi, M.J.; Kondrat, F.D.; Lai, S.; Kolander, K.D.; Kwok, W.M.; Dai, Q.; Grzybowski, M.N.; Zhang, H.; et al. The Human 343delT HSPB5 Chaperone Associated with Early-Onset Skeletal Myopathy Causes Defects in Protein Solubility. J. Biol. Chem. 2016, 291, 14939–14953. [Google Scholar] [CrossRef] [PubMed]
- Meehan, S.; Berry, Y.; Luisi, B.; Dobson, C.M.; Carver, J.A.; MacPhee, C.E. Amyloid Fibril Formation by Lens Crystallin Proteins and its Implications for Cataract Formation. J. Biol. Chem. 2004, 279, 3413–3419. [Google Scholar] [CrossRef] [PubMed]
- Meehan, S.; Knowles, T.P.; Baldwin, A.J.; Smith, J.F.; Squires, A.M.; Clements, P.; Treweek, T.M.; Ecroyd, H.; Tartaglia, G.G.; Vendruscolo, M.; et al. Characterisation of Amyloid Fibril Formation by Small Heat-Shock Chaperone Proteins Human αA-, αB- and R120G αB-Crystallins. J. Mol. Biol. 2007, 372, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Maloyan, A.; Gulick, J.; Glabe, C.G.; Kayed, R.; Robbins, J. Exercise Reverses Preamyloid Oligomer and Prolongs Survival in αB-Crystallin-Based Desmin-Related Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 5995–6000. [Google Scholar] [CrossRef]
- Sanbe, A.; Yamauchi, J.; Miyamoto, Y.; Fujiwara, Y.; Murabe, M.; Tanoue, A. Interruption of CryAB-Amyloid Oligomer Formation by HSP22. J. Biol. Chem. 2007, 282, 555–563. [Google Scholar] [CrossRef]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular Mechanisms of Amyloid Oligomers Toxicity. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S67–S78. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, J.B.; Horak, K.M.; Zheng, H.; Kumarapeli, A.R.; Li, J.; Li, F.; Gerdes, A.M.; Wawrousek, E.F.; Wang, X. Intrasarcoplasmic Amyloidosis Impairs Proteolytic Function of Proteasomes in Cardiomyocytes by Compromising Substrate Uptake. Circ. Res. 2005, 97, 1018–1026. [Google Scholar] [CrossRef]
- Pattison, J.S.; Osinska, H.; Robbins, J. Atg7 Induces Basal Autophagy and Rescues Autophagic Deficiency in CryABR120G Cardiomyocytes. Circ. Res. 2011, 109, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Maloyan, A.; Sayegh, J.; Osinska, H.; Chua, B.H.; Robbins, J. Manipulation of Death Pathways in Desmin-Related Cardiomyopathy. Circ. Res. 2010, 106, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.S.; Pattison, J.S.; Osinska, H.; James, J.; Gulick, J.; McLendon, P.M.; Hill, J.A.; Sadoshima, J.; Robbins, J. Enhanced Autophagy Ameliorates Cardiac Proteinopathy. J. Clin. Investig. 2013, 123, 5284–5297. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Zhang, H.; Cui, T.; Wang, X. TFEB Activation Protects Against Cardiac Proteotoxicity Via Increasing Autophagic Flux. J. Mol. Cell. Cardiol. 2017, 113, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Horak, K.M.; Su, H.; Sanbe, A.; Robbins, J.; Wang, X. Enhancement of Proteasomal Function Protects Against Cardiac Proteinopathy and ischemia/reperfusion Injury in Mice. J. Clin. Investig. 2011, 121, 3689–3700. [Google Scholar] [CrossRef]
- Zheng, H.; Tang, M.; Zheng, Q.; Kumarapeli, A.R.; Horak, K.M.; Tian, Z.; Wang, X. Doxycycline Attenuates Protein Aggregation in Cardiomyocytes and Improves Survival of a Mouse Model of Cardiac Proteinopathy. J. Am. Coll. Cardiol. 2010, 56, 1418–1426. [Google Scholar] [CrossRef]
- Claeys, K.G.; van der Ven, P.F.; Behin, A.; Stojkovic, T.; Eymard, B.; Dubourg, O.; Laforêt, P.; Faulkner, G.; Richard, P.; Vicart, P.; et al. Differential Involvement of Sarcomeric Proteins in Myofibrillar Myopathies: A Morphological and Immunohistochemical Study. Acta Neuropathol. 2009, 117, 293–307. [Google Scholar] [CrossRef]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin Cytoskeleton Linked to Muscle Mitochondrial Distribution and Respiratory Function. J. Cell Biol. 2000, 150, 1283–1298. [Google Scholar] [CrossRef]
- Winter, L.; Wittig, I.; Peeva, V.; Eggers, B.; Heidler, J.; Chevessier, F.; Kley, R.A.; Barkovits, K.; Strecker, V.; Berwanger, C.; et al. Mutant Desmin Substantially Perturbs Mitochondrial Morphology, Function and Maintenance in Skeletal Muscle Tissue. Acta Neuropathol. 2016, 132, 453–473. [Google Scholar] [CrossRef]
- Diokmetzidou, A.; Soumaka, E.; Kloukina, I.; Tsikitis, M.; Makridakis, M.; Varela, A.; Davos, C.H.; Georgopoulos, S.; Anesti, V.; Vlahou, A.; et al. Desmin and αB-Crystallin Interplay in the Maintenance of Mitochondrial Homeostasis and Cardiomyocyte Survival. J. Cell. Sci. 2016, 129, 3705–3720. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Varadharaj, S.; Khanderao, G.D.; Davidson, C.J.; Kannan, S.; Firpo, M.A.; Zweier, J.L.; Benjamin, I.J. Sustained Activation of Nuclear Erythroid 2-Related Factor 2/antioxidant Response Element Signaling Promotes Reductive Stress in the Human Mutant Protein Aggregation Cardiomyopathy in Mice. Antioxid. Redox Signal. 2011, 14, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, N.S.; Firpo, M.A.; Milash, B.A.; Weiss, R.B.; Benjamin, I.J. Global Expression Profiling Identifies a Novel Biosignature for Protein Aggregation R120GCryAB Cardiomyopathy in Mice. Physiol. Genom. 2008, 35, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Banerjee Mustafi, S.; Grose, J.H.; Zhang, H.; Pratt, G.W.; Sadoshima, J.; Christians, E.S.; Benjamin, I.J. Aggregate-Prone R120GCRYAB Triggers Multifaceted Modifications of the Thioredoxin System. Antioxid. Redox Signal. 2014, 20, 2891–2906. [Google Scholar] [CrossRef] [PubMed]
- Perng, M.D.; Muchowski, P.J.; van Den IJssel, P.; Wu, G.J.; Hutcheson, A.M.; Clark, J.I.; Quinlan, R.A. The Cardiomyopathy and Lens Cataract Mutation in αB-Crystallin Alters its Protein Structure, Chaperone Activity, and Interaction with Intermediate Filaments in Vitro. J. Biol. Chem. 1999, 274, 33235–33243. [Google Scholar] [CrossRef]
- van der Smagt, J.J.; Vink, A.; Kirkels, J.H.; Nelen, M.; ter Heide, H.; Molenschot, M.M.; Weger, R.A.; Schellekens, P.A.; Hoogendijk, J.; Dooijes, D. Congenital Posterior Pole Cataract and Adult Onset Dilating Cardiomyopathy: Expanding the Phenotype of αB-Crystallinopathies. Clin. Genet. 2014, 85, 381–385. [Google Scholar] [CrossRef]
- Vos, M.J.; Zijlstra, M.P.; Kanon, B.; van Waarde-Verhagen, M.A.; Brunt, E.R.; Oosterveld-Hut, H.M.; Carra, S.; Sibon, O.C.; Kampinga, H.H. HSPB7 is the most Potent polyQ Aggregation Suppressor within the HSPB Family of Molecular Chaperones. Hum. Mol. Genet. 2010, 19, 4677–4693. [Google Scholar] [CrossRef]
- Jovcevski, B.; Kelly, M.A.; Rote, A.P.; Berg, T.; Gastall, H.Y.; Benesch, J.L.; Aquilina, J.A.; Ecroyd, H. Phosphomimics Destabilize Hsp27 Oligomeric Assemblies and Enhance Chaperone Activity. Chem. Biol. 2015, 22, 186–195. [Google Scholar] [CrossRef]
- Hayes, D.; Napoli, V.; Mazurkie, A.; Stafford, W.F.; Graceffa, P. Phosphorylation Dependence of hsp27 Multimeric Size and Molecular Chaperone Function. J. Biol. Chem. 2009, 284, 18801–18807. [Google Scholar] [CrossRef]
- Sha, E.; Nakamura, M.; Ankai, K.; Yamamoto, Y.Y.; Oka, T.; Yohda, M. Functional and Structural Characterization of HspB1/Hsp27 from Chinese Hamster Ovary Cells. FEBS Open Bio 2019, 9, 1826–1834. [Google Scholar] [CrossRef]
- Mehlen, P.; Hickey, E.; Weber, L.A.; Arrigo, A.P. Large Unphosphorylated Aggregates as the Active Form of hsp27 which Controls Intracellular Reactive Oxygen Species and Glutathione Levels and Generates a Protection Against TNFalpha in NIH-3T3-Ras Cells. Biochem. Biophys. Res. Commun. 1997, 241, 187–192. [Google Scholar] [CrossRef]
- Eaton, P.; Fuller, W.; Shattock, M.J. S-Thiolation of HSP27 Regulates its Multimeric Aggregate Size Independently of Phosphorylation. J. Biol. Chem. 2002, 277, 21189–21196. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Shinohara, H.; Goto, S.; Inaguma, Y.; Morishita, R.; Asano, T. Copurification of Small Heat Shock Protein with αB Crystallin from Human Skeletal Muscle. J. Biol. Chem. 1992, 267, 7718–7725. [Google Scholar] [PubMed]
- Zantema, A.; Verlaan-De Vries, M.; Maasdam, D.; Bol, S.; van der Eb, A. Heat Shock Protein 27 and αB-Crystallin can Form a Complex, which Dissociates by Heat Shock. J. Biol. Chem. 1992, 267, 12936–12941. [Google Scholar] [PubMed]
- Zavialov, A.; Benndorf, R.; Ehrnsperger, M.; Zav’yalov, V.; Dudich, I.; Buchner, J.; Gaestel, M. The Effect of the Intersubunit Disulfide Bond on the Structural and Functional Properties of the Small Heat Shock Protein Hsp25. Int. J. Biol. Macromol. 1998, 22, 163–173. [Google Scholar] [CrossRef]
- Diaz-Latoud, C.; Buache, E.; Javouhey, E.; Arrigo, A.P. Substitution of the Unique Cysteine Residue of Murine Hsp25 Interferes with the Protective Activity of this Stress Protein through Inhibition of Dimer Formation. Antioxid. Redox Signal. 2005, 7, 436–445. [Google Scholar] [CrossRef]
- Chalova, A.S.; Sudnitsyna, M.V.; Semenyuk, P.I.; Orlov, V.N.; Gusev, N.B. Effect of Disulfide Crosslinking on Thermal Transitions and Chaperone-Like Activity of Human Small Heat Shock Protein HspB1. Cell Stress Chaperones 2014, 19, 963–972. [Google Scholar] [CrossRef]
- Rajagopal, P.; Liu, Y.; Shi, L.; Clouser, A.F.; Klevit, R.E. Structure of the α-Crystallin Domain from the Redox-Sensitive Chaperone, HSPB1. J. Biomol. NMR 2015, 63, 223–228. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Virot, S.; Chaufour, S.; Firdaus, W.; Kretz-Remy, C.; Diaz-Latoud, C. Hsp27 Consolidates Intracellular Redox Homeostasis by Upholding Glutathione in its Reduced Form and by Decreasing Iron Intracellular Levels. Antioxid. Redox Signal. 2005, 7, 414–422. [Google Scholar] [CrossRef]
- Paul, C.; Simon, S.; Gibert, B.; Virot, S.; Manero, F.; Arrigo, A.P. Dynamic Processes that Reflect Anti-Apoptotic Strategies Set Up by HspB1 (Hsp27). Exp. Cell Res. 2010, 316, 1535–1552. [Google Scholar] [CrossRef]
- Mehlen, P.; Kretz-Remy, C.; Préville, X.; Arrigo, A.P. Human hsp27, Drosophila hsp27 and Human αB-Crystallin Expression-Mediated Increase in Glutathione is Essential for the Protective Activity of these Proteins Against TNFα-Induced Cell Death. EMBO J. 1996, 15, 2695–2706. [Google Scholar] [CrossRef]
- Préville, X.; Salvemini, F.; Giraud, S.; Chaufour, S.; Paul, C.; Stepien, G.; Ursini, M.V.; Arrigo, A.P. Mammalian Small Stress Proteins Protect Against Oxidative Stress through their Ability to Increase Glucose-6-Phosphate Dehydrogenase Activity and by Maintaining Optimal Cellular Detoxifying Machinery. Exp. Cell Res. 1999, 247, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Min, J.N.; Park, E.M.; Han, M.Y.; Lee, Y.S.; Lee, Y.J.; Park, Y.M. Role of Small Heat Shock Protein HSP25 in Radioresistance and Glutathione-Redox Cycle. J. Cell. Physiol. 2000, 183, 100–107. [Google Scholar] [CrossRef]
- Escobedo, J.; Pucci, A.M.; Koh, T.J. HSP25 Protects Skeletal Muscle Cells Against Oxidative Stress. Free Radic. Biol. Med. 2004, 37, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zheng, C.; Zhang, Y.; Chang, Y.Z.; Qian, Z.M.; Shen, X. Heat Shock Protein 27 Downregulates the Transferrin Receptor 1-Mediated Iron Uptake. Int. J. Biochem. Cell Biol. 2006, 38, 1402–1416. [Google Scholar] [CrossRef]
- Mehlen, P.; Schulze-Osthoff, K.; Arrigo, A.P. Small Stress Proteins as Novel Regulators of Apoptosis. Heat Shock Protein 27 Blocks Fas/APO-1- and Staurosporine-Induced Cell Death. J. Biol. Chem. 1996, 271, 16510–16514. [Google Scholar] [CrossRef]
- Garrido, C.; Bruey, J.M.; Fromentin, A.; Hammann, A.; Arrigo, A.P.; Solary, E. HSP27 Inhibits Cytochrome c-Dependent Activation of Procaspase-9. FASEB J. 1999, 13, 2061–2070. [Google Scholar] [CrossRef]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. Hsp27 Negatively Regulates Cell Death by Interacting with Cytochrome c. Nat. Cell Biol. 2000, 2, 645–652. [Google Scholar] [CrossRef]
- Charette, S.J.; Lavoie, J.N.; Lambert, H.; Landry, J. Inhibition of Daxx-Mediated Apoptosis by Heat Shock Protein 27. Mol. Cell. Biol. 2000, 20, 7602–7612. [Google Scholar] [CrossRef]
- Concannon, C.G.; Orrenius, S.; Samali, A. Hsp27 Inhibits Cytochrome c-Mediated Caspase Activation by Sequestering both Pro-Caspase-3 and Cytochrome c. Gene Expr. 2001, 9, 195–201. [Google Scholar] [CrossRef]
- Parcellier, A.; Schmitt, E.; Gurbuxani, S.; Seigneurin-Berny, D.; Pance, A.; Chantôme, A.; Plenchette, S.; Khochbin, S.; Solary, E.; Garrido, C. HSP27 is a Ubiquitin-Binding Protein Involved in I-κBα Proteasomal Degradation. Mol. Cell. Biol. 2003, 23, 5790–5802. [Google Scholar] [CrossRef]
- Katsogiannou, M.; Andrieu, C.; Baylot, V.; Baudot, A.; Dusetti, N.J.; Gayet, O.; Finetti, P.; Garrido, C.; Birnbaum, D.; Bertucci, F.; et al. The Functional Landscape of Hsp27 Reveals New Cellular Processes such as DNA Repair and Alternative Splicing and Proposes Novel Anticancer Targets. Mol. Cell. Proteomics 2014, 13, 3585–3601. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.; Mnich, K.; Oommen, D.; Chakravarthy, R.; Almeida-Souza, L.; Krols, M.; Saveljeva, S.; Doyle, K.; Gupta, S.; Timmerman, V.; et al. HSPB1 Facilitates ERK-Mediated Phosphorylation and Degradation of BIM to Attenuate Endoplasmic Reticulum Stress-Induced Apoptosis. Cell. Death Dis. 2017, 8, e3026. [Google Scholar] [CrossRef] [PubMed]
- Bryantsev, A.L.; Loktionova, S.A.; Ilyinskaya, O.P.; Tararak, E.M.; Kampinga, H.H.; Kabakov, A.E. Distribution, Phosphorylation, and Activities of Hsp25 in Heat-Stressed H9c2 Myoblasts: A Functional Link to Cytoprotection. Cell Stress Chaperones 2002, 7, 146–155. [Google Scholar] [CrossRef]
- Benndorf, R.; Hayess, K.; Ryazantsev, S.; Wieske, M.; Behlke, J.; Lutsch, G. Phosphorylation and Supramolecular Organization of Murine Small Heat Shock Protein HSP25 Abolish its Actin Polymerization-Inhibiting Activity. J. Biol. Chem. 1994, 269, 20780–20784. [Google Scholar] [PubMed]
- Landry, J.; Huot, J. Modulation of Actin Dynamics during Stress and Physiological Stimulation by a Signaling Pathway Involving p38 MAP Kinase and Heat-Shock Protein 27. Biochem. Cell Biol. 1995, 73, 703–707. [Google Scholar] [CrossRef]
- Lavoie, J.N.; Lambert, H.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of Cellular Thermoresistance and Actin Filament Stability Accompanies Phosphorylation-Induced Changes in the Oligomeric Structure of Heat Shock Protein 27. Mol. Cell. Biol. 1995, 15, 505–516. [Google Scholar] [CrossRef]
- Pivovarova, A.V.; Chebotareva, N.A.; Chernik, I.S.; Gusev, N.B.; Levitsky, D.I. Small Heat Shock Protein Hsp27 Prevents Heat-Induced Aggregation of F-Actin by Forming Soluble Complexes with Denatured Actin. FEBS J. 2007, 274, 5937–5948. [Google Scholar] [CrossRef]
- Clarke, J.P.; Mearow, K.M. Cell Stress Promotes the Association of Phosphorylated HspB1 with F-Actin. PLoS ONE 2013, 8, e68978. [Google Scholar] [CrossRef]
- Hoffman, L.; Jensen, C.C.; Yoshigi, M.; Beckerle, M. Mechanical Signals Activate p38 MAPK Pathway-Dependent Reinforcement of Actin Via Mechanosensitive HspB1. Mol. Biol. Cell 2017, 28, 2661–2675. [Google Scholar] [CrossRef]
- Kayser, J.; Haslbeck, M.; Dempfle, L.; Krause, M.; Grashoff, C.; Buchner, J.; Herrmann, H.; Bausch, A.R. The Small Heat Shock Protein Hsp27 Affects Assembly Dynamics and Structure of Keratin Intermediate Filament Networks. Biophys. J. 2013, 105, 1778–1785. [Google Scholar] [CrossRef]
- Nefedova, V.V.; Sudnitsyna, M.V.; Gusev, N.B. Interaction of Small Heat Shock Proteins with Light Component of Neurofilaments (NFL). Cell Stress Chaperones 2017, 22, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Souza, L.; Asselbergh, B.; De Winter, V.; Goethals, S.; Timmerman, V.; Janssens, S. HSPB1 Facilitates the Formation of Non-Centrosomal Microtubules. PLoS ONE 2013, 8, e66541. [Google Scholar] [CrossRef] [PubMed]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant Small Heat-Shock Protein 27 Causes Axonal Charcot-Marie-Tooth Disease and Distal Hereditary Motor Neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Laura, M.; Wavrant-De Vrieze, F.; Blake, J.; Wood, N.; Reilly, M.M. Mutations in the HSP27 (HSPB1) Gene Cause Dominant, Recessive, and Sporadic Distal HMN/CMT Type 2. Neurology 2008, 71, 1660–1668. [Google Scholar] [CrossRef]
- Luigetti, M.; Fabrizi, G.M.; Madia, F.; Ferrarini, M.; Conte, A.; Del Grande, A.; Tasca, G.; Tonali, P.A.; Sabatelli, M. A Novel HSPB1 Mutation in an Italian Patient with CMT2/dHMN Phenotype. J. Neurol. Sci. 2010, 298, 114–117. [Google Scholar] [CrossRef]
- Capponi, S.; Geroldi, A.; Fossa, P.; Grandis, M.; Ciotti, P.; Gulli, R.; Schenone, A.; Mandich, P.; Bellone, E. HSPB1 and HSPB8 in Inherited Neuropathies: Study of an Italian Cohort of dHMN and CMT2 Patients. J. Peripher. Nerv. Syst. 2011, 16, 287–294. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Geuens, T.; Petiot, P.; Péréon, Y.; Adriaenssens, E.; Haidar, M.; Capponi, S.; Maisonobe, T.; Fournier, E.; Dubourg, O.; et al. Axonal Neuropathies due to Mutations in Small Heat Shock Proteins: Clinical, Genetic, and Functional Insights into Novel Mutations. Hum. Mutat. 2017, 38, 556–568. [Google Scholar] [CrossRef]
- Rossor, A.M.; Morrow, J.M.; Polke, J.M.; Murphy, S.M.; Houlden, H.; INC-RDCRC; Laura, M.; Manji, H.; Blake, J.; Reilly, M.M. Pilot Phenotype and Natural History Study of Hereditary Neuropathies Caused by Mutations in the HSPB1 Gene. Neuromuscul. Disord. 2017, 27, 50–56. [Google Scholar] [CrossRef]
- Bugiardini, E.; Rossor, A.M.; Lynch, D.S.; Swash, M.; Pittman, A.M.; Blake, J.C.; Hanna, M.G.; Houlden, H.; Holton, J.L.; Reilly, M.M.; et al. Homozygous Mutation in HSPB1 Causing Distal Vacuolar Myopathy and Motor Neuropathy. Neurol. Genet. 2017, 3, e168. [Google Scholar] [CrossRef]
- DiVincenzo, C.; Elzinga, C.D.; Medeiros, A.C.; Karbassi, I.; Jones, J.R.; Evans, M.C.; Braastad, C.D.; Bishop, C.M.; Jaremko, M.; Wang, Z.; et al. The Allelic Spectrum of Charcot-Marie-Tooth Disease in Over 17,000 Individuals with Neuropathy. Mol. Genet. Genom. Med. 2014, 2, 522–529. [Google Scholar] [CrossRef]
- Adriaenssens, E.; Geuens, T.; Baets, J.; Echaniz-Laguna, A.; Timmerman, V. Novel Insights in the Disease Biology of Mutant Small Heat Shock Proteins in Neuromuscular Diseases. Brain 2017, 140, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Scarlato, M.; Viganò, F.; Carrera, P.; Previtali, S.C.; Bolino, A. A Novel Heat Shock Protein 27 Homozygous Mutation: Widening of the Continuum between MND/dHMN/CMT2. J. Peripher. Nerv. Syst. 2015, 20, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Capponi, S.; Geuens, T.; Geroldi, A.; Origone, P.; Verdiani, S.; Cichero, E.; Adriaenssens, E.; De Winter, V.; Bandettini di Poggio, M.; Barberis, M.; et al. Molecular Chaperones in the Pathogenesis of Amyotrophic Lateral Sclerosis: The Role of HSPB1. Hum. Mutat. 2016, 37, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Dierick, I.; Irobi, J.; Janssens, S.; Theuns, J.; Lemmens, R.; Jacobs, A.; Corsmit, E.; Hersmus, N.; Van Den Bosch, L.; Robberecht, W.; et al. Genetic Variant in the HSPB1 Promoter Region Impairs the HSP27 Stress Response. Hum. Mutat. 2007, 28, 830. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Smith, D.J.; Duff, J.; Pyle, A.; Griffin, H.; Polvikoski, T.; Birchall, D.; Horvath, R.; Chinnery, P.F. Novel HSPB1 Mutation Causes both Motor Neuronopathy and Distal Myopathy. Neurol. Genet. 2016, 2, e110. [Google Scholar] [CrossRef]
- Nefedova, V.V.; Datskevich, P.N.; Sudnitsyna, M.V.; Strelkov, S.V.; Gusev, N.B. Physico-Chemical Properties of R140G and K141Q Mutants of Human Small Heat Shock Protein HspB1 Associated with Hereditary Peripheral Neuropathies. Biochimie 2013, 95, 1582–1592. [Google Scholar] [CrossRef]
- Weeks, S.D.; Muranova, L.K.; Heirbaut, M.; Beelen, S.; Strelkov, S.V.; Gusev, N.B. Characterization of Human Small Heat Shock Protein HSPB1 α-Crystallin Domain Localized Mutants Associated with Hereditary Motor Neuron Diseases. Sci. Rep. 2018, 8, 688. [Google Scholar] [CrossRef]
- Nefedova, V.V.; Sudnitsyna, M.V.; Strelkov, S.V.; Gusev, N.B. Structure and Properties of G84R and L99M Mutants of Human Small Heat Shock Protein HspB1 Correlating with Motor Neuropathy. Arch. Biochem. Biophys. 2013, 538, 16–24. [Google Scholar] [CrossRef]
- Chalova, A.S.; Sudnitsyna, M.V.; Strelkov, S.V.; Gusev, N.B. Characterization of Human Small Heat Shock Protein HspB1 that Carries C-Terminal Domain Mutations Associated with Hereditary Motor Neuron Diseases. Biochim. Biophys. Acta 2014, 1844, 2116–2126. [Google Scholar] [CrossRef]
- Muranova, L.K.; Weeks, S.D.; Strelkov, S.V.; Gusev, N.B. Characterization of Mutants of Human Small Heat Shock Protein HspB1 Carrying Replacements in the N-Terminal Domain and 180Associated with Hereditary Motor Neuron Diseases. PLoS ONE 2015, 10, e0126248. [Google Scholar] [CrossRef]
- Ackerley, S.; James, P.A.; Kalli, A.; French, S.; Davies, K.E.; Talbot, K. A Mutation in the Small Heat-Shock Protein HSPB1 Leading to Distal Hereditary Motor Neuronopathy Disrupts Neurofilament Assembly and the Axonal Transport of Specific Cellular Cargoes. Hum. Mol. Genet. 2006, 15, 347–354. [Google Scholar] [CrossRef] [PubMed]
- D’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 Inhibitors Reverse Axonal Loss in a Mouse Model of Mutant HSPB1-Induced Charcot-Marie-Tooth Disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, S.C.; Joo, J.; Choi, Y.R.; Moon, H.W.; Kwak, G.; Yeo, H.K.; Lee, J.S.; Ahn, H.J.; Jung, N.; et al. Overexpression of Mutant HSP27 Causes Axonal Neuropathy in Mice. J. Biomed. Sci. 2015, 22, 43. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Renusch, S.R.; Naiman, N.E.; Gu, S.; Sneh, A.; Arnold, W.D.; Sahenk, Z.; Kolb, S.J. Mutant HSPB1 Overexpression in Neurons is Sufficient to Cause Age-Related Motor Neuronopathy in Mice. Neurobiol. Dis. 2012, 47, 163–173. [Google Scholar] [CrossRef]
- Bouhy, D.; Geuens, T.; De Winter, V.; Almeida-Souza, L.; Katona, I.; Weis, J.; Hochepied, T.; Goossens, S.; Haigh, J.J.; Janssens, S.; et al. Characterization of New Transgenic Mouse Models for Two Charcot-Marie-Tooth-Causing HspB1 Mutations using the Rosa26 Locus. J. Neuromuscul Dis. 2016, 3, 183–200. [Google Scholar] [CrossRef]
- Zhai, J.; Lin, H.; Julien, J.P.; Schlaepfer, W.W. Disruption of Neurofilament Network with Aggregation of Light Neurofilament Protein: A Common Pathway Leading to Motor Neuron Degeneration due to Charcot-Marie-Tooth Disease-Linked Mutations in NFL and HSPB1. Hum. Mol. Genet. 2007, 16, 3103–3116. [Google Scholar] [CrossRef]
- Holmgren, A.; Bouhy, D.; De Winter, V.; Asselbergh, B.; Timmermans, J.P.; Irobi, J.; Timmerman, V. Charcot-Marie-Tooth Causing HSPB1 Mutations Increase Cdk5-Mediated Phosphorylation of Neurofilaments. Acta Neuropathol. 2013, 126, 93–108. [Google Scholar] [CrossRef]
- Almeida-Souza, L.; Asselbergh, B.; d’Ydewalle, C.; Moonens, K.; Goethals, S.; de Winter, V.; Azmi, A.; Irobi, J.; Timmermans, J.P.; Gevaert, K.; et al. Small Heat-Shock Protein HSPB1 Mutants Stabilize Microtubules in Charcot-Marie-Tooth Neuropathy. J. Neurosci. 2011, 31, 15320–15328. [Google Scholar] [CrossRef]
- Kim, J.Y.; Woo, S.Y.; Hong, Y.B.; Choi, H.; Kim, J.; Choi, H.; Mook-Jung, I.; Ha, N.; Kyung, J.; Koo, S.K.; et al. HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with HSPB1 Mutation. Stem Cells Int. 2016, 2016, 9475981. [Google Scholar] [CrossRef]
- Kalmar, B.; Innes, A.; Wanisch, K.; Kolaszynska, A.K.; Pandraud, A.; Kelly, G.; Abramov, A.Y.; Reilly, M.M.; Schiavo, G.; Greensmith, L. Mitochondrial Deficits and Abnormal Mitochondrial Retrograde Axonal Transport Play a Role in the Pathogenesis of Mutant Hsp27-Induced Charcot Marie Tooth Disease. Hum. Mol. Genet. 2017, 26, 3313–3326. [Google Scholar] [CrossRef]
- Shen, S.; Benoy, V.; Bergman, J.A.; Kalin, J.H.; Frojuello, M.; Vistoli, G.; Haeck, W.; Van Den Bosch, L.; Kozikowski, A.P. Bicyclic-Capped Histone Deacetylase 6 Inhibitors with Improved Activity in a Model of Axonal Charcot-Marie-Tooth Disease. ACS Chem. Neurosci. 2016, 7, 240–258. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.U.; Linzer, R.W.; Truman, J.P.; Gurevich, M.; Hannun, Y.A.; Senkal, C.E.; Obeid, L.M. Decreased Ceramide Underlies Mitochondrial Dysfunction in Charcot-Marie-Tooth 2F. FASEB J. 2018, 32, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Min, J.N.; Masters, S.; Mivechi, N.F.; Moskophidis, D. Insights into Function and Regulation of Small Heat Shock Protein 25 (HSPB1) in a Mouse Model with Targeted Gene Disruption. Genesis 2007, 45, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Heilman, P.L.; Song, S.; Miranda, C.J.; Meyer, K.; Srivastava, A.K.; Knapp, A.; Wier, C.G.; Kaspar, B.K.; Kolb, S.J. HSPB1 Mutations Causing Hereditary Neuropathy in Humans Disrupt Non-Cell Autonomous Protection of Motor Neurons. Exp. Neurol. 2017, 297, 101–109. [Google Scholar] [CrossRef]
- Ylikallio, E.; Konovalova, S.; Dhungana, Y.; Hilander, T.; Junna, N.; Partanen, J.V.; Toppila, J.P.; Auranen, M.; Tyynismaa, H. Truncated HSPB1 Causes Axonal Neuropathy and Impairs Tolerance to Unfolded Protein Stress. BBA Clin. 2015, 3, 233–242. [Google Scholar] [CrossRef]
- Geuens, T.; De Winter, V.; Rajan, N.; Achsel, T.; Mateiu, L.; Almeida-Souza, L.; Asselbergh, B.; Bouhy, D.; Auer-Grumbach, M.; Bagni, C.; et al. Mutant HSPB1 Causes Loss of Translational Repression by Binding to PCBP1, an RNA Binding Protein with a Possible Role in Neurodegenerative Disease. Acta Neuropathol. Commun. 2017, 5, 5. [Google Scholar] [CrossRef]
- Haidar, M.; Asselbergh, B.; Adriaenssens, E.; De Winter, V.; Timmermans, J.P.; Auer-Grumbach, M.; Juneja, M.; Timmerman, V. Neuropathy-Causing Mutations in HSPB1 Impair Autophagy by Disturbing the Formation of SQSTM1/p62 Bodies. Autophagy 2019, 15, 1051–1068. [Google Scholar] [CrossRef]
- Ikeda, Y.; Abe, A.; Ishida, C.; Takahashi, K.; Hayasaka, K.; Yamada, M. A Clinical Phenotype of Distal Hereditary Motor Neuronopathy Type II with a Novel HSPB1 Mutation. J. Neurol. Sci. 2009, 277, 9–12. [Google Scholar] [CrossRef]
- Lin, K.P.; Soong, B.W.; Yang, C.C.; Huang, L.W.; Chang, M.H.; Lee, I.H.; Antonellis, A.; Lee, Y.C. The Mutational Spectrum in a Cohort of Charcot-Marie-Tooth Disease Type 2 among the Han Chinese in Taiwan. PLoS ONE 2011, 6, e29393. [Google Scholar] [CrossRef]
- Kijima, K.; Numakura, C.; Goto, T.; Takahashi, T.; Otagiri, T.; Umetsu, K.; Hayasaka, K. Small Heat Shock Protein 27 Mutation in a Japanese Patient with Distal Hereditary Motor Neuropathy. J. Hum. Genet. 2005, 50, 473–476. [Google Scholar] [CrossRef]
- Boelens, W.C.; Van Boekel, M.A.; De Jong, W.W. HspB3, the most Deviating of the Six Known Human Small Heat Shock Proteins. Biochim. Biophys. Acta 1998, 1388, 513–516. [Google Scholar] [CrossRef]
- Molyneaux, B.J.; Arlotta, P.; Fame, R.M.; MacDonald, J.L.; MacQuarrie, K.L.; Macklis, J.D. Novel Subtype-Specific Genes Identify Distinct Subpopulations of Callosal Projection Neurons. J. Neurosci. 2009, 29, 12343–12354. [Google Scholar] [CrossRef] [PubMed]
- Kirbach, B.B.; Golenhofen, N. Differential Expression and Induction of Small Heat Shock Proteins in Rat Brain and Cultured Hippocampal Neurons. J. Neurosci. Res. 2011, 89, 162–175. [Google Scholar] [CrossRef]
- Kondaurova, E.M.; Naumenko, V.S.; Sinyakova, N.A.; Kulikov, A.V. Map3k1, Il6st, Gzmk, and Hspb3 Gene Coexpression Network in the Mechanism of Freezing Reaction in Mice. J. Neurosci. Res. 2011, 89, 267–273. [Google Scholar] [CrossRef] [PubMed]
- La Padula, V.; Staszewski, O.; Nestel, S.; Busch, H.; Boerries, M.; Roussa, E.; Prinz, M.; Krieglstein, K. HSPB3 Protein is Expressed in Motoneurons and Induces their Survival After Lesion-Induced Degeneration. Exp. Neurol. 2016, 286, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Sugiyama, Y.; Hayashi, Y.; Nyu-i, N.; Yoshida, M.; Nonaka, I.; Ishiura, S.; Arahata, K.; Ohno, S. MKBP, a Novel Member of the Small Heat Shock Protein Family, Binds and Activates the Myotonic Dystrophy Protein Kinase. J. Cell Biol. 1998, 140, 1113–1124. [Google Scholar] [CrossRef]
- den Engelsman, J.; Boros, S.; Dankers, P.Y.; Kamps, B.; Vree Egberts, W.T.; Böde, C.S.; Lane, L.A.; Aquilina, J.A.; Benesch, J.L.; Robinson, C.V.; et al. The Small Heat-Shock Proteins HSPB2 and HSPB3 Form Well-Defined Heterooligomers in a Unique 3 to 1 Subunit Ratio. J. Mol. Biol. 2009, 393, 1022–1032. [Google Scholar] [CrossRef]
- Clark, A.R.; Vree Egberts, W.; Kondrat, F.D.L.; Hilton, G.R.; Ray, N.J.; Cole, A.R.; Carver, J.A.; Benesch, J.L.P.; Keep, N.H.; Boelens, W.C.; et al. Terminal Regions Confer Plasticity to the Tetrameric Assembly of Human HspB2 and HspB3. J. Mol. Biol. 2018, 430, 3297–3310. [Google Scholar] [CrossRef]
- Shama, K.M.; Suzuki, A.; Harada, K.; Fujitani, N.; Kimura, H.; Ohno, S.; Yoshida, K. Transient Up-Regulation of Myotonic Dystrophy Protein Kinase-Binding Protein, MKBP, and HSP27 in the Neonatal Myocardium. Cell Struct. Funct. 1999, 24, 1–4. [Google Scholar] [CrossRef]
- Rusmini, P.; Polanco, M.J.; Cristofani, R.; Cicardi, M.E.; Meroni, M.; Galbiati, M.; Piccolella, M.; Messi, E.; Giorgetti, E.; Lieberman, A.P.; et al. Aberrant Autophagic Response in the Muscle of A Knock-in Mouse Model of Spinal and Bulbar Muscular Atrophy. Sci. Rep. 2015, 5, 15174. [Google Scholar] [CrossRef]
- Bruinsma, I.B.; Bruggink, K.A.; Kinast, K.; Versleijen, A.A.; Segers-Nolten, I.M.; Subramaniam, V.; Kuiperij, H.B.; Boelens, W.; de Waal, R.M.; Verbeek, M.M. Inhibition of Alpha-Synuclein Aggregation by Small Heat Shock Proteins. Proteins 2011, 79, 2956–2967. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.; Raman, B.; Ramakrishna, T.; Rao, C. HspB2/myotonic Dystrophy Protein Kinase Binding Protein (MKBP) as a Novel Molecular Chaperone: Structural and Functional Aspects. PLoS ONE 2012, 7, e29810. [Google Scholar] [CrossRef] [PubMed]
- Asthana, A.; Raman, B.; Ramakrishna, T.; Rao, C. Structural Aspects and Chaperone Activity of Human HspB3: Role of the "C-Terminal Extension". Cell Biochem. Biophys. 2012, 64, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Langston, K.; Wang, X.; Squires, S.; Mustafi, S.B.; Hayes, W.; Neubert, J.; Fischer, S.K.; Fasano, M.; Saunders, G.M.; et al. Characterization of the Cardiac Overexpression of HSPB2 Reveals Mitochondrial and Myogenic Roles Supported by a Cardiac HspB2 Interactome. PLoS ONE 2015, 10, e0133994. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Tsujimoto, N.; Nakagawa, H.; Iwaki, T.; Fukumaki, Y.; Iwaki, A. Association of HSPB2, a Member of the Small Heat Shock Protein Family, with Mitochondria. Exp. Cell Res. 2001, 271, 161–168. [Google Scholar] [CrossRef]
- Ishiwata, T.; Orosz, A.; Wang, X.; Mustafi, S.B.; Pratt, G.W.; Christians, E.S.; Boudina, S.; Abel, E.D.; Benjamin, I.J. HSPB2 is Dispensable for the Cardiac Hypertrophic Response but Reduces Mitochondrial Energetics Following Pressure Overload in Mice. PLoS ONE 2012, 7, e42118. [Google Scholar] [CrossRef]
- Yoshida, K.; Aki, T.; Harada, K.; Shama, K.M.; Kamoda, Y.; Suzuki, A.; Ohno, S. Translocation of HSP27 and MKBP in Ischemic Heart. Cell Struct. Funct. 1999, 24, 181–185. [Google Scholar] [CrossRef]
- Verschuure, P.; Croes, Y.; van den IJssel, P.R.; Quinlan, R.A.; de Jong, W.W.; Boelens, W.C. Translocation of Small Heat Shock Proteins to the Actin Cytoskeleton upon Proteasomal Inhibition. J. Mol. Cell. Cardiol. 2002, 34, 117–128. [Google Scholar] [CrossRef]
- Morelli, F.F.; Verbeek, D.S.; Bertacchini, J.; Vinet, J.; Mediani, L.; Marmiroli, S.; Cenacchi, G.; Nasi, M.; De Biasi, S.; Brunsting, J.F.; et al. Aberrant Compartment Formation by HSPB2 Mislocalizes Lamin A and Compromises Nuclear Integrity and Function. Cell. Rep. 2017, 20, 2100–2115. [Google Scholar] [CrossRef]
- Markiewicz, E.; Ledran, M.; Hutchison, C.J. Remodelling of the Nuclear Lamina and Nucleoskeleton is Required for Skeletal Muscle Differentiation in Vitro. J. Cell. Sci. 2005, 118, 409–420. [Google Scholar] [CrossRef]
- Fontaine, J.M.; Sun, X.; Benndorf, R.; Welsh, M.J. Interactions of HSP22 (HSPB8) with HSP20, αB-Crystallin, and HSPB3. Biochem. Biophys. Res. Commun. 2005, 337, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Snyder, P.J.; Poi, E.J.; Renard, E.A.; Bartlett, A.; Gu, S.; Sutton, S.; Arnold, W.D.; Freimer, M.L.; Lawson, V.H.; et al. Mutant Small Heat Shock Protein B3 Causes Motor Neuropathy: Utility of a Candidate Gene Approach. Neurology 2010, 74, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.E.; Nam, S.H.; Lee, A.J.; Hong, Y.B.; Choi, B.O.; Chung, K.W. Small Heat Shock Protein B3 (HSPB3) Mutation in an Axonal Charcot-Marie-Tooth Disease Family. J. Peripher. Nerv. Syst. 2018, 23, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Benndorf, R.; Sun, X.; Gilmont, R.R.; Biederman, K.J.; Molloy, M.P.; Goodmurphy, C.W.; Cheng, H.; Andrews, P.C.; Welsh, M.J. HSP22, a New Member of the Small Heat Shock Protein Superfamily, Interacts with Mimic of Phosphorylated HSP27 ((3D)HSP27). J. Biol. Chem. 2001, 276, 26753–26761. [Google Scholar] [CrossRef]
- Kappé, G.; Verschuure, P.; Philipsen, R.L.; Staalduinen, A.A.; Van de Boogaart, P.; Boelens, W.C.; De Jong, W.W. Characterization of Two Novel Human Small Heat Shock Proteins: Protein Kinase-Related HspB8 and Testis-Specific HspB9. Biochim. Biophys. Acta 2001, 1520, 1–6. [Google Scholar] [CrossRef]
- Sanbe, A.; Marunouchi, T.; Abe, T.; Tezuka, Y.; Okada, M.; Aoki, S.; Tsumura, H.; Yamauchi, J.; Tanonaka, K.; Nishigori, H.; et al. Phenotype of Cardiomyopathy in Cardiac-Specific Heat Shock Protein B8 K141N Transgenic Mouse. J. Biol. Chem. 2013, 288, 8910–8921. [Google Scholar] [CrossRef]
- Crippa, V.; Sau, D.; Rusmini, P.; Boncoraglio, A.; Onesto, E.; Bolzoni, E.; Galbiati, M.; Fontana, E.; Marino, M.; Carra, S.; et al. The Small Heat Shock Protein B8 (HspB8) Promotes Autophagic Removal of Misfolded Proteins Involved in Amyotrophic Lateral Sclerosis (ALS). Hum. Mol. Genet. 2010, 19, 3440–3456. [Google Scholar] [CrossRef]
- Seidel, K.; Vinet, J.; Dunnen, W.F.; Brunt, E.R.; Meister, M.; Boncoraglio, A.; Zijlstra, M.P.; Boddeke, H.W.; Rub, U.; Kampinga, H.H.; et al. The HSPB8-BAG3 Chaperone Complex is Upregulated in Astrocytes in the Human Brain Affected by Protein Aggregation Diseases. Neuropathol. Appl. Neurobiol. 2012, 38, 39–53. [Google Scholar] [CrossRef]
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. BAG3 Induces the Sequestration of Proteasomal Clients into Cytoplasmic Puncta: Implications for a Proteasome-to-Autophagy Switch. Autophagy 2014, 10, 1603–1621. [Google Scholar] [CrossRef]
- Crippa, V.; D’Agostino, V.G.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Loffredo, R.; Pancher, M.; Piccolella, M.; Galbiati, M.; et al. Transcriptional Induction of the Heat Shock Protein B8 Mediates the Clearance of Misfolded Proteins Responsible for Motor Neuron Diseases. Sci. Rep. 2016, 6, 22827. [Google Scholar] [CrossRef]
- Nivon, M.; Fort, L.; Muller, P.; Richet, E.; Simon, S.; Guey, B.; Fournier, M.; Arrigo, A.P.; Hetz, C.; Atkin, J.D.; et al. NFκB is a Central Regulator of Protein Quality Control in Response to Protein Aggregation Stresses Via Autophagy Modulation. Mol. Biol. Cell 2016, 27, 1712–1727. [Google Scholar] [CrossRef]
- Cristofani, R.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Ferrari, V.; Tedesco, B.; Casarotto, E.; Chierichetti, M.; Messi, E.; Piccolella, M.; et al. The Regulation of the Small Heat Shock Protein B8 in Misfolding Protein Diseases Causing Motoneuronal and Muscle Cell Death. Front. Neurosci. 2019, 13, 796. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Fontaine, J.M.; Rest, J.S.; Shelden, E.A.; Welsh, M.J.; Benndorf, R. Interaction of Human HSP22 (HSPB8) with Other Small Heat Shock Proteins. J. Biol. Chem. 2004, 279, 2394–2402. [Google Scholar] [CrossRef] [PubMed]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Heterooligomeric Complexes of Human Small Heat Shock Proteins. Cell Stress Chaperones 2012, 17, 157–169. [Google Scholar] [CrossRef]
- Datskevich, P.N.; Mymrikov, E.V.; Gusev, N.B. Utilization of Fluorescent Chimeras for Investigation of Heterooligomeric Complexes Formed by Human Small Heat Shock Proteins. Biochimie 2012, 94, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of-Function Mutations in Co-Chaperone BAG3 Destabilize Small HSPs and Cause Cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef]
- Guilbert, S.M.; Lambert, H.; Rodrigue, M.A.; Fuchs, M.; Landry, J.; Lavoie, J.N. HSPB8 and BAG3 Cooperate to Promote Spatial Sequestration of Ubiquitinated Proteins and Coordinate the Cellular Adaptive Response to Proteasome Insufficiency. FASEB J. 2018, 32, 3518–3535. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to Autophagy and Proteasome Ameliorates its Aggregation in Amyotrophic Lateral Sclerosis Target Cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef]
- Chowdary, T.K.; Raman, B.; Ramakrishna, T.; Rao, C.M. Mammalian Hsp22 is a Heat-Inducible Small Heat-Shock Protein with Chaperone-Like Activity. Biochem. J. 2004, 381, 379–387. [Google Scholar] [CrossRef]
- Carra, S.; Sivilotti, M.; Chávez Zobel, A.T.; Lambert, H.; Landry, J. HspB8, a Small Heat Shock Protein Mutated in Human Neuromuscular Disorders, has in Vivo Chaperone Activity in Cultured Cells. Hum. Mol. Genet. 2005, 14, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Shemetov, A.A.; Seit-Nebi, A.S.; Gusev, N.B. Phosphorylation of Human Small Heat Shock Protein HspB8 (Hsp22) by ERK1 Protein Kinase. Mol. Cell. Biochem. 2011, 355, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, A.S.; Markov, D.I.; Gusev, N.B.; Levitsky, D.I. Thermally Induced Structural Changes of Intrinsically Disordered Small Heat Shock Protein Hsp22. Biophys. Chem. 2009, 145, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Crippa, V.; Giorgetti, E.; Boncoraglio, A.; Cristofani, R.; Carra, S.; Poletti, A. Clearance of the Mutant Androgen Receptor in Motoneuronal Models of Spinal and Bulbar Muscular Atrophy. Neurobiol. Aging 2013, 34, 2585–2603. [Google Scholar] [CrossRef]
- Hamouda, M.A.; Belhacene, N.; Puissant, A.; Colosetti, P.; Robert, G.; Jacquel, A.; Mari, B.; Auberger, P.; Luciano, F. The Small Heat Shock Protein B8 (HSPB8) Confers Resistance to Bortezomib by Promoting Autophagic Removal of Misfolded Proteins in Multiple Myeloma Cells. Oncotarget 2014, 5, 6252–6266. [Google Scholar] [CrossRef]
- Crippa, V.; Cicardi, M.E.; Ramesh, N.; Seguin, S.J.; Ganassi, M.; Bigi, I.; Diacci, C.; Zelotti, E.; Baratashvili, M.; Gregory, J.M.; et al. The Chaperone HSPB8 Reduces the Accumulation of Truncated TDP-43 Species in Cells and Protects Against TDP-43-Mediated Toxicity. Hum. Mol. Genet. 2016, 25, 3908–3924. [Google Scholar] [CrossRef]
- Cristofani, R.; Crippa, V.; Rusmini, P.; Cicardi, M.E.; Meroni, M.; Licata, N.V.; Sala, G.; Giorgetti, E.; Grunseich, C.; Galbiati, M.; et al. Inhibition of Retrograde Transport Modulates Misfolded Protein Accumulation and Clearance in Motoneuron Diseases. Autophagy 2017, 13, 1280–1303. [Google Scholar] [CrossRef]
- Cristofani, R.; Crippa, V.; Vezzoli, G.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Tedesco, B.; Piccolella, M.; et al. The Small Heat Shock Protein B8 (HSPB8) Efficiently Removes Aggregating Species of Dipeptides Produced in C9ORF72-Related Neurodegenerative Diseases. Cell Stress Chaperones 2018, 23, 1–12. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Cristofani, R.; Crippa, V.; Ferrari, V.; Tedesco, B.; Casarotto, E.; Chierichetti, M.; Galbiati, M.; Piccolella, M.; Messi, E.; et al. Autophagic and Proteasomal Mediated Removal of Mutant Androgen Receptor in Muscle Models of Spinal and Bulbar Muscular Atrophy. Front. Endocrinol. 2019, 10, 569. [Google Scholar] [CrossRef]
- Carra, S.; Seguin, S.J.; Landry, J. HspB8 and Bag3: A New Chaperone Complex Targeting Misfolded Proteins to Macroautophagy. Autophagy 2008, 4, 237–239. [Google Scholar] [CrossRef]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-Assisted Selective Autophagy is Essential for Muscle Maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Brunsting, J.F.; Lambert, H.; Landry, J.; Kampinga, H.H. HspB8 Participates in Protein Quality Control by a Non-Chaperone-Like Mechanism that Requires eIF2α phosphorylation. J. Biol. Chem. 2009, 284, 5523–5532. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tan, J.; Zhou, F.; Hu, Z.; Yang, B. Heat Shock Protein B8 (HSPB8) Reduces Oxygen-Glucose Deprivation/Reperfusion Injury Via the Induction of Mitophagy. Cell. Physiol. Biochem. 2018, 48, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Wang, L.; Sui, X.; Qiu, H.; Hong, C.; Hedhli, N.; Ginion, A.; Shah, A.; Pelat, M.; Bertrand, L.; et al. H11 Kinase Prevents Myocardial Infarction by Preemptive Preconditioning of the Heart. Circ. Res. 2006, 98, 280–288. [Google Scholar] [CrossRef]
- Chen, L.; Lizano, P.; Zhao, X.; Sui, X.; Dhar, S.K.; Shen, Y.T.; Vatner, D.E.; Vatner, S.F.; Depre, C. Preemptive Conditioning of the Swine Heart by H11 kinase/Hsp22 Provides Cardiac Protection through Inducible Nitric Oxide Synthase. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1303–H1310. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, H.; Mo, X.; Xiao, H.; Hu, Z. HspB8 is Neuroprotective during Oxygen Glucose Deprivation and Reperfusion. Curr. Neurovasc Res. 2015, 12, 63–72. [Google Scholar] [CrossRef]
- Jo, H.S.; Kim, D.W.; Shin, M.J.; Cho, S.B.; Park, J.H.; Lee, C.H.; Yeo, E.J.; Choi, Y.J.; Yeo, H.J.; Sohn, E.J.; et al. Tat-HSP22 Inhibits Oxidative Stress-Induced Hippocampal Neuronal Cell Death by Regulation of the Mitochondrial Pathway. Mol. Brain 2017, 10, 1. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Knezevic, T.; Gupta, M.K.; Gordon, J.; Cheung, J.Y.; Feldman, A.M.; Khalili, K. Evidence for the Role of BAG3 in Mitochondrial Quality Control in Cardiomyocytes. J. Cell. Physiol. 2017, 232, 797–805. [Google Scholar] [CrossRef]
- Qiu, H.; Lizano, P.; Laure, L.; Sui, X.; Rashed, E.; Park, J.Y.; Hong, C.; Gao, S.; Holle, E.; Morin, D.; et al. H11 kinase/heat Shock Protein 22 Deletion Impairs both Nuclear and Mitochondrial Functions of STAT3 and Accelerates the Transition into Heart Failure on Cardiac Overload. Circulation 2011, 124, 406–415. [Google Scholar] [CrossRef]
- Rashed, E.; Lizano, P.; Dai, H.; Thomas, A.; Suzuki, C.K.; Depre, C.; Qiu, H. Heat Shock Protein 22 (Hsp22) Regulates Oxidative Phosphorylation upon its Mitochondrial Translocation with the Inducible Nitric Oxide Synthase in Mammalian Heart. PLoS ONE 2015, 10, e0119537. [Google Scholar] [CrossRef]
- Laure, L.; Long, R.; Lizano, P.; Zini, R.; Berdeaux, A.; Depre, C.; Morin, D. Cardiac H11 kinase/Hsp22 Stimulates Oxidative Phosphorylation and Modulates Mitochondrial Reactive Oxygen Species Production: Involvement of a Nitric Oxide-Dependent Mechanism. Free Radic. Biol. Med. 2012, 52, 2168–2176. [Google Scholar] [CrossRef] [PubMed]
- Morrow, G.; Inaguma, Y.; Kato, K.; Tanguay, R.M. The Small Heat Shock Protein Hsp22 of Drosophila Melanogaster is a Mitochondrial Protein Displaying Oligomeric Organization. J. Biol. Chem. 2000, 275, 31204–31210. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Boncoraglio, A.; Kanon, B.; Brunsting, J.F.; Minoia, M.; Rana, A.; Vos, M.J.; Seidel, K.; Sibon, O.C.; Kampinga, H.H. Identification of the Drosophila Ortholog of HSPB8: Implication of HSPB8 Loss of Function in Protein Folding Diseases. J. Biol. Chem. 2010, 285, 37811–37822. [Google Scholar] [CrossRef] [PubMed]
- Hedhli, N.; Wang, L.; Wang, Q.; Rashed, E.; Tian, Y.; Sui, X.; Madura, K.; Depre, C. Proteasome Activation during Cardiac Hypertrophy by the Chaperone H11 Kinase/Hsp22. Cardiovasc. Res. 2008, 77, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Irobi, J.; Van Impe, K.; Seeman, P.; Jordanova, A.; Dierick, I.; Verpoorten, N.; Michalik, A.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; et al. Hot-Spot Residue in Small Heat-Shock Protein 22 Causes Distal Motor Neuropathy. Nat. Genet. 2004, 36, 597–601. [Google Scholar] [CrossRef]
- Tang, B.S.; Zhao, G.H.; Luo, W.; Xia, K.; Cai, F.; Pan, Q.; Zhang, R.X.; Zhang, F.F.; Liu, X.M.; Chen, B.; et al. Small Heat-Shock Protein 22 Mutated in Autosomal Dominant Charcot-Marie-Tooth Disease Type 2L. Hum. Genet. 2005, 116, 222–224. [Google Scholar] [CrossRef]
- Nakhro, K.; Park, J.M.; Kim, Y.J.; Yoon, B.R.; Yoo, J.H.; Koo, H.; Choi, B.O.; Chung, K.W. A Novel Lys141Thr Mutation in Small Heat Shock Protein 22 (HSPB8) Gene in Charcot-Marie-Tooth Disease Type 2L. Neuromuscul. Disord. 2013, 23, 656–663. [Google Scholar] [CrossRef]
- Ghaoui, R.; Palmio, J.; Brewer, J.; Lek, M.; Needham, M.; Evilä, A.; Hackman, P.; Jonson, P.H.; Penttilä, S.; Vihola, A.; et al. Mutations in HSPB8 Causing a New Phenotype of Distal Myopathy and Motor Neuropathy. Neurology 2016, 86, 391–398. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Lornage, X.; Lannes, B.; Schneider, R.; Bierry, G.; Dondaine, N.; Boland, A.; Deleuze, J.F.; Böhm, J.; Thompson, J.; et al. HSPB8 Haploinsufficiency Causes Dominant Adult-Onset Axial and Distal Myopathy. Acta Neuropathol. 2017, 134, 163–165. [Google Scholar] [CrossRef]
- Cortese, A.; Laurà, M.; Casali, C.; Nishino, I.; Hayashi, Y.K.; Magri, S.; Taroni, F.; Stuani, C.; Saveri, P.; Moggio, M.; et al. Altered TDP-43-Dependent Splicing in HSPB8-Related Distal Hereditary Motor Neuropathy and Myofibrillar Myopathy. Eur. J. Neurol. 2018, 25, 154–163. [Google Scholar] [CrossRef]
- Al-Tahan, S.; Weiss, L.; Yu, H.; Tang, S.; Saporta, M.; Vihola, A.; Mozaffar, T.; Udd, B.; Kimonis, V. New Family with HSPB8-Associated Autosomal Dominant Rimmed Vacuolar Myopathy. Neurol. Genet. 2019, 5, e349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, F.; Li, X.; Huang, S.; Zi, X.; Liu, T.; Liu, S.; Li, X.; Xia, K.; Pan, Q.; et al. A Novel Transgenic Mouse Model of Chinese Charcot-Marie-Tooth Disease Type 2L. Neural Regen. Res. 2014, 9, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Bouhy, D.; Juneja, M.; Katona, I.; Holmgren, A.; Asselbergh, B.; De Winter, V.; Hochepied, T.; Goossens, S.; Haigh, J.J.; Libert, C.; et al. A Knock-in/knock-Out Mouse Model of HSPB8-Associated Distal Hereditary Motor Neuropathy and Myopathy Reveals Toxic Gain-of-Function of Mutant Hspb8. Acta Neuropathol. 2018, 135, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska, J.; Dubińska-Magiera, M.; Jagla, T.; Jagla, K.; Daczewska, M. Drosophila Hsp67Bc Hot-Spot Variants Alter Muscle Structure and Function. Cell Mol. Life Sci. 2018, 75, 4341–4356. [Google Scholar] [CrossRef]
- Kwok, A.S.; Phadwal, K.; Turner, B.J.; Oliver, P.L.; Raw, A.; Simon, A.K.; Talbot, K.; Agashe, V.R. HspB8 Mutation Causing Hereditary Distal Motor Neuropathy Impairs Lysosomal Delivery of Autophagosomes. J. Neurochem. 2011, 119, 1155–1161. [Google Scholar] [CrossRef]
- Irobi, J.; Almeida-Souza, L.; Asselbergh, B.; De Winter, V.; Goethals, S.; Dierick, I.; Krishnan, J.; Timmermans, J.P.; Robberecht, W.; De Jonghe, P.; et al. Mutant HSPB8 Causes Motor Neuron-Specific Neurite Degeneration. Hum. Mol. Genet. 2010, 19, 3254–3265. [Google Scholar] [CrossRef]
- Yang, X.D.; Cen, Z.D.; Cheng, H.P.; Shi, K.; Bai, J.; Xie, F.; Wu, H.W.; Li, B.B.; Luo, W. L-3-n-Butylphthalide Protects HSPB8 K141N Mutation-Induced Oxidative Stress by Modulating the Mitochondrial Apoptotic and Nrf2 Pathways. Front. Neurosci. 2017, 11, 402. [Google Scholar] [CrossRef]
- Irobi, J.; Holmgren, A.; De Winter, V.; Asselbergh, B.; Gettemans, J.; Adriaensen, D.; Ceuterick-de Groote, C.; Van Coster, R.; De Jonghe, P.; Timmerman, V. Mutant HSPB8 Causes Protein Aggregates and a Reduced Mitochondrial Membrane Potential in Dermal Fibroblasts from Distal Hereditary Motor Neuropathy Patients. Neuromuscul. Disord. 2012, 22, 699–711. [Google Scholar] [CrossRef]
- Sun, X.; Fontaine, J.M.; Hoppe, A.D.; Carra, S.; DeGuzman, C.; Martin, J.L.; Simon, S.; Vicart, P.; Welsh, M.J.; Landry, J.; et al. Abnormal Interaction of Motor Neuropathy-Associated Mutant HspB8 (Hsp22) Forms with the RNA Helicase Ddx20 (gemin3). Cell Stress Chaperones 2010, 15, 567–582. [Google Scholar] [CrossRef]
- Curmi, F.; Cauchi, R.J. The Multiple Lives of DEAD-Box RNA Helicase DP103/DDX20/Gemin3. Biochem. Soc. Trans. 2018, 46, 329–341. [Google Scholar] [CrossRef]
- Cacciottolo, R.; Ciantar, J.; Lanfranco, M.; Borg, R.M.; Vassallo, N.; Bordonné, R.; Cauchi, R.J. SMN Complex Member Gemin3 Self-Interacts and has a Functional Relationship with ALS-Linked Proteins TDP-43, FUS and Sod1. Sci. Rep. 2019, 9, 18666. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jonson, P.H.; Sarparanta, J.; Palmio, J.; Sarkar, M.; Vihola, A.; Evilä, A.; Suominen, T.; Penttilä, S.; Savarese, M.; et al. TIA1 Variant Drives Myodegeneration in Multisystem Proteinopathy with SQSTM1 Mutations. J. Clin. Investig. 2018, 128, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol. Sci. 2016, 37, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Reed, J.C. Molecular Chaperone Targeting and Regulation by BAG Family Proteins. Nat. Cell Biol. 2001, 3, E237–E241. [Google Scholar] [CrossRef] [PubMed]
- Stürner, E.; Behl, C. The Role of the Multifunctional BAG3 Protein in Cellular Protein Quality Control and in Disease. Front. Mol. Neurosci. 2017, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Mock, J.Y.; Chartron, J.W.; Zaslaver, M.; Xu, Y.; Ye, Y.; Clemons, W.M., Jr. Bag6 Complex Contains a Minimal Tail-Anchor-Targeting Module and a Mock BAG Domain. Proc. Natl. Acad. Sci. USA 2015, 112, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Minami, R.; Hayakawa, A.; Kagawa, H.; Yanagi, Y.; Yokosawa, H.; Kawahara, H. BAG-6 is Essential for Selective Elimination of Defective Proteasomal Substrates. J. Cell Biol. 2010, 190, 637–650. [Google Scholar] [CrossRef]
- Rauch, J.N.; Gestwicki, J.E. Binding of Human Nucleotide Exchange Factors to Heat Shock Protein 70 (Hsp70) Generates Functionally Distinct Complexes in Vitro. J. Biol. Chem. 2014, 289, 1402–1414. [Google Scholar] [CrossRef]
- Rauch, J.N.; Zuiderweg, E.R.; Gestwicki, J.E. Non-Canonical Interactions between Heat Shock Cognate Protein 70 (Hsc70) and Bcl2-Associated Anthanogene (BAG) Co-Chaperones are Important for Client Release. J. Biol. Chem. 2016, 291, 19848–19857. [Google Scholar] [CrossRef]
- Lee, J.H.; Takahashi, T.; Yasuhara, N.; Inazawa, J.; Kamada, S.; Tsujimoto, Y. Bis, a Bcl-2-Binding Protein that Synergizes with Bcl-2 in Preventing Cell Death. Oncogene 1999, 18, 6183–6190. [Google Scholar] [CrossRef]
- Franceschelli, S.; Rosati, A.; Lerose, R.; De Nicola, S.; Turco, M.C.; Pascale, M. Bag3 Gene Expression is Regulated by Heat Shock Factor 1. J. Cell. Physiol. 2008, 215, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Rosati, A.; Ammirante, M.; Gentilella, A.; Basile, A.; Festa, M.; Pascale, M.; Marzullo, L.; Belisario, M.A.; Tosco, A.; Franceschelli, S.; et al. Apoptosis Inhibition in Cancer Cells: A Novel Molecular Pathway that Involves BAG3 Protein. Int. J. Biochem. Cell. Biol. 2007, 39, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Chen, B. BAG3 Regulates Multiple Myeloma Cell Proliferation through FOXM1/Rb/E2F Axis. Cancer Gene Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Pegoraro, E.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in BAG3 Causes Severe Dominant Childhood Muscular Dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, L.N.; Cheng, L.; Tu, S.; Guo, S.J.; Le, H.Y.; Xiong, Q.; Mo, R.; Li, C.Y.; Jeong, J.S.; et al. Bcl2-Associated Athanogene 3 Interactome Analysis Reveals a New Role in Modulating Proteasome Activity. Mol. Cell Proteom. 2013, 12, 2804–2819. [Google Scholar] [CrossRef]
- Ulbricht, A.; Eppler, F.J.; Tapia, V.E.; van der Ven, P.F.; Hampe, N.; Hersch, N.; Vakeel, P.; Stadel, D.; Haas, A.; Saftig, P.; et al. Cellular Mechanotransduction Relies on Tension-Induced and Chaperone-Assisted Autophagy. Curr. Biol. 2013, 23, 430–435. [Google Scholar] [CrossRef]
- Sun, C.; De Mello, V.; Mohamed, A.; Ortuste Quiroga, H.P.; Garcia-Munoz, A.; Al Bloshi, A.; Tremblay, A.M.; von Kriegsheim, A.; Collie-Duguid, E.; Vargesson, N.; et al. Common and Distinctive Functions of the Hippo Effectors Taz and Yap in Skeletal Muscle Stem Cell Function. Stem Cells 2017, 35, 1958–1972. [Google Scholar] [CrossRef]
- Chevessier, F.; Schuld, J.; Orfanos, Z.; Plank, A.C.; Wolf, L.; Maerkens, A.; Unger, A.; Schlötzer-Schrehardt, U.; Kley, R.A.; Von Horsten, S.; et al. Myofibrillar Instability Exacerbated by Acute Exercise in Filaminopathy. Hum. Mol. Genet. 2015, 24, 7207–7220. [Google Scholar] [CrossRef]
- Xu, Z.; Graham, K.; Foote, M.; Liang, F.; Rizkallah, R.; Hurt, M.; Wang, Y.; Wu, Y.; Zhou, Y. 14-3-3 Protein Targets Misfolded Chaperone-Associated Proteins to Aggresomes. J. Cell. Sci. 2013, 126, 4173–4186. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Kaya, A.M.; Wolfrum, U.; Clement, A.M.; Behl, C. BAG3 Mediates Chaperone-Based Aggresome-Targeting and Selective Autophagy of Misfolded Proteins. EMBO Rep. 2011, 12, 149–156. [Google Scholar] [CrossRef]
- Ravi Chandra, B.; Gowthaman, R.; Akhouri, R.R.; Gupta, D.; Sharma, A. Distribution of Proline-Rich (PxxP) Motifs in Distinct Proteomes: Functional and Therapeutic Implications for Malaria and Tuberculosis. Protein Eng. Des. Sel. 2004, 17, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Myers, V.D.; Tomar, D.; Madesh, M.; Wang, J.; Song, J.; Zhang, X.Q.; Gupta, M.K.; Tahrir, F.G.; Gordon, J.; McClung, J.M.; et al. Haplo-Insufficiency of Bcl2-Associated Athanogene 3 in Mice Results in Progressive Left Ventricular Dysfunction, β-Adrenergic Insensitivity, and Increased Apoptosis. J. Cell. Physiol. 2018, 233, 6319–6326. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Ahn, S.G.; Kim, S.A. BAG3 Affects the Nucleocytoplasmic Shuttling of HSF1 upon Heat Stress. Biochem. Biophys. Res. Commun. 2015, 464, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Impens, F.; Radoshevich, L.; Cossart, P.; Ribet, D. Mapping of SUMO Sites and Analysis of SUMOylation Changes Induced by External Stimuli. Proc. Natl. Acad. Sci. USA 2014, 111, 12432–12437. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.; Nielsen, M.L. Site-Specific Mapping of the Human SUMO Proteome Reveals Co-Modification with Phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Meng, X.; Gao, Y.Y.; Liu, B.Q.; Niu, X.F.; Zhang, H.Y.; Du, Z.X. Characterization of BAG3 Cleavage during Apoptosis of Pancreatic Cancer Cells. J. Cell. Physiol. 2010, 224, 94–100. [Google Scholar] [CrossRef]
- Rosati, A.; Graziano, V.; De Laurenzi, V.; Pascale, M.; Turco, M.C. BAG3: A Multifaceted Protein that Regulates Major Cell Pathways. Cell. Death Dis. 2011, 2, e141. [Google Scholar] [CrossRef]
- Gentilella, A.; Khalili, K. Autoregulation of Co-Chaperone BAG3 Gene Transcription. J. Cell. Biochem. 2009, 108, 1117–1124. [Google Scholar] [CrossRef]
- Nivon, M.; Abou-Samra, M.; Richet, E.; Guyot, B.; Arrigo, A.P.; Kretz-Remy, C. NF-κB Regulates Protein Quality Control After Heat Stress through Modulation of the BAG3-HspB8 Complex. J. Cell. Sci. 2012, 125, 1141–1151. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 Rescues Neurodegeneration and Provides an Essential Link between Autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef]
- Du, Z.X.; Zhang, H.Y.; Meng, X.; Gao, Y.Y.; Zou, R.L.; Liu, B.Q.; Guan, Y.; Wang, H.Q. Proteasome Inhibitor MG132 Induces BAG3 Expression through Activation of Heat Shock Factor 1. J. Cell. Physiol. 2009, 218, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, A.; Gehlert, S.; Leciejewski, B.; Schiffer, T.; Bloch, W.; Höhfeld, J. Induction and Adaptation of Chaperone-Assisted Selective Autophagy CASA in Response to Resistance Exercise in Human Skeletal Muscle. Autophagy 2015, 11, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Varlet, A.A.; Fuchs, M.; Luthold, C.; Lambert, H.; Landry, J.; Lavoie, J.N. Fine-Tuning of Actin Dynamics by the HSPB8-BAG3 Chaperone Complex Facilitates Cytokinesis and Contributes to its Impact on Cell Division. Cell Stress Chaperones 2017, 22, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Felzen, V.; Hiebel, C.; Stürner, E.; Perumal, N.; Manicam, C.; Sehn, E.; Grus, F.; Wolfrum, U.; Behl, C. Enhanced Autophagic-Lysosomal Activity and Increased BAG3-Mediated Selective Macroautophagy as Adaptive Response of Neuronal Cells to Chronic Oxidative Stress. Redox Biol. 2019, 24, 101181. [Google Scholar] [CrossRef]
- Myers, V.D.; McClung, J.M.; Wang, J.; Tahrir, F.G.; Gupta, M.K.; Gordon, J.; Kontos, C.H.; Khalili, K.; Cheung, J.Y.; Feldman, A.M. The Multifunctional Protein BAG3: A Novel Therapeutic Target in Cardiovascular Disease. JACC Basic Transl. Sci. 2018, 3, 122–131. [Google Scholar] [CrossRef]
- Kathage, B.; Gehlert, S.; Ulbricht, A.; Lüdecke, L.; Tapia, V.E.; Orfanos, Z.; Wenzel, D.; Bloch, W.; Volkmer, R.; Fleischmann, B.K.; et al. The Cochaperone BAG3 Coordinates Protein Synthesis and Autophagy Under Mechanical Strain through Spatial Regulation of mTORC1. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 62–75. [Google Scholar] [CrossRef]
- Meriin, A.B.; Narayanan, A.; Meng, L.; Alexandrov, I.; Varelas, X.; Cissé, I.I.; Sherman, M.Y. Hsp70-Bag3 Complex is a Hub for Proteotoxicity-Induced Signaling that Controls Protein Aggregation. Proc. Natl. Acad. Sci. USA 2018, 115, E7043–E7052. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein Quality Control during Aging Involves Recruitment of the Macroautophagy Pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Ulbricht, A.; Arndt, V.; Höhfeld, J. Chaperone-Assisted Proteostasis is Essential for Mechanotransduction in Mammalian Cells. Commun. Integr. Biol. 2013, 6, e24925. [Google Scholar] [CrossRef]
- Yuan, Y.; Pan, S.S.; Shen, Y.J. Cardioprotection of Exercise Preconditioning Involving Heat Shock Protein 70 and Concurrent Autophagy: A Potential Chaperone-Assisted Selective Macroautophagy Effect. J. Physiol. Sci. 2018, 68, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.E.; Sarkar, S.; Rubinsztein, D.C. Trehalose Reduces Aggregate Formation and Delays Pathology in a Transgenic Mouse Model of Oculopharyngeal Muscular Dystrophy. Hum. Mol. Genet. 2006, 15, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a Novel mTOR-Independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and α-Synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose Induces Autophagy Via Lysosomal-Mediated TFEB Activation in Models of Motoneuron Degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]
- Klimek, C.; Jahnke, R.; Wördehoff, J.; Kathage, B.; Stadel, D.; Behrends, C.; Hergovich, A.; Höhfeld, J. The Hippo Network Kinase STK38 Contributes to Protein Homeostasis by Inhibiting BAG3-Mediated Autophagy. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 10, 1556–1566. [Google Scholar] [CrossRef]
- Hackman, P.; Sarparanta, J.; Lehtinen, S.; Vihola, A.; Evilä, A.; Jonson, P.H.; Luque, H.; Kere, J.; Screen, M.; Chinnery, P.F.; et al. Welander Distal Myopathy is Caused by a Mutation in the RNA-Binding Protein TIA1. Ann. Neurol. 2013, 73, 500–509. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in Prion-Like Domains in hnRNPA2B1 and hnRNPA1 Cause Multisystem Proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Gordon, D.; Dafinca, R.; Scaber, J.; Alegre-Abarrategui, J.; Farrimond, L.; Scott, C.; Biggs, D.; Kent, L.; Oliver, P.L.; Davies, B.; et al. Single-Copy Expression of an Amyotrophic Lateral Sclerosis-Linked TDP-43 Mutation (M337V) in BAC Transgenic Mice Leads to Altered Stress Granule Dynamics and Progressive Motor Dysfunction. Neurobiol. Dis. 2019, 121, 148–162. [Google Scholar] [CrossRef]
- Duggan, M.; Torkzaban, B.; Ahooyi, T.M.; Khalili, K.; Gordon, J. Age-Related Neurodegenerative Diseases. J. Cell. Physiol. 2019, 235, 3131–3141. [Google Scholar] [CrossRef]
- Hishiya, A.; Kitazawa, T.; Takayama, S. BAG3 and Hsc70 Interact with Actin Capping Protein CapZ to Maintain Myofibrillar Integrity Under Mechanical Stress. Circ. Res. 2010, 107, 1220–1231. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Gordon, J.; Feldman, A.M.; Cheung, J.; Khalili, K.; Mohseni Ahooyi, T. Evidence for the Impact of BAG3 on Electrophysiological Activity of Primary Culture of Neonatal Cardiomyocytes. J. Cell. Physiol. 2019, 234, 18371–18381. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.M.; Gordon, J.; Wang, J.; Song, J.; Zhang, X.Q.; Myers, V.D.; Tilley, D.G.; Gao, E.; Hoffman, N.E.; Tomar, D.; et al. BAG3 Regulates Contractility and Ca(2+) Homeostasis in Adult Mouse Ventricular Myocytes. J. Mol. Cell. Cardiol. 2016, 92, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Semmler, A.L.; Sacconi, S.; Bach, J.E.; Liebe, C.; Bürmann, J.; Kley, R.A.; Ferbert, A.; Anderheiden, R.; Van den Bergh, P.; Martin, J.J.; et al. Unusual Multisystemic Involvement and a Novel BAG3 Mutation Revealed by NGS Screening in a Large Cohort of Myofibrillar Myopathies. Orphanet J. Rare Dis. 2014, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, C.; Dawson, D.B.; Thorland, E.C.; Lundquist, P.A.; Eckloff, B.W.; Wu, Y.; Baheti, S.; Evans, J.M.; Scherer, S.S.; et al. Target-Enrichment Sequencing and Copy Number Evaluation in Inherited Polyneuropathy. Neurology 2016, 86, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Odgerel, Z.; Sarkozy, A.; Lee, H.S.; McKenna, C.; Rankin, J.; Straub, V.; Lochmuller, H.; Paola, F.; D’Amico, A.; Bertini, E.; et al. Inheritance Patterns and Phenotypic Features of Myofibrillar Myopathy Associated with a BAG3 Mutation. Neuromuscul. Disord. 2010, 20, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Cherk, S.W.; Chan, S.K.; Wong, S.; Tong, T.W.; Ho, W.S.; Chan, A.Y.; Lee, K.C.; Mak, C.M. BAG3-Related Myofibrillar Myopathy in a Chinese Family. Clin. Genet. 2012, 81, 394–398. [Google Scholar] [CrossRef]
- Jaffer, F.; Murphy, S.M.; Scoto, M.; Healy, E.; Rossor, A.M.; Brandner, S.; Phadke, R.; Selcen, D.; Jungbluth, H.; Muntoni, F.; et al. BAG3 Mutations: Another Cause of Giant Axonal Neuropathy. J. Peripher. Nerv. Syst. 2012, 17, 210–216. [Google Scholar] [CrossRef]
- Kim, S.J.; Nam, S.H.; Kanwal, S.; Nam, D.E.; Yoo, D.H.; Chae, J.H.; Suh, Y.L.; Chung, K.W.; Choi, B.O. BAG3 Mutation in a Patient with Atypical Phenotypes of Myofibrillar Myopathy and Charcot-Marie-Tooth Disease. Genes Genom. 2018, 40, 1269–1277. [Google Scholar] [CrossRef]
- Andersen, A.G.; Fornander, F.; Schrøder, H.D.; Krag, T.; Straub, V.; Duno, M.; Vissing, J. BAG3 Myopathy is Not always Associated with Cardiomyopathy. Neuromuscul. Disord. 2018, 28, 798–801. [Google Scholar] [CrossRef]
- Konersman, C.G.; Bordini, B.J.; Scharer, G.; Lawlor, M.W.; Zangwill, S.; Southern, J.F.; Amos, L.; Geddes, G.C.; Kliegman, R.; Collins, M.P. BAG3 Myofibrillar Myopathy Presenting with Cardiomyopathy. Neuromuscul. Disord. 2015, 25, 418–422. [Google Scholar] [CrossRef]
- Kostera-Pruszczyk, A.; Suszek, M.; Płoski, R.; Franaszczyk, M.; Potulska-Chromik, A.; Pruszczyk, P.; Sadurska, E.; Karolczak, J.; Kamińska, A.M.; Rędowicz, M.J. BAG3-Related Myopathy, Polyneuropathy and Cardiomyopathy with Long QT Syndrome. J. Muscle Res. Cell. Motil. 2015, 36, 423–432. [Google Scholar] [CrossRef]
- Noury, J.B.; Maisonobe, T.; Richard, P.; Delague, V.; Malfatti, E.; Stojkovic, T. Rigid Spine Syndrome Associated with Sensory-Motor Axonal Neuropathy Resembling Charcot-Marie-Tooth Disease is Characteristic of Bcl-2-Associated Athanogene-3 Gene Mutations Even without Cardiac Involvement. Muscle Nerve 2018, 57, 330–334. [Google Scholar] [CrossRef] [PubMed]
- D’Avila, F.; Meregalli, M.; Lupoli, S.; Barcella, M.; Orro, A.; De Santis, F.; Sitzia, C.; Farini, A.; D’Ursi, P.; Erratico, S.; et al. Exome Sequencing Identifies Variants in Two Genes Encoding the LIM-Proteins NRAP and FHL1 in an Italian Patient with BAG3 Myofibrillar Myopathy. J. Muscle Res. Cell. Motil. 2016, 37, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Schänzer, A.; Rupp, S.; Gräf, S.; Zengeler, D.; Jux, C.; Akintürk, H.; Gulatz, L.; Mazhari, N.; Acker, T.; Van Coster, R.; et al. Dysregulated Autophagy in Restrictive Cardiomyopathy due to Pro209Leu Mutation in BAG3. Mol. Genet. Metab. 2018, 123, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Bogomolovas, J.; Trexler, C.; Chen, J. The BAG3-Dependent and -Independent Roles of Cardiac Small Heat Shock Proteins. JCI Insight 2019, 4, e126464. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, K.; Zhang, X.; Tang, Y.; Xu, D. Advances in the Role and Mechanism of BAG3 in Dilated Cardiomyopathy. Heart Fail. Rev. 2019. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Züchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-Wide Studies of Copy Number Variation and Exome Sequencing Identify Rare Variants in BAG3 as a Cause of Dilated Cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef]
- Franaszczyk, M.; Bilinska, Z.T.; Sobieszczańska-Małek, M.; Michalak, E.; Sleszycka, J.; Sioma, A.; Małek, Ł.A.; Kaczmarska, D.; Walczak, E.; Włodarski, P.; et al. The BAG3 Gene Variants in Polish Patients with Dilated Cardiomyopathy: Four Novel Mutations and a Genotype-Phenotype Correlation. J. Transl. Med. 2014, 12, 192. [Google Scholar] [CrossRef]
- Shy, M.; Rebelo, A.P.; Feely, S.M.; Abreu, L.A.; Tao, F.; Swenson, A.; Bacon, C.; Zuchner, S. Mutations in BAG3 Cause Adult-Onset Charcot-Marie-Tooth Disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 313–315. [Google Scholar] [CrossRef]
- Fu, J.; Ma, M.; Song, J.; Pang, M.; Li, G.; Zhang, J. BAG3 p.Pro209Ser Mutation Identified in a Chinese Family with Charcot-Marie-Tooth Disease. J. Neurol. 2019. [Google Scholar] [CrossRef]
- Arimura, T.; Ishikawa, T.; Nunoda, S.; Kawai, S.; Kimura, A. Dilated Cardiomyopathy-Associated BAG3 Mutations Impair Z-Disc Assembly and Enhance Sensitivity to Apoptosis in Cardiomyocytes. Hum. Mutat. 2011, 32, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, E.; Gréen, A.; Gustavsson, P.; Liedén, A.; Nordenskjöld, M.; Papadogiannakis, N.; Pettersson, K.; Nilsson, D.; Jonasson, J.; Iwarsson, E. Identification of Putative Pathogenic Single Nucleotide Variants (SNVs) in Genes Associated with Heart Disease in 290 Cases of Stillbirth. PLoS ONE 2019, 14, e0210017. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Pérez-Serra, A.; Campuzano, O.; Moncayo-Arlandi, J.; Allegue, C.; Iglesias, A.; Mangas, A.; Brugada, R. Familial Dilated Cardiomyopathy Caused by a Novel Frameshift in the BAG3 Gene. PLoS ONE 2016, 11, e0158730. [Google Scholar] [CrossRef] [PubMed]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A Genome-Wide Association Study Identifies Two Loci Associated with Heart Failure due to Dilated Cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef]
- Rafiq, M.A.; Chaudhry, A.; Care, M.; Spears, D.A.; Morel, C.F.; Hamilton, R.M. Whole Exome Sequencing Identified 1 Base Pair Novel Deletion in BCL2-Associated Athanogene 3 (BAG3) Gene Associated with Severe Dilated Cardiomyopathy (DCM) Requiring Heart Transplant in Multiple Family Members. Am. J. Med. Genet. A 2017, 173, 699–705. [Google Scholar] [CrossRef]
- Ruparelia, A.A.; Oorschot, V.; Vaz, R.; Ramm, G.; Bryson-Richardson, R.J. Zebrafish Models of BAG3 Myofibrillar Myopathy Suggest a Toxic Gain of Function Leading to BAG3 Insufficiency. Acta Neuropathol. 2014, 128, 821–833. [Google Scholar] [CrossRef]
- Ding, Y.; Dvornikov, A.V.; Ma, X.; Zhang, H.; Wang, Y.; Lowerison, M.; Packard, R.R.; Wang, L.; Chen, J.; Zhang, Y.; et al. Haploinsufficiency of Mechanistic Target of Rapamycin Ameliorates bag3 Cardiomyopathy in Adult Zebrafish. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef]
- Youn, D.Y.; Lee, D.H.; Lim, M.H.; Yoon, J.S.; Lim, J.H.; Jung, S.E.; Yeum, C.E.; Park, C.W.; Youn, H.J.; Lee, J.S.; et al. Bis Deficiency Results in Early Lethality with Metabolic Deterioration and Involution of Spleen and Thymus. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1349–E1357. [Google Scholar] [CrossRef][Green Version]
- Fang, X.; Bogomolovas, J.; Zhou, P.S.; Mu, Y.; Ma, X.; Chen, Z.; Zhang, L.; Zhu, M.; Veevers, J.; Ouyang, K.; et al. P209L Mutation in Bag3 does Not Cause Cardiomyopathy in Mice. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H392–H399. [Google Scholar] [CrossRef]
- Harrison, A.F.; Shorter, J. RNA-Binding Proteins with Prion-Like Domains in Health and Disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef]








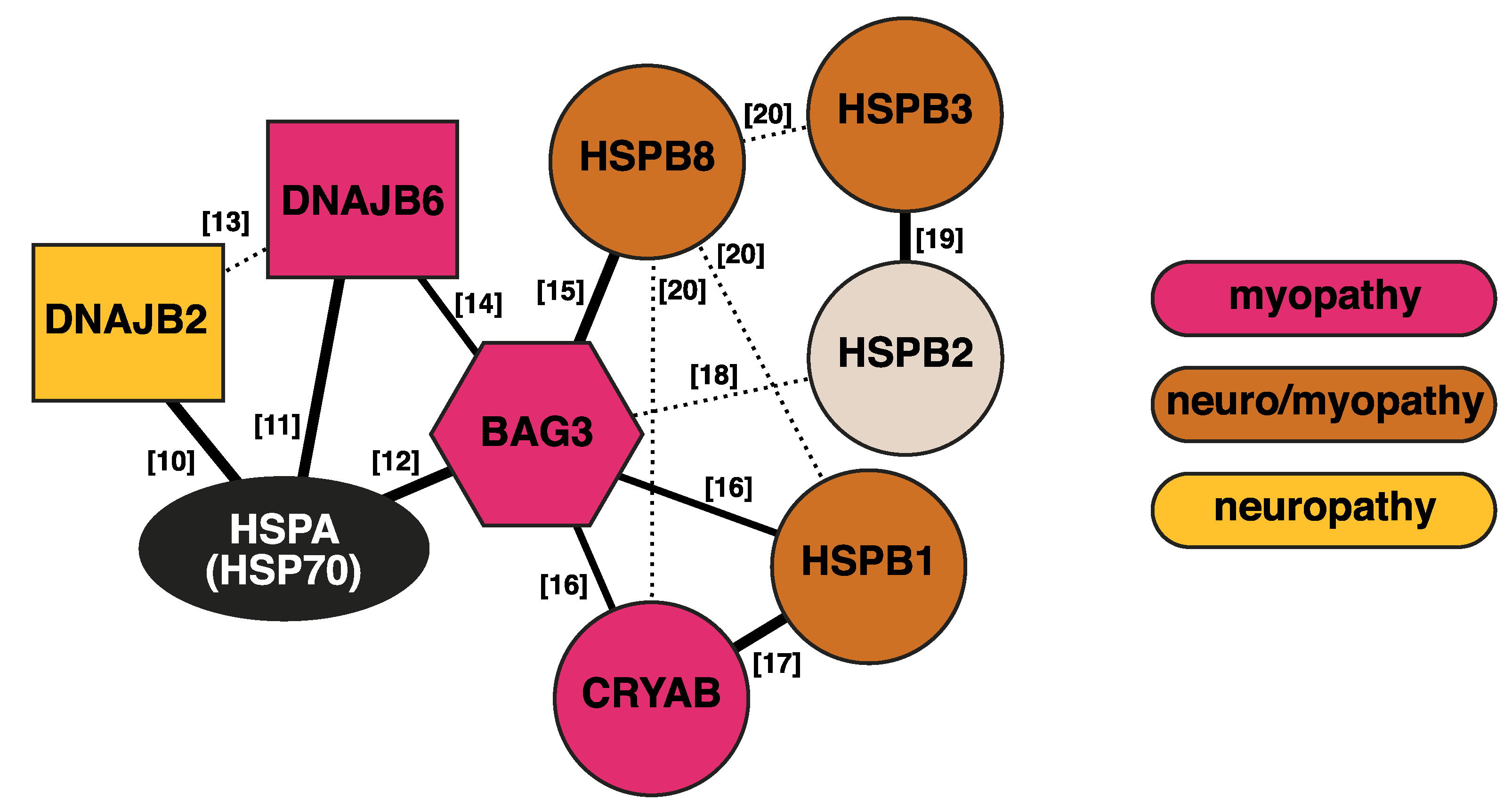{kind=link}
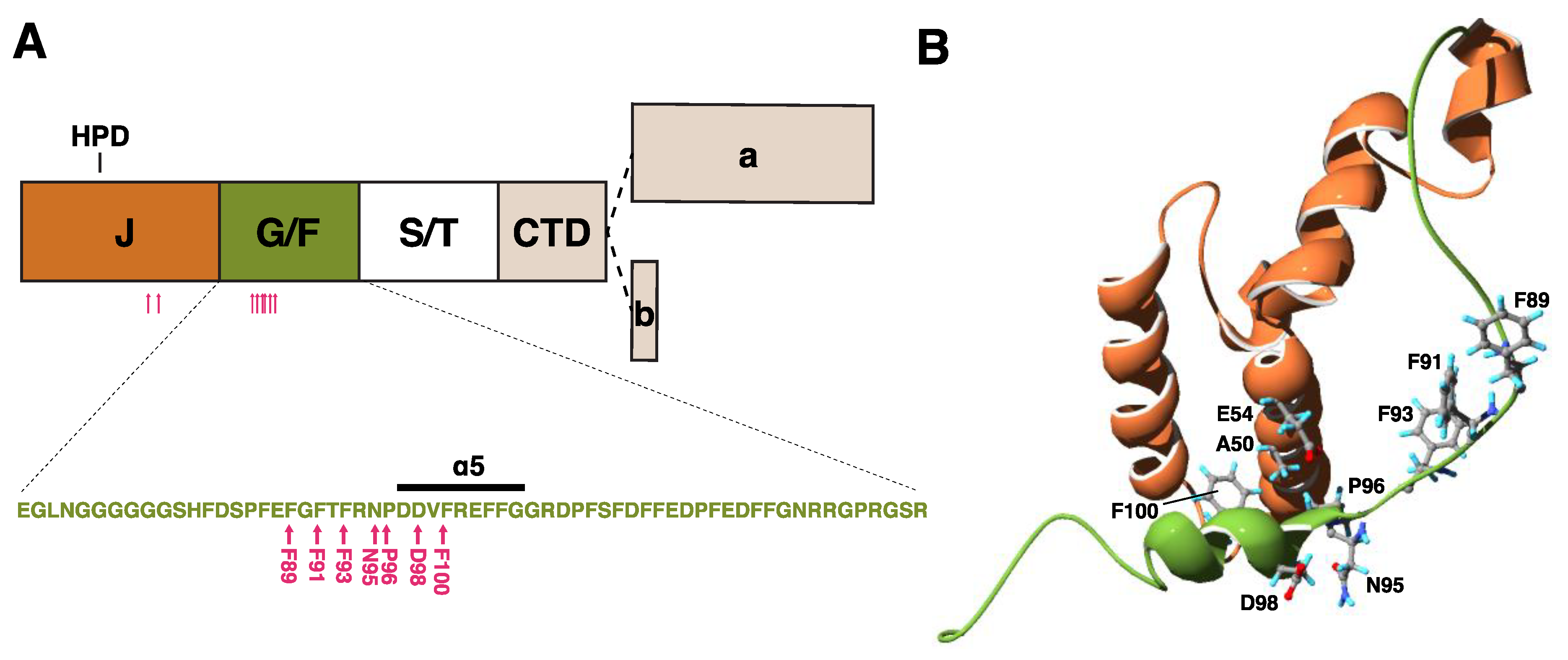{kind=link}
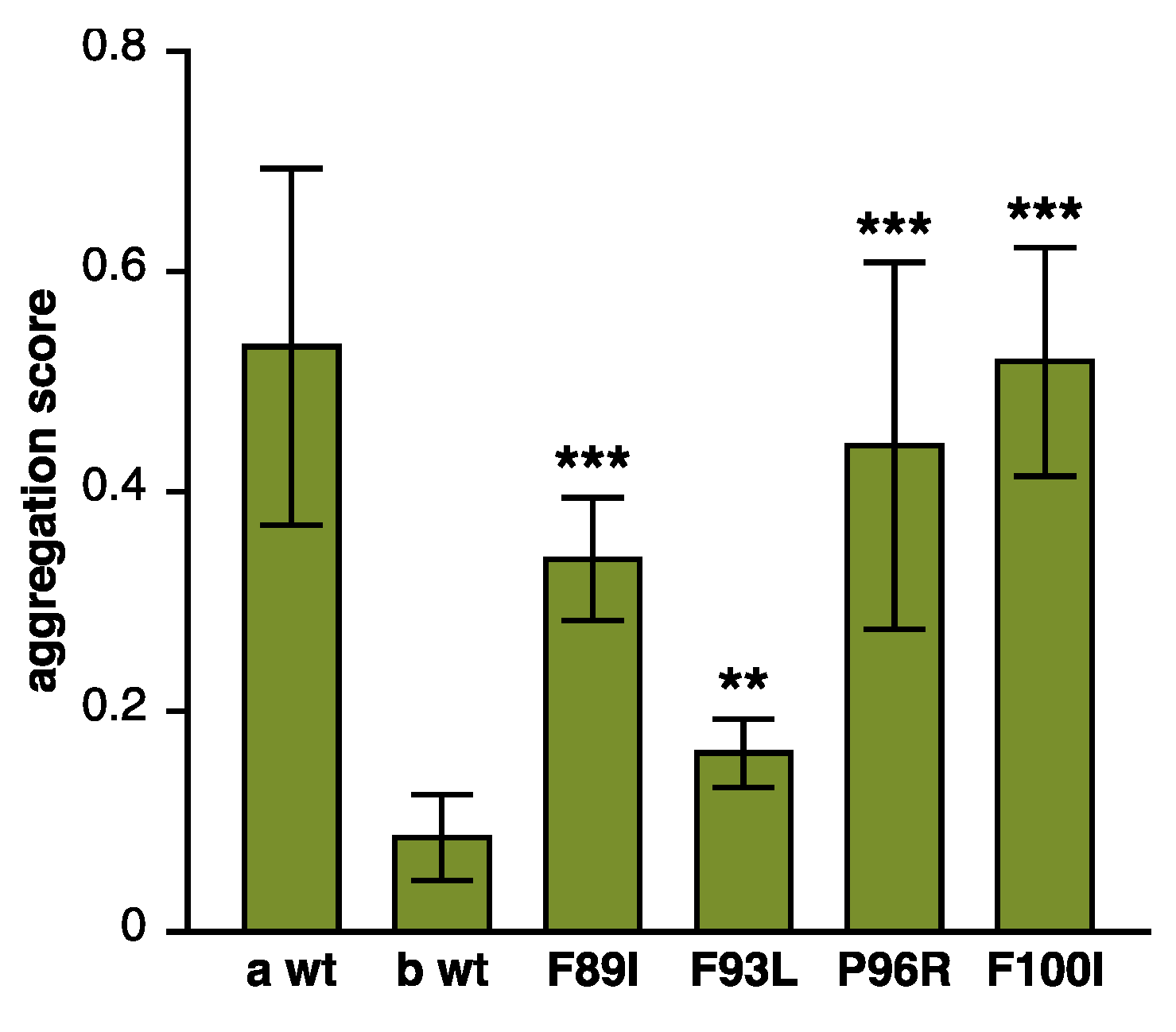{kind=link}
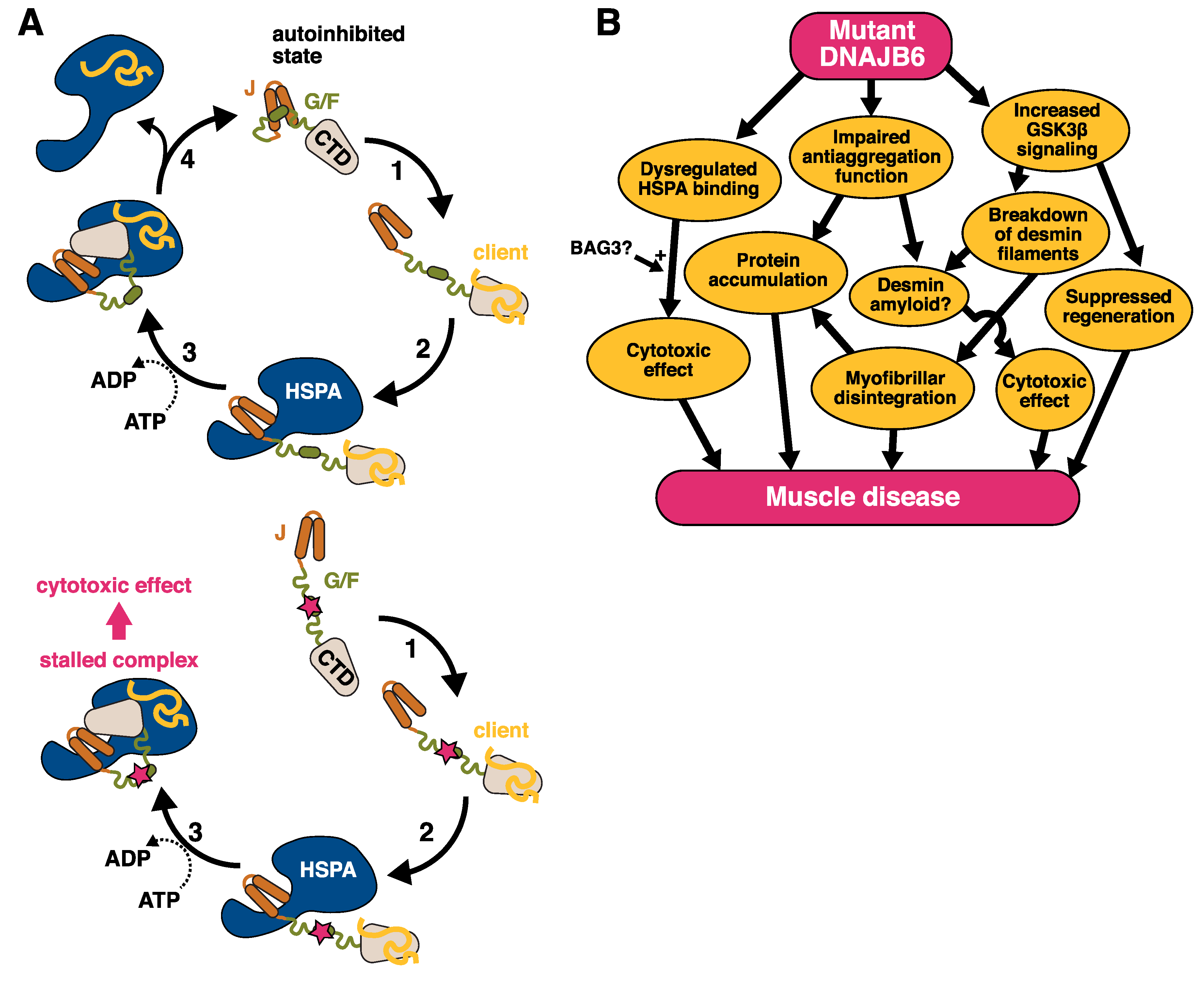{kind=link}
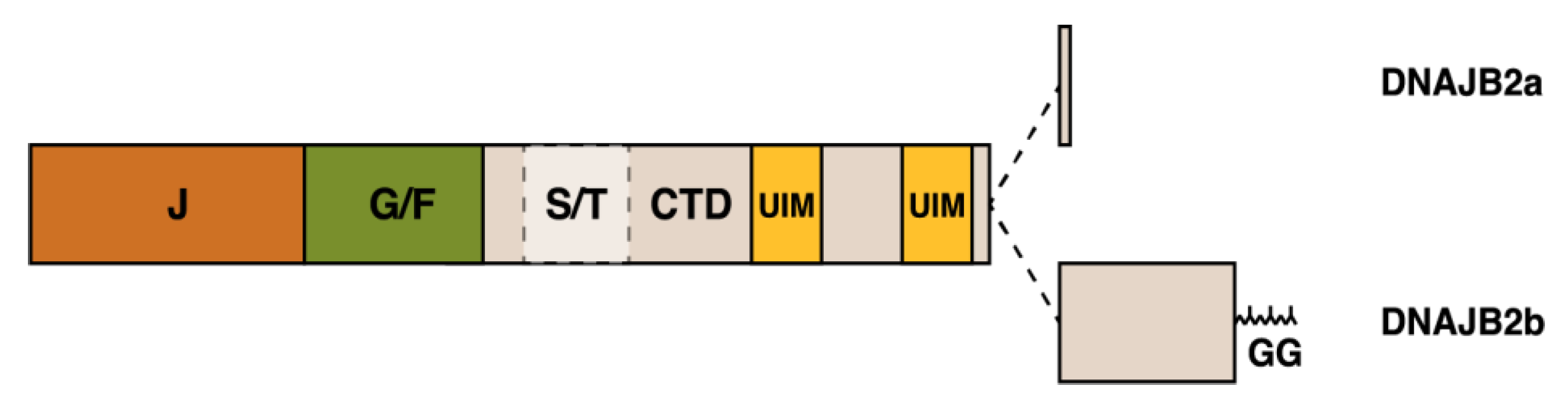{kind=link}
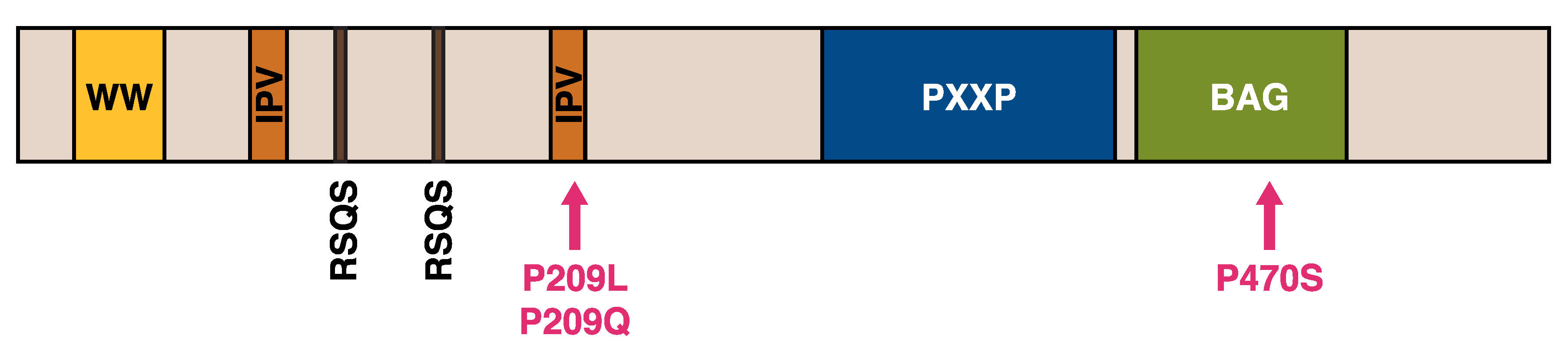{kind=link}
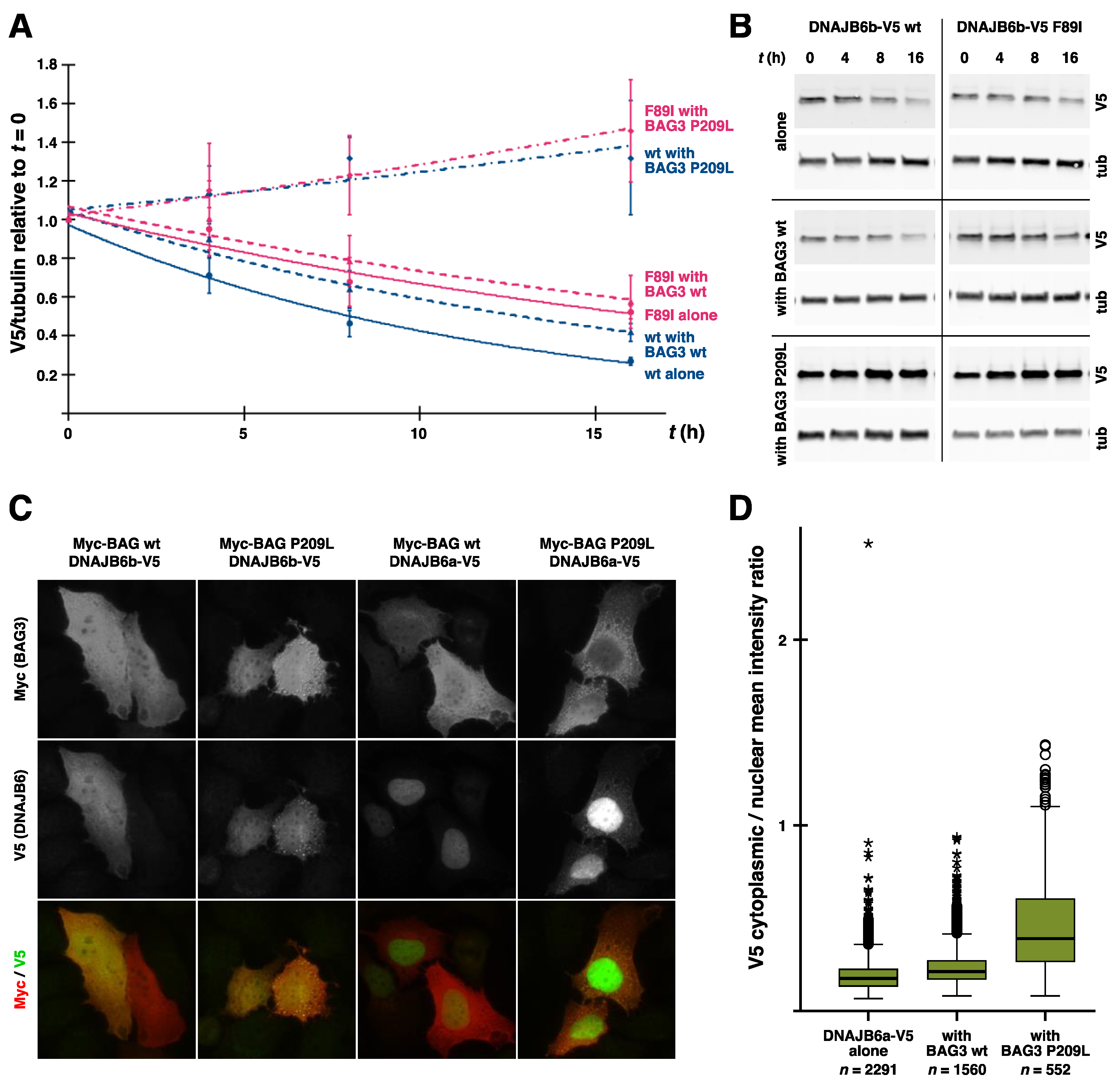{kind=link}
| Gene Symbol | Neuromuscular Disorder(s) (MIM1 Number When Available) |
|---|---|
| BAG3 | MFM6 (#612954); CMD1HH (#613881); CMT2 |
| CCT5 | Hereditary sensory neuropathy with spastic paraplegia (#256840) |
| CRYAB | MFM2 (#608810); CMD1II (#615184); Fatal infantile hypertonic myofibrillar myopathy (#613869) |
| DNAJB2 | DSMA5 (#614881); CMT2 |
| DNAJB6 | LGMD D1 DNAJB6-related (#603511); Distal myopathy with rimmed vacuoles |
| HSPB1 | dHMN2B (#608634); CMT2F (#606595) |
| HSPB3 | dHMN2C (#613376); CMT2; (neuro)myopathy |
| HSPB8 | dHMN2A (#158590); CMT2L (#608673); Neuromyopathy with rimmed vacuoles |
| HSPD1 | Spastic paraplegia 13, autosomal dominant (#605280) |
| SACS | Spastic ataxia, Charlevoix–Saguenay type (#270550) |
| SIL1 | Marinesco–Sjögren syndrome (#248800) |
| STUB1 | Spinocerebellar ataxia, autosomal recessive 16 (#615768) |
| TOR1A | Torsion dystonia, early onset (#128100) |
| VCP | Scapuloperoneal muscular dystrophy and dropped head syndrome; Distal myopathy; IBMPFD (# 167320); ALS14 (#613954); CMT2Y (#616687) |
| VMA21 | X-linked myopathy with excessive autophagy (XMEA) (#310440) |
| Domain | cDNA Change | Protein Change | Phenotype | References |
|---|---|---|---|---|
| J | c.149C>T | p.A50V | distal | [90] |
| c.161A>C | p.E54A | proximo-distal | [90] | |
| G/F | c.265T>A | p.F89I | LGMD | [14,92,93,94] |
| c.271T>A | p.F91I | LGMD (severe) | [88,95] | |
| c.271T>G | p.F91V | mild | [92,96] | |
| c.271T>C | p.F91L | LGMD (severe) | [95,97] | |
| c.273C>G | [88,92] | |||
| c.277T>A | p.F93I | LGMD | [98] | |
| c.277T>C | p.F93L | LGMD | [14,86] | |
| c.279C>A | [14,92] | |||
| c.279C>G | [14,88,89,92,98,99] | |||
| c.284A>T | p.N95I | LGMD | [89] | |
| c.287C>G | p.P96R | distal–proximal | [86] | |
| c.287C>T | p.P96L | [100,101,102] | ||
| c.293_295delATG | p.D98del | distal | [89] | |
| c.298T>A | p.F100I | [103] | ||
| c.298C>A | p.F100V | distal onset | [88] | |
| c.346+5G>A | p.G79_F115del | severe, early onset | [88] |
| Protein Change | PolyQ Aggregation | PARK p.C289G Aggregation | Sis1 Complementation 1 | [RNQ+] Propagation 2 | [PSI+] Propagation/Solubility 2 | TDP-43 Aggregation | hnRNPA2 p.D290V Aggregation 3 | Hrb98DE Localization & interaction 3 | Myotoxicity in Zebrafish | References |
|---|---|---|---|---|---|---|---|---|---|---|
| A50V | ††††† | † | [90] | |||||||
| E54A | †††† | † | [90] | |||||||
| F89I | †††† | †† | ††/† | †† | † | † | † | ††† | [14,59,105] | |
| F91I | †† | [95] | ||||||||
| F91L | ††† | ††† | [95] | |||||||
| F93L | † | † | – | – | – | † | † | ††(†) | [14,31,59,90,105] | |
| N95I | †††† | [89] | ||||||||
| P96R | ††††† | ††† | †/– | † | † | † | [105], this paper | |||
| P96L | ††† | [102] | ||||||||
| D98del | †††† | [89] | ||||||||
| F100I | ††††† | this paper |
| Mutation 1 | Phenotype | References |
|---|---|---|
| c.14A>G (p.Y5C) | CMT2 | [113] |
| c.229+1G>A (splice) | dHMN | [113] |
| c.310delC (p.R104Gfs*97) | CMT2 | [37] |
| c.352+1G>A (splice) | dHMN, CMT2, parkinsonism | [112,127,128] |
| c.619-1G>A (splice) | CMT2 | [37] |
| g.219277938_219281781del2 | dHMN, parkinsonism | [129] |
| Mutation 1 | Phenotype 2 | Inheritance 3 | Thermal Stability | Aggregation | Hyperphosphorylation | Oligomer Size | Hetero-oligom/HSPB1 | Hetero-oligom/HSPB6 | HSPB8 Interaction | Chaperone Activity | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| c.3G>A (p.M1?) | infantile MFM | R | [234] | ||||||||
| c.60delC (p.S21Afs*24) | infantile MFM | R | [232] | ||||||||
| c.325G>C (p.D109H) | MFM + DCM + cat | D | [225] | ||||||||
| c.2326A>C (p.D109A) | MFM (+ DCM) + cat | D | [226] | ||||||||
| c.326A>G (p.D109G) | AxM + RCM | D | + | [227] | |||||||
| c.343delT (p.S115Pfs*14) | infantile MFM | R | + | [233,274] | |||||||
| c.358A>G (p.R120G) | MFM + HCM + cat | D | – | ++ | + | + | + | – – – | + | – | [167,222,250,253,257,267,272,294] |
| c.451C>T (p.Q151*) | MFM | D | – | + | + | – – – | – | – | +/– | [223,258,259,272] | |
| c.460G>A (p.G154S) | DM/DCM | D | – | = | = | = | +/– | [224,229,261] | |||
| c.464_465delCT (p.P155Rfs*9) | MFM | D | – | ++ | + | – – | – | – | +/– | [223,258,259,272] | |
| c.470G>A (p.R157H) | DCM | D | – | + | – | = | – | +/= | [228,261] | ||
| c.527A>G (p.*176Wext*19) | DCM + cat | D | [295] |
| Domain | Mutation 1 | Inheritance 2 | Oligomer Size | Oligomer Stability | Oligomer Sensitivity to Phosphorylation | Dimerization | Chaperone Function | Client Binding | Thermal Stability | Aggregation | Cytotoxicity | Cell Stress Tolerance | Effects on Neurofilaments | Effects on Microtubules | Susceptibility to Proteasomal Degradation Mutant HSPB1 | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NTD | P7S | D | = | – | + | [337] | ||||||||||
| G34R | D | + | + | – | – | [336,350] | ||||||||||
| P39L | D | + | + | – | ± | + | [334,350,360] | |||||||||
| E41K | D | + | + | – | – | [336,350] | ||||||||||
| G53D | R | = | – | + | [337] | |||||||||||
| L58Afs*105 | D | = | + | [337,340] | ||||||||||||
| A61Rfs*100 | D | = | + | [337] | ||||||||||||
| G84R | D | + | – | + | – | (+) | [331,334,348] | |||||||||
| S86L | R | [342] | ||||||||||||||
| ACD | L99M | R | + | – | + | – | (+) | [331,334,348] | ||||||||
| R127W | D | + | – | + | – | + | + | – | + | + | [170,333,347,358] | |||||
| R127L | D | – | [365] | |||||||||||||
| Q128R | D | = | – | + | [337] | |||||||||||
| D129E | D | [345] | ||||||||||||||
| S135F | D | + | – | + | – | + | + | = | + | + | + | + | [170,333,347,356,358,360] | |||
| R136W | D | + | – | – | + | + | = | = | + | [170,333,347,358] | ||||||
| R140G | SD | ++/– | – | ≈ | – | – | + | + | (+) | [210,331,334,339,346,360] | ||||||
| K141Q | D | (+) | – | – | – | (+) | [331,346,368] | |||||||||
| T151I | D | = | = | = | = | = | [170,333,358] | |||||||||
| T164A | D | – | = | [349,369] | ||||||||||||
| M169Cfs*2 | D | = | – | – | [365] | |||||||||||
| CTD | T180I | D | ≈ | [335,349] | ||||||||||||
| P182S | D | + | + | = | – | + | (+) | [331,349,370] | ||||||||
| P182L | D | + | = | = | + | – | + | = | [170,333,351,358] | |||||||
| S187L | D | = | + | – | [337] | |||||||||||
| R188W | D | – | – | = | [336,349] | |||||||||||
| A204Gfs*6 | D | + | – | [343] |
| Mutation 1 | Phenotype | Effects | Ref |
|---|---|---|---|
| c.21 G>T (p.R7S) | dHMN | slightly altered oligomerization | [378,392] |
| p.L34Ffs*50 (p.A33Afs*50 in [389]) | myopathy | unstable protein, loss of HSPB2 regulation | [389] |
| p.R116P | myopathy with axonal neuropathy | aggregation, loss of HSPB2 interaction and regulation | [389] |
| c.352T>C (p.Y118H) | CMT2 | not determined, likely loss of HSPB2 interaction and regulation | [393] |
| Mutation 1 | Phenotype 2 | Self-Interaction | HSPB1 Interaction | CRYAB Interaction | BAG3 Interaction | In Vitro Chaperone Act. | In Vivo Chaperone Act. | Aggregation | Cytotoxicity | References |
|---|---|---|---|---|---|---|---|---|---|---|
| P90L | NP | = | (–) | (+) | [337] | |||||
| N138T | NP | = | (–) | (+) | [337] | |||||
| K141E | NP/NMP | + | + | + | (–) | +/– | – | + | ++ | [20,397,411,433,435,438] |
| K141M | NP | + | (–) | (+) | [337] | |||||
| K141T | NP | [437] | ||||||||
| K141N | NP | ++ | ++ | + | +/– | – | +/(–) | + | [20,337,396,397,411,433,435,448] | |
| Q170Gfs*45 | MP | [439] | ||||||||
| P173Sfs*43 | NMP | [438] |
| Mutation 1 | Phenotype 2 | References |
|---|---|---|
| c.211C>T (p.R71W) | DCM | [517] |
| c.268C>T (p.R90*) | DCM | [517] |
| c.326A>G (p.H109R) | DCM | [517] |
| c.367C>T (p.R123*) | DCM | [517] |
| delEx3-4 | DCM | [518] |
| delEx4 | DCM | [517] |
| c.625C>T (p.P209S) | CMT | [519,520] |
| c.626C>T (p.P209L) | MFM | [464,505,506,507,508,509,510,511,512,513,514] |
| c.626C>A (p.P209Q) | MFM | [503,504] |
| c.652C>T (p.R218W) | DCM | [521,522] |
| c.652delC (p.R218Gfs*89) | DCM | [517] |
| c.727delC (p.H243Tfs*64) | DCM | [523] |
| c.752delA (p.Q251Rfs*56) | DCM | [524] |
| c.784G>A (p.A262T) | DCM | [517] |
| c.913delC (p.M306*) | DCM | [525] |
| c.925C>T (p.R309*) | DCM | [524] |
| c.1055delC (p.Q353Rfs*10) | DCM | [518] |
| c.1135delG (p.G379Afs*45) | DCM | [518] |
| c.1153_1160delTCTTCCCC (p.S385Qfs*56) | DCM | [524] |
| c.1181_1182delGA (p.R395fs*48) | DCM | [524] |
| c.1353C>A (p.T451*) | DCM | [518] |
| c.1363G>A (p.E455K) | DCM | [518,524] |
| c.1385T>C (p.L462P) | DCM | [521] |
| c. 1402G>A (p.V468M) | DCM | [524] |
| c.1408C>T (p.P470S) | MFM | [106] |
| c.1430G>A (p.R477H) | DCM | [517] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarparanta, J.; Jonson, P.H.; Kawan, S.; Udd, B. Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results. Int. J. Mol. Sci. 2020, 21, 1409. https://doi.org/10.3390/ijms21041409
Sarparanta J, Jonson PH, Kawan S, Udd B. Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results. International Journal of Molecular Sciences. 2020; 21(4):1409. https://doi.org/10.3390/ijms21041409
Chicago/Turabian StyleSarparanta, Jaakko, Per Harald Jonson, Sabita Kawan, and Bjarne Udd. 2020. "Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results" International Journal of Molecular Sciences 21, no. 4: 1409. https://doi.org/10.3390/ijms21041409
APA StyleSarparanta, J., Jonson, P. H., Kawan, S., & Udd, B. (2020). Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results. International Journal of Molecular Sciences, 21(4), 1409. https://doi.org/10.3390/ijms21041409