Early Pro-Inflammatory Remodeling of HDL Proteome in a Model of Diet-Induced Obesity: 2H2O-Metabolic Labeling-Based Kinetic Approach


Abstract

1. Introduction
2. Results
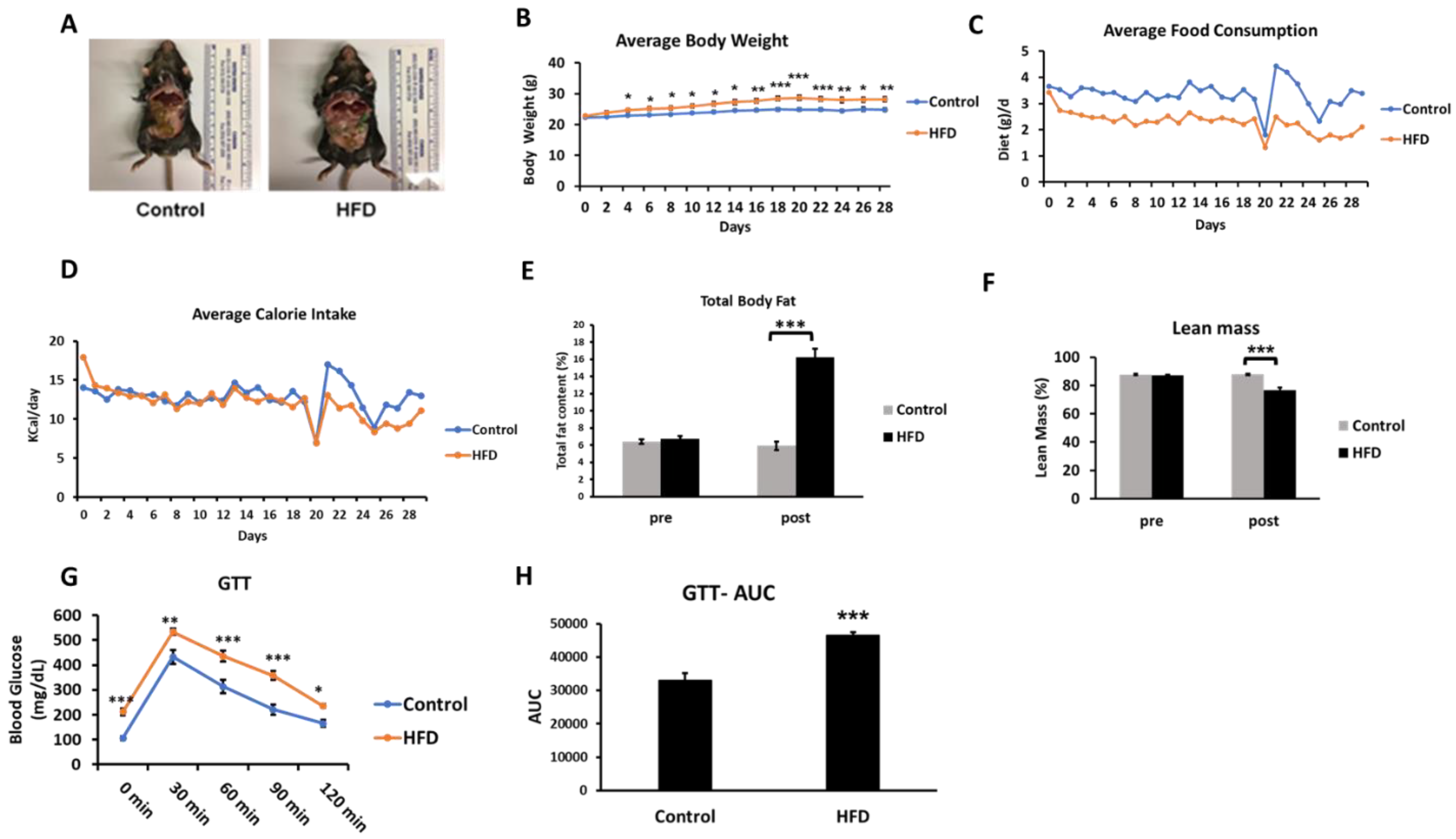
2.1. HFD Induces Partial Perturbation of Metabolic Profile at Four Weeks of Feeding
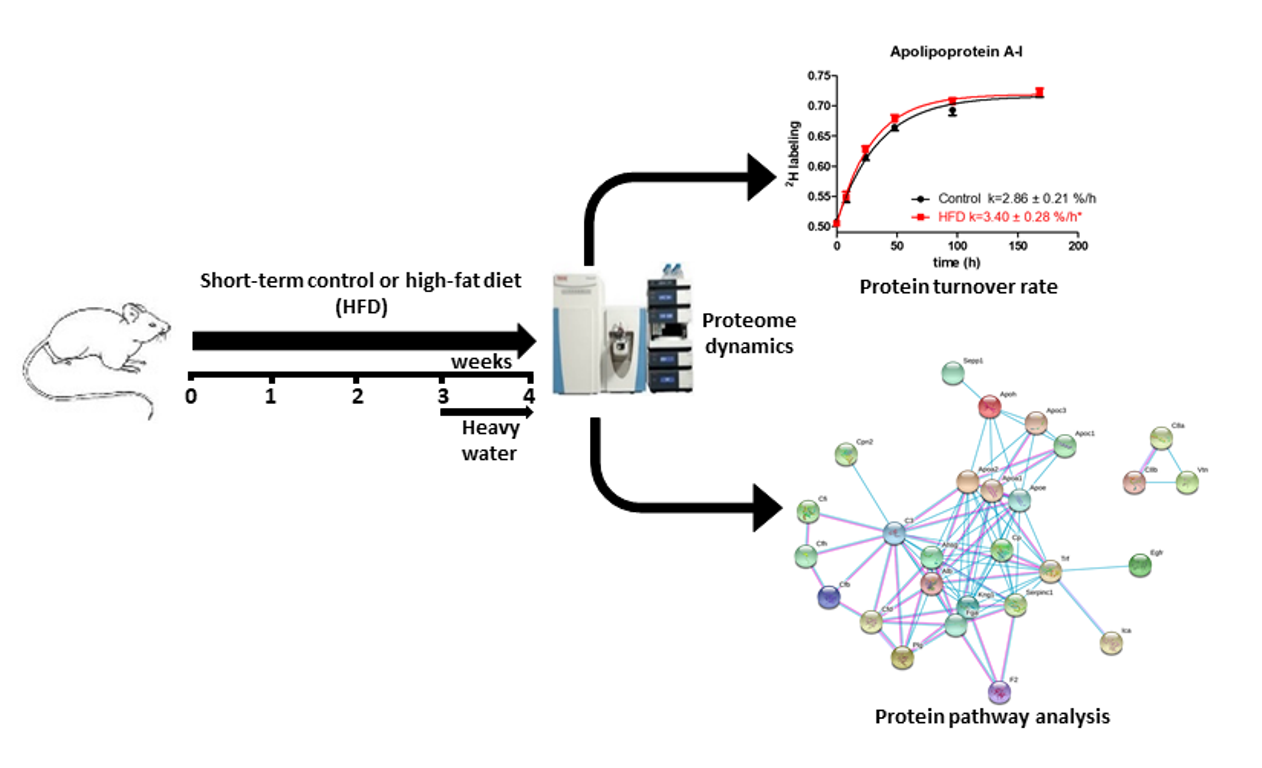
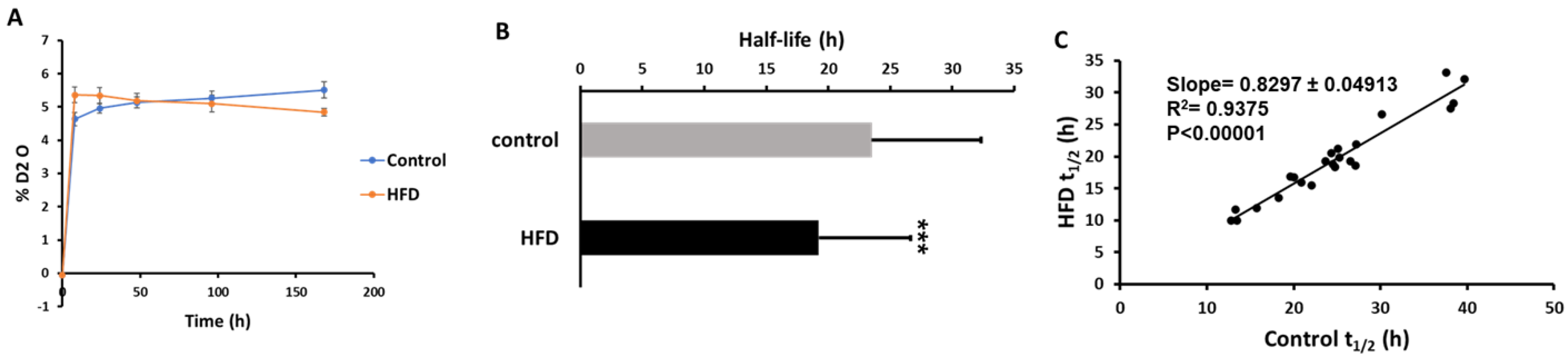
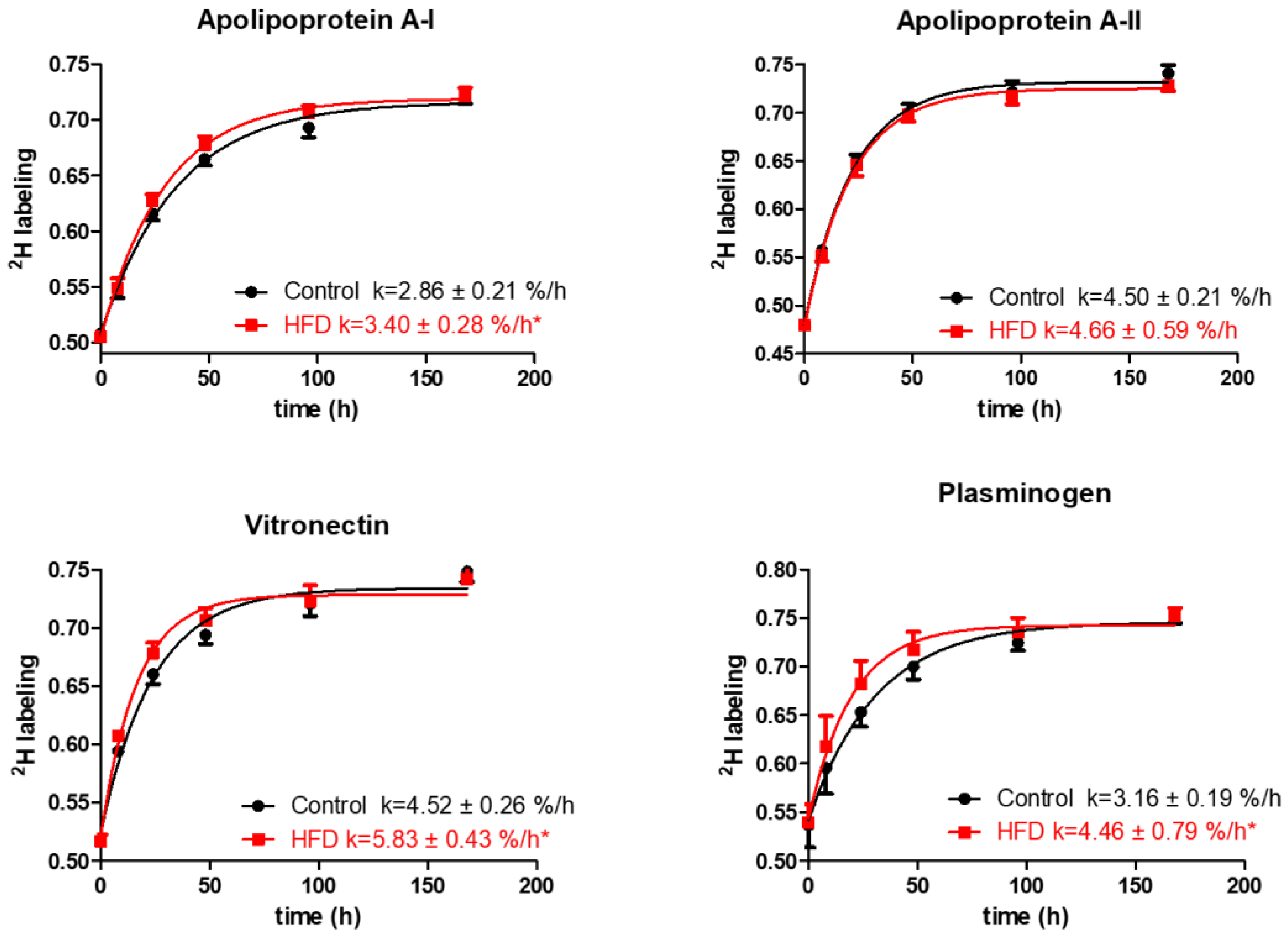
2.2. Effect of Short-Term HFD on Plasma HDL Proteome Dynamics
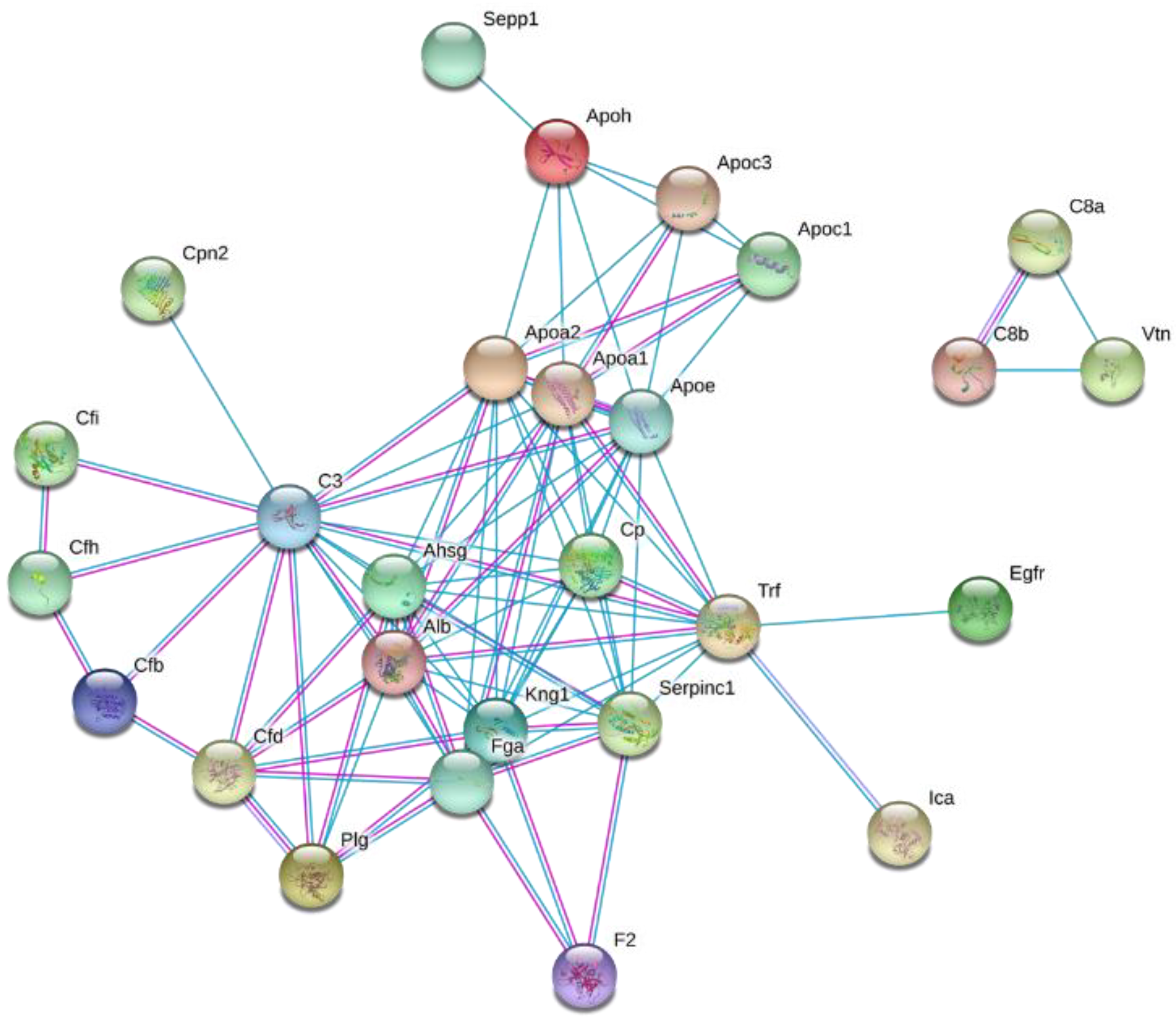
2.3. Pathways Associated with the HFD-Induced Alterations in Plasma HDL Proteome Dynamics
3. Discussion
4. Materials and Methods
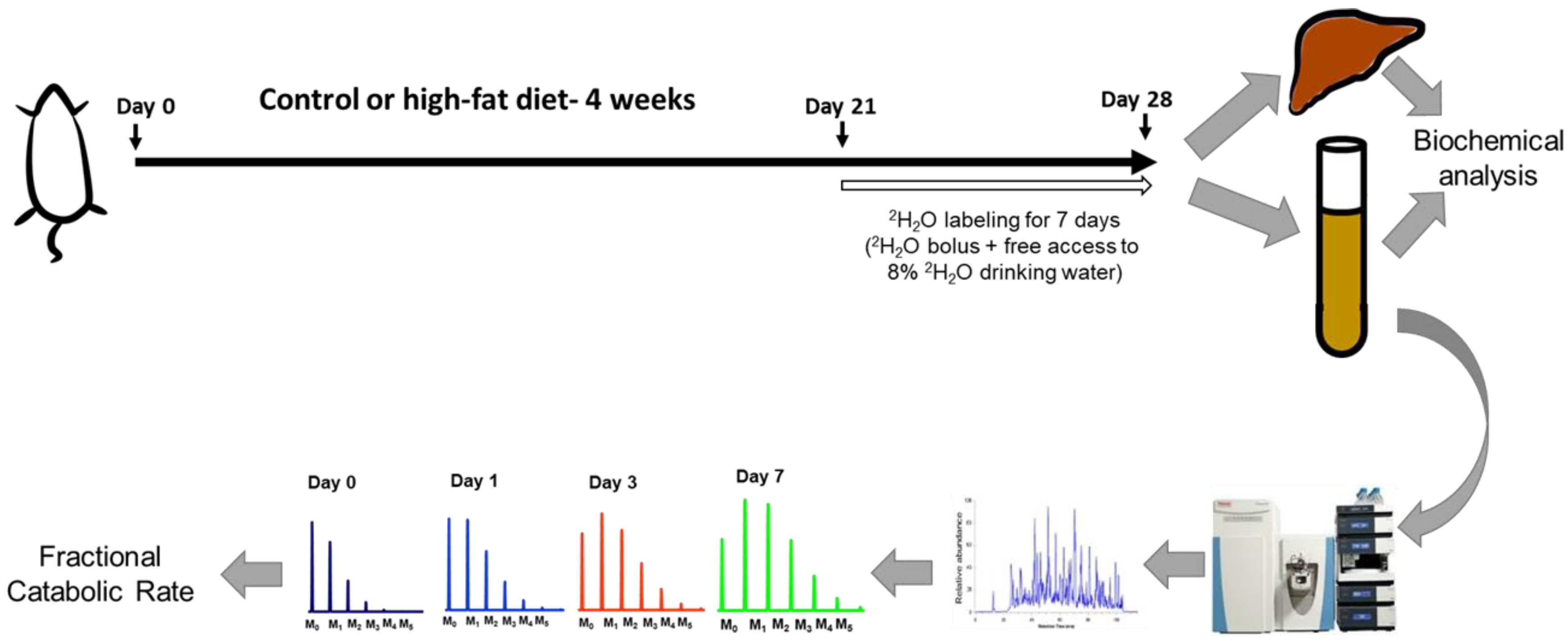
4.1. Animal Experiments
4.2. In Vivo LPL Activity
4.3. Lipid Measurements
4.4. RNA Isolation, cDNA Synthesis, and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5. Total Body Water Enrichment Measurements
4.6. Isolation of HDL Proteins
4.7. Tryptic Digestion of Proteins
4.8. Tandem Mass Spectrometry Analysis
4.9. Proteome Dynamics Analysis
4.10. Turnover Rate Constant and Half-Life Calculation
4.11. Network Analysis
4.12. Data Accession
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Atawia, R.T.; Bunch, K.L.; Toque, H.A.; Caldwell, R.B.; Caldwell, R.W. Mechanisms of obesity-induced metabolic and vascular dysfunctions. Front. Biosci. 2019, 24, 890–934. [Google Scholar]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.K. Emerging risk biomarkers in cardiovascular diseases and disorders. J. Lipids 2015, 2015, 971453. [Google Scholar] [CrossRef]
- Thomas, M.R.; Lip, G.Y. Novel risk markers and risk assessments for cardiovascular disease. Circ. Res. 2017, 120, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Liao, J.K. A mouse model of diet-induced obesity and insulin resistance. Methods Mol. Biol. 2012, 821, 421–433. [Google Scholar]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef]
- Ronsein, G.E.; Vaisar, T. Deepening our understanding of HDL proteome. Expert Rev. Proteom. 2019, 16, 749–760. [Google Scholar] [CrossRef]
- Ronsein, G.E.; Vaisar, T. Inflammation, remodeling, and other factors affecting HDL cholesterol efflux. Curr. Opin. Lipidol. 2017, 28, 52–59. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Effect of inflammation on HDL structure and function. Curr. Opin. Lipidol. 2016, 27, 521–530. [Google Scholar] [CrossRef]
- Jovicic, N.; Jeftic, I.; Jovanovic, I.; Radosavljevic, G.; Arsenijevic, N.; Lukic, M.L.; Pejnovic, N. Differential immunometabolic phenotype in Th1 and Th2 dominant mouse strains in response to high-fat feeding. PLoS ONE 2015, 10, e0134089. [Google Scholar] [CrossRef] [PubMed]
- Hariri, N.; Thibault, L. High-fat diet-induced obesity in animal models. Nutr. Res. Rev. 2010, 23, 270–299. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, M.; Rim, J.S.; Koza, R.A.; Kozak, L.P. Variation in type 2 diabetes-related traits in mouse strains susceptible to diet-induced obesity. Diabetes 2003, 52, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Coppey, L.J.; Davidson, E.P.; Yorek, M.A. Rat models of diet-induced obesity and high fat/low dose streptozotocin type 2 diabetes: Effect of reversal of high fat diet compared to treatment with enalapril or menhaden oil on glucose utilization and neuropathic endpoints. J. Diabetes Res. 2015, 2015, 307285. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A.; Woods, S.C. Overview of animal models of obesity. Curr. Protoc. Pharmacol. 2012, 58, 5.61.1–5.61.18. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.M.; Campbell, F.M.; Drew, J.E.; Koch, C.; Hoggard, N.; Rees, W.D.; Kamolrat, T.; Thi Ngo, H.; Steffensen, I.L.; Gray, S.R.; et al. The development of diet-induced obesity and glucose intolerance in c57bl/6 mice on a high-fat diet consists of distinct phases. PLoS ONE 2014, 9, e106159. [Google Scholar] [CrossRef]
- Lai, Y.S.; Chen, W.C.; Kuo, T.C.; Ho, C.T.; Kuo, C.H.; Tseng, Y.J.; Lu, K.H.; Lin, S.H.; Panyod, S.; Sheen, L.Y. Mass-spectrometry-based serum metabolomics of a c57bl/6j mouse model of high-fat-diet-induced non-alcoholic fatty liver disease development. J. Agric. Food Chem. 2015, 63, 7873–7884. [Google Scholar] [CrossRef]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation reduces the stability of apoai and increases HDL dysfunction in diet-controlled type 2 diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- McCullough, A.; Previs, S.F.; Dasarathy, J.; Lee, K.; Osme, A.; Kim, C.; Ilchenko, S.; Lorkowski, S.W.; Smith, J.D.; Dasarathy, S.; et al. HDL flux is higher in patients with nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E852–E862. [Google Scholar] [CrossRef] [PubMed]
- Sadygov, R.G.; Avva, J.; Rahman, M.; Lee, K.; Ilchenko, S.; Kasumov, T.; Borzou, A. D2ome, software for in vivo protein turnover analysis using heavy water labeling and lc-ms, reveals remodeling of hepatic proteome dynamics in a mouse model of NAFLD. J. Proteome Res. 2018, 17, 3740–3748. [Google Scholar] [CrossRef]
- Kasumov, T.; Li, L.; Li, M.; Gulshan, K.; Kirwan, J.P.; Liu, X.; Previs, S.; Willard, B.; Smith, J.D.; McCullough, A. Ceramide as a mediator of non-alcoholic fatty liver disease and associated atherosclerosis. PLoS ONE 2015, 10, e0126910. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bebek, G.; Previs, S.F.; Smith, J.D.; Sadygov, R.G.; McCullough, A.J.; Willard, B.; Kasumov, T. Proteome dynamics reveals pro-inflammatory remodeling of plasma proteome in a mouse model of NAFLD. J. Proteome Res. 2016, 15, 3388–3404. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Haddad, A.; Osme, A.; Kim, C.; Borzou, A.; Ilchenko, S.; Allende, D.; Dasarathy, S.; McCullough, A.; Sadygov, R.G.; et al. Hepatic mitochondrial defects in a nonalcoholic fatty liver disease mouse model are associated with increased degradation of oxidative phosphorylation subunits. Mol. Cell Proteom. 2018, 17, 2371–2386. [Google Scholar] [CrossRef]
- Fajardo, R.J.; Karim, L.; Calley, V.I.; Bouxsein, M.L. A review of rodent models of type 2 diabetic skeletal fragility. J. Bone Miner. Res. 2014, 29, 1025–1040. [Google Scholar] [CrossRef]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 2012, 53, 51–65. [Google Scholar] [CrossRef]
- Hayek, T.; Ito, Y.; Azrolan, N.; Verdery, R.B.; Aalto-Setala, K.; Walsh, A.; Breslow, J.L. Dietary fat increases high density lipoprotein (HDL) levels both by increasing the transport rates and decreasing the fractional catabolic rates of HDL cholesterol ester and apolipoprotein (apo) a-i. Presentation of a new animal model and mechanistic studies in human apo a-i transgenic and control mice. J. Clin. Investig. 1993, 91, 1665–1671. [Google Scholar]
- Brunner, F.J.; Waldeyer, C.; Ojeda, F.; Salomaa, V.; Kee, F.; Sans, S.; Thorand, B.; Giampaoli, S.; Brambilla, P.; Tunstall-Pedoe, H.; et al. Application of non-HDL cholesterol for population-based cardiovascular risk stratification: Results from the multinational cardiovascular risk consortium. Lancet 2019, 394, 2173–2183. [Google Scholar] [CrossRef]
- Pan, X.; Zhang, Y.; Kim, H.G.; Liangpunsakul, S.; Dong, X.C. FOXO transcription factors protect against the diet-induced fatty liver disease. Sci. Rep. 2017, 7, 44597. [Google Scholar] [CrossRef]
- Davidson, W.S.; Heink, A.; Sexmith, H.; Melchior, J.T.; Gordon, S.M.; Kuklenyik, Z.; Woollett, L.; Barr, J.R.; Jones, J.I.; Toth, C.A.; et al. The effects of apolipoprotein B depletion on HDL subspecies composition and function. J. Lipid Res. 2016, 57, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Kheniser, K.G.; Osme, A.; Kim, C.; Ilchenko, S.; Kasumov, T.; Kashyap, S.R. Temporal dynamics of high-density lipoprotein proteome in diet-controlled subjects with type 2 diabetes. Biomolecules 2020, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Willard, B.; Li, L.; Li, M.; Conger, H.; Buffa, J.A.; Previs, S.; McCullough, A.; Hazen, S.L.; Smith, J.D. 2H2O-based high-density lipoprotein turnover method for the assessment of dynamic high-density lipoprotein function in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; Tan, L.; Long, J.L.; Davidson, W.S. Proteomic diversity of high density lipoproteins: Our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 2013, 54, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Preissner, K.T.; Reuning, U. Vitronectin in vascular context: Facets of a multitalented matricellular protein. Semin. Thromb. Hemost. 2011, 37, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Ekmekci, O.B.; Ekmekci, H. Vitronectin in atherosclerotic disease. Clin. Chim. Acta 2006, 368, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Salama, Y.; Takahashi, S.; Osada, T.; Hattori, K. The multifaceted role of plasminogen in inflammation. Cell Signal. 2020, 75, 109761. [Google Scholar] [CrossRef]
- Heller, M.; Stalder, D.; Schlappritzi, E.; Hayn, G.; Matter, U.; Haeberli, A. Mass spectrometry-based analytical tools for the molecular protein characterization of human plasma lipoproteins. Proteomics 2005, 5, 2619–2630. [Google Scholar] [CrossRef]
- Vaisar, T.; Pennathur, S.; Green, P.S.; Gharib, S.A.; Hoofnagle, A.N.; Cheung, M.C.; Byun, J.; Vuletic, S.; Kassim, S.; Singh, P.; et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Investig. 2007, 117, 746–756. [Google Scholar] [CrossRef]
- Davidson, W.S.; Silva, R.A.; Chantepie, S.; Lagor, W.R.; Chapman, M.J.; Kontush, A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 870–876. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The string database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Willard, B.; Rachdaoui, N.; Kirwan, J.P.; Sadygov, R.G.; Stanley, W.C.; Previs, S.; McCullough, A.J.; Kasumov, T. Plasma proteome dynamics: Analysis of lipoproteins and acute phase response proteins with 2H2O metabolic labeling. Mol. Cell Proteom. 2012, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.Y.; Zhao, Y.; Heng, C.K. Sequential responses to high-fat and high-calorie feeding in an obese mouse model. Obesity (Silver Spring) 2008, 16, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wang, M.Y.; Kakuma, T.; Wang, Z.W.; Babcock, E.; McCorkle, K.; Higa, M.; Zhou, Y.T.; Unger, R.H. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J. Biol. Chem. 2001, 276, 5629–5635. [Google Scholar] [CrossRef]
- Sun, B.; Jia, Y.; Hong, J.; Sun, Q.; Gao, S.; Hu, Y.; Zhao, N.; Zhao, R. Sodium butyrate ameliorates high-fat-diet-induced non-alcoholic fatty liver disease through peroxisome proliferator-activated receptor alpha-mediated activation of beta oxidation and suppression of inflammation. J. Agric. Food Chem. 2018, 66, 7633–7642. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; de Beer, M.C.; Noffsinger, V.; Tannock, L.R.; Ramaiah, C.; Webb, N.R.; van der Westhuyzen, D.R.; de Beer, F.C. Hdl remodeling during the acute phase response. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 261–267. [Google Scholar] [CrossRef]
- Navab, M.; Hama-Levy, S.; Van Lenten, B.J.; Fonarow, G.C.; Cardinez, C.J.; Castellani, L.W.; Brennan, M.L.; Lusis, A.J.; Fogelman, A.M.; La Du, B.N. Mildly oxidized ldl induces an increased apolipoprotein j/paraoxonase ratio. J. Clin. Investig. 1997, 99, 2005–2019. [Google Scholar] [CrossRef]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum amyloid a impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef]
- Barbu, A.; Hamad, O.A.; Lind, L.; Ekdahl, K.N.; Nilsson, B. The role of complement factor c3 in lipid metabolism. Mol. Immunol. 2015, 67, 101–107. [Google Scholar] [CrossRef]
- Engstrom, G.; Hedblad, B.; Eriksson, K.F.; Janzon, L.; Lindgarde, F. Complement. c3 is a risk factor for the development of diabetes: A population-based cohort study. Diabetes 2005, 54, 570–575. [Google Scholar] [CrossRef]
- Kolev, M.; Kemper, C. Keeping it all going-complement meets metabolism. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Onat, A.; Hergenc, G.; Can, G.; Kaya, Z.; Yuksel, H. Serum complement c3: A determinant of cardiometabolic risk, additive to the metabolic syndrome, in middle-aged population. Metabolism 2010, 59, 628–634. [Google Scholar]
- Hertle, E.; Stehouwer, C.D.; van Greevenbroek, M.M. The complement system in human cardiometabolic disease. Mol. Immunol. 2014, 61, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Arisqueta, L.; Navarro-Imaz, H.; Labiano, I.; Rueda, Y.; Fresnedo, O. High.-fat diet overfeeding promotes nondetrimental liver steatosis in female mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G772–G780. [Google Scholar] [CrossRef]
- Wolf, G. High-fat, high-cholesterol diet raises plasma HDL cholesterol: Studies on the mechanism of this effect. Nutr. Rev. 1996, 54, 34–35. [Google Scholar] [CrossRef]
- Siri-Tarino, P.W. Effects of diet on high-density lipoprotein cholesterol. Curr. Atheroscler. Rep. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Lackey, D.E.; Olefsky, J.M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 2016, 12, 15–28. [Google Scholar] [CrossRef]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef]
- Grant, R.W.; Dixit, V.D. Mechanisms of disease: Inflammasome activation and the development of type 2 diabetes. Front. Immunol. 2013, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fu, J. Novel insights into the nlrp 3 inflammasome in atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [PubMed]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid a-mediated reactive oxygen species generation and nlrp3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, H.; Taheri, S.; Mondy, W.; Bonilha, L.; Magwood, G.S.; Lackland, D.; Adams, R.J.; Kindy, M.S. Serum amyloid a-mediated inflammasome activation of microglial cells in cerebral ischemia. J. Neurosci. 2019, 39, 9465–9476. [Google Scholar] [CrossRef]
- Bian, F.; Yang, X.Y.; Xu, G.; Zheng, T.; Jin, S. CRP-induced NLRP3 inflammasome activation increases ldl transcytosis across endothelial cells. Front. Pharmacol. 2019, 10, 40. [Google Scholar] [CrossRef]
- Hwang, L.L.; Wang, C.H.; Li, T.L.; Chang, S.D.; Lin, L.C.; Chen, C.P.; Chen, C.T.; Liang, K.C.; Ho, I.K.; Yang, W.S.; et al. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring) 2010, 18, 463–469. [Google Scholar] [CrossRef]
- Pettersson, U.S.; Walden, T.B.; Carlsson, P.O.; Jansson, L.; Phillipson, M. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory t cell population in adipose tissue. PLoS ONE 2012, 7, e46057. [Google Scholar] [CrossRef]
- Tower, J.; Pomatto, L.C.D.; Davies, K.J.A. Sex differences in the response to oxidative and proteolytic stress. Redox Biol. 2020, 31, 101488. [Google Scholar] [CrossRef]
- Congdon, E.E. Sex differences in autophagy contribute to female vulnerability in alzheimer’s disease. Front. Neurosci. 2018, 12, 372. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Mahsut, A.; Wang, S.P.; McLaren, D.G.; Bhat, G.; Herath, K.; Miller, P.L.; Hubbard, B.K.; Johns, D.G.; Previs, S.F.; Roddy, T.P. Headspace analyses of 2H labeling of acetone: Enabling studies of fatty acid oxidation in vivo. Anal. Biochem. 2011, 408, 351–353. [Google Scholar] [CrossRef]
- Walsh, M.T.; Atkinson, D. Solubilization of low-density lipoprotein with sodium deoxycholate and recombination of apoprotein b with dimyristoylphosphatidylcholine. Biochemistry 1983, 22, 3170–3178. [Google Scholar] [CrossRef]
- Kasumov, T.; Ilchenko, S.; Li, L.; Rachdaoui, N.; Sadygov, R.G.; Willard, B.; McCullough, A.J.; Previs, S. Measuring protein synthesis using metabolic 2H labeling, high-resolution mass spectrometry, and an algorithm. Anal. Biochem. 2011, 412, 47–55. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the pride database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Diet | High-Fat Diet | p-Value | |
|---|---|---|---|
| Body weight (g) | 25.4 ± 0.4 | 28.2 ± 0.7 | <0.005 |
| Liver weight (g) | 1.04 ± 0.03 | 0.85 ± 0.02 | <0.005 |
| Liver weight/Body weight (%) | 4.27 ± 0.10 | 3.01 ± 0.06 | <0.005 |
| Blood glucose (mg/dL) | 106.5 ± 7.5 | 211.7 ± 13.5 | <0005 |
| Plasma TG (mg/dL) | 35.5 ± 2.1 | 36.7 ± 2.4 | 0.718 |
| Plasma LPL activity (nmol FFA/min/mL) | 362.6 ± 11.5 | 312.6 ± 7.7 | <0.005 |
| Plasma TC (mg/dL) | 87.3 ± 2.7 | 139.9 ± 6.3 | <0.005 |
| Plasma HDL-C (mg/dL) | 69.9 ± 3.0 | 97.3 ± 6.5 | <0.005 |
| Plasma non-HDL-C (mg/dL) (calculated) | 17.5 ± 4.2 | 49.3 ± 10.9 | <0.05 |
| TC/HDL-C ratio | 1.26 ± 0.06 | 1.54 ± 0.14 | 0.120 |
| Hepatic TG (μg/mg liver tissue) | 11.3 ± 0.4 | 14.3 ± 0.9 | <0.05 |
| Accession (Uniprot) | Protein | Molecular Weight (kDa) | Function # | Control | HFD |
|---|---|---|---|---|---|
| Mean t½ (hours ±SD) | Mean t½ (hours ±SD) | ||||
| Q546G4 | Albumin | 68.7 | Chaperone binding; DNA binding; drug binding; enzyme binding; fatty acid binding; identical protein binding | 76.93 ± 7.49 | 66.09 ± 6.13 * |
| P29699 | Alpha-2-HS-glycoprotein | 37.3 | Cysteine-type endopeptidase inhibitor activity; endopeptidase inhibitor activity; receptor signaling protein tyrosine kinase inhibitor activity | 20.92 ± 1.60 | 15.92 ± 2.74 * |
| A0A0A6YWH7 | Antithrombin-III (fragment) | 52.0 | Heparin binding; peptidase inhibitor activity; protease binding; serine-type endopeptidase inhibitor activity | 24.77 ± 2.13 | 18.34 ± 1.16 * |
| Q3V2G1 | Apolipoprotein A-I | 30.6 | Amyloid-beta binding; apolipoprotein A-I receptor binding; apolipoprotein receptor binding; chemorepellent activity; cholesterol binding | 24.33 ± 1.76 | 20.51 ± 1.64 * |
| Q6LD55 | Apolipoprotein A-II | 11.3 | Apolipoprotein receptor binding; cholesterol binding; cholesterol transporter activity; heat shock protein binding; high-density lipoprotein particle binding | 15.43 ± 0.69 | 15.09 ± 1.68 |
| Q6LAL7 | Beta-2-glycoprotein 1 | 38.6 | Heparin binding; identical protein binding; lipid binding; phospholipid binding | 30.88 ± 3.46 | 20.75 ± 4.67 * |
| Q9DBB9 | Carboxypeptidase N subunit 2 | 60.5 | Enzyme regulator activity; metallocarboxypeptidase activity | 30.14 ± 2.26 | 26.60 ± 2.00 * |
| P01027 | Complement C3 | 186.5 | C5L2 anaphylatoxin chemotactic receptor binding; cofactor binding; endopeptidase inhibitor activity; lipid binding; protein binding | 12.77 ± 0.81 | 9.98 ± 1.20 * |
| Q3UEG8 | Complement Factor B | 85.5 | Complement binding; protein binding; serine-type endopeptidase activity | 12.71 ± 1.01 | 10.85 ± 1.04 |
| D6RGQ0 | Complement Factor H | 139.1 | Complement component C3b binding; heparan sulfate proteoglycan binding; heparin binding; protein binding | 19.60 ± 1.84 | 16.80 ± 1.58 * |
| Q61129 | Complement Factor I | 67.3 | Hydrolase activity; metal ion binding; peptidase activity; scavenger receptor activity; serine-type endopeptidase activity | 25.25 ± 1.29 | 19.88 ± 1.50 * |
| Q91X72 | Hemopexin | 51.3 | Cellular iron ion homeostasis; heme metabolic process; heme transport; hemoglobin metabolic process; positive regulation of humoral immune response mediated by circulating immunoglobulin | 39.82 ± 6.57 | 39.92 ± 9.65 |
| A0A0R4J039 | Histidine-rich Glycoprotein | 59.6 | Cysteine-type endopeptidase inhibitor activity; heme binding; heparan sulfate proteoglycan binding; heparin binding; immunoglobulin binding | 23.63 ± 1.99 | 19.28 ± 2.83 * |
| A0A0R4J038 | Kininogen-1 | 73.1 | Cysteine-type endopeptidase inhibitor activity; peptidase inhibitor activity; receptor binding | 25.09 ± 1.59 | 21.18 ± 0.91 * |
| P28665 | Murinoglobulin-1 | 165.3 | Endopeptidase inhibitor activity; peptidase inhibitor activity; serine-type endopeptidase inhibitor activity | 39.67 ± 3.22 | 32.08 ± 1.73 * |
| Q3V1T9 | Plasminogen | 90.8 | Apolipoprotein binding; chaperone binding; endopeptidase activity; enzyme binding; hydrolase activity | 22.04 ± 1.25 | 15.45 ± 3.42 * |
| Q61838 | Pregnancy Zone Protein | 165.8 | Brain-derived neurotrophic factor binding; endopeptidase inhibitor activity; nerve growth factor binding; peptidase inhibitor activity; protein complex binding | 41.71 ± 3.50 | 38.91 ± 2.29 |
| Q07456 | Protein AMBP | 39.0 | Heme binding; IgA binding; peptidase inhibitor activity; protein homodimerization activity; serine-type endopeptidase inhibitor activity | 14.23 ± 1.97 | 13.21 ± 1.09 |
| Q3TJ94 | Prothrombin | 70.3 | Calcium ion binding; heparin binding; hydrolase activity; lipopolysaccharide binding; peptidase activity | 27.20 ± 6.04 | 21.87 ± 4.66 * |
| Q921I1 | Serotransferrin | 77.0 | Ferric iron binding; ferric iron transmembrane transporter activity; ferrous iron binding; protein binding | 37.62 ± 1.74 | 33.12 ± 6.63 * |
| P21614 | Vitamin D-binding Protein | 53.6 | Actin binding; vitamin D binding; vitamin transporter activity | 13.48 ± 0.50 | 10.00 ± 0.84 * |
| G3X8Q5 | Ceruloplasmin | 121.1 | Chaperone binding; copper ion binding; ferroxidase activity; metal ion binding; oxidoreductase activity | 27.10 ± 6.12 | 18.55 ± 3.41 * |
| P34928 | Apolipoprotein C-I | 9.3 | Fatty acid binding; lipase inhibitor activity; phosphatidylcholine binding; phospholipase inhibitor activity | 17.66 ± 6.52 | 10.39 ± 8.06 |
| P33622 | Apolipoprotein C-III | 10.9 | Lipase inhibitor activity; lipid binding; phospholipid binding | 10.16 ± 1.57 | 11.30 ± 2.18 |
| P08226 | Apolipoprotein E | 35.9 | Amyloid-beta binding; antioxidant activity; cholesterol binding; cholesterol transporter activity; heparin binding | 10.50 ± 5.53 | 8.09 ± 1.95 |
| A2A997 | Complement component C8 Alpha Chain | 66.1 | Complement binding; protein complex binding | 26.50 ± 2.22 | 19.24 ± 3.85 * |
| Q9QWK4 | CD5 Antigen-Like | 38.9 | Scavenger receptor activity | 32.66 ± 8.35 | 28.42 ± 3.43 |
| Q8BH35 | Complement component C8 beta chain | 66.2 | Constituent of the membrane attack complex (MAC) that plays a key role in the innate and adaptive immune response by forming pores in the plasma membrane of target cells | 24.51 ± 4.68 | 18.83 ± 1.95 * |
| Q9EP98 | Epidermal growth factor receptor | 138.4 | Protein tyrosine kinase activity | 38.05 ± 3.82 | 27.58 ± 3.90 * |
| P03953 | Complement Factor D | 28.1 | actor D cleaves factor B when the latter is complexed with factor C3b, activating the C3bbb complex, which then becomes the C3 convertase of the alternate pathway. Its function is homologous to that of C1s in the classical pathway | 20.06 ± 1.19 | 16.71 ± 0.61 * |
| A0A075B5P6 | Ig mu chain C region (fragments) | 50.1 | Immunoglobin receptor binding | 44.95 ± 8.23 | 35.34 ± 11.15 |
| Q9DBD0 | Inhibitor of Carbonic Anhydrase | 76.7 | Inhibitor for carbonic anhydrase 2 (CA2). Does not bind iron ions | 18.24 ± 1.36 | 13.52 ± 0.41 * |
| P29788 | Vitronectin | 54.9 | Interact with glycosaminoglycans and proteoglycans. Is recognized by certain members of the integrin family and serves as a cell-to-substrate adhesion molecule. Inhibitor of the membrane-damaging effect of the terminal cytolytic complement pathway. | 15.71 ± 2.27 | 11.95 ± 0.88 * |
| E9PV24 | Fibrinogen alpha chain | 87.4 | 40.15 ± 7.35 | 41.78 ± 3.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadana, P.; Lin, L.; Aghayev, M.; Ilchenko, S.; Kasumov, T. Early Pro-Inflammatory Remodeling of HDL Proteome in a Model of Diet-Induced Obesity: 2H2O-Metabolic Labeling-Based Kinetic Approach. Int. J. Mol. Sci. 2020, 21, 7472. https://doi.org/10.3390/ijms21207472
Sadana P, Lin L, Aghayev M, Ilchenko S, Kasumov T. Early Pro-Inflammatory Remodeling of HDL Proteome in a Model of Diet-Induced Obesity: 2H2O-Metabolic Labeling-Based Kinetic Approach. International Journal of Molecular Sciences. 2020; 21(20):7472. https://doi.org/10.3390/ijms21207472
Chicago/Turabian StyleSadana, Prabodh, Li Lin, Mirjavid Aghayev, Serguei Ilchenko, and Takhar Kasumov. 2020. "Early Pro-Inflammatory Remodeling of HDL Proteome in a Model of Diet-Induced Obesity: 2H2O-Metabolic Labeling-Based Kinetic Approach" International Journal of Molecular Sciences 21, no. 20: 7472. https://doi.org/10.3390/ijms21207472
APA StyleSadana, P., Lin, L., Aghayev, M., Ilchenko, S., & Kasumov, T. (2020). Early Pro-Inflammatory Remodeling of HDL Proteome in a Model of Diet-Induced Obesity: 2H2O-Metabolic Labeling-Based Kinetic Approach. International Journal of Molecular Sciences, 21(20), 7472. https://doi.org/10.3390/ijms21207472