Abstract
Nitrones are a significant class of compounds highly useful in organic synthesis; in particular, they are key intermediates for the synthesis of new biologically active nitrogen compounds. The first part of this review aims to provide a structured and concise overview of nitrones, summarizing their synthetic methodologies and highlighting the environmentally friendly approaches. Their fundamental transformations and a thorough explanation of their reactivities are addressed, either in rearrangements to similar compounds or as fragments/intermediates for more complex molecules. Lastly, physicochemical properties, therapeutic potential, and industrial applications are also addressed. This review gives an update on the scientific discoveries in the field of nitrones, focusing on organizing the existing information and highlighting subtle details to enhance chemical comprehension.
1. Introduction
The nitrones (Figure 1) are a family of organic compounds known since 1890, when they were first synthesized by Beckmann [1]. Since then, lots of scientific material has been published and research into them is intense. They were first named by Pfeiffer in 1916, due to their similarity with ketones, in terms of reactivity [2]. Then, they became famous for their good enantioselectivity in 1,3-dipolar cycloadditions, and nowadays there is a huge repertoire of syntheses where they can be implemented as intermediates. Furthermore, the nitrones themselves can be applied as spin traps, and they have anti-oxidative properties, useful in the production of polymers and in biological systems [3].
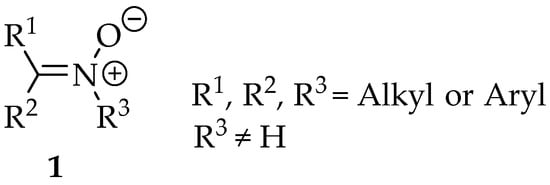
Figure 1.
Nitrone’s structure.
In early days, the nitrones were named according to their family’s name, with the substituents allocated to their α- or N- positions. For example, nitrone 2 (Figure 2) was named N,α diphenylnitrone 2 [4]. However, this nomenclature is no longer recommended by IUPAC, according to the rules in the blue book of 2013 [5]. It is even misleading, as the α-carbon should not be the sp2 carbon bound to the nitrogen, but the one next to it, like in ketones. The correct name, or the preferred IUPAC name (PIN), is now (Z)-N,1-diphenylmethanimine oxide 2. They are named after their imine analog, followed by the term oxide. For the structures 3 and 4, their names are (Z)-N-phenylpentan-2-imine oxide and 2,2,6 trimethyl 2,3,4,5 tetrahydropyridine oxide, respectively.
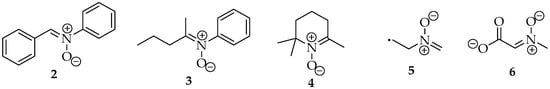
Figure 2.
Five nitrones’ examples: (Z)-N,1-diphenylmethanimine oxide 2; (Z)-N-phenylpentan-2-imine oxide 3; 2,2,6 trimethyl-2,3,4,5 tetrahydropyridine oxide 4; 2-(methylenoxo-λ5-azanyl)ethyl 5; and (Z)-2-(methyloxo-λ5-azanylidene)ethanoate 6.
In terms of priority among other compound classes, following IUPAC rules, they rank fifth because they are zwitterions [5]. Therefore, this group is named as substituent only when there are radicals or anions in the same structure. In those cases, the expression “oxo-λ5-azanyl” is used. For example, in structure 5, the preferred name should be 2-(methylenoxo-λ5-azanyl)ethyl, whereas for structure 6, the suffix -idene must be added, as the nitrone group is linked to the main chain by the double bond, so the name is (Z)-2-(methyloxo-λ5-azanylidene)ethanoate. When naming these compounds, one should not forget the diastereoisomerism of the double bond. Usually, they are more stable in the Z configuration due to steric effects from the substituents.
Finally, one distinction often made is between aldonitrones and ketonitrones (Scheme 1) [4]. In the first case, only one substituent is present in the sp2 carbon, whereas in the second case the nitrone is fully substituted. These designations should result from the reaction between hydroxylamine derivatives and aldehydes or ketones, respectively (Scheme 1).
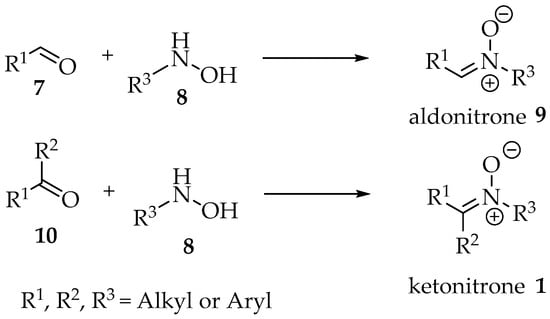
Scheme 1.
Reaction between hydroxylamine derivatives 8 and aldehydes 7 or ketones 10 as justification for the terms aldonitrone 9 and ketonitrone 1.
Some less recent reviews focus on the reactions of nitrones, particularly 1,3-dipolar cycloadditions and the synthesis of heterocyclic compounds [2,3,4,6,7,8,9,10], and the newest one is dedicated to describing the results of a single research group. This review aims to summarize that content while emphasizing mechanistic details, including the latest developments. It also clearly organizes the key aspects of nitrone chemistry. Additionally, there is a dedicated section discussing the applications of nitrones beyond organic synthesis.
2. Nitrone Synthesis
Various methods have been employed to prepare nitrones, which are hygroscopic products. These products can exist as oils, typically for aliphatic structures, or as solid compounds, generally for aromatic structures, and they have a transparent or white appearance for simpler products [11,12]. In the first case, there is even a trend for the nitrones to form dimers, though structures with higher complexity can also be formed (Figure 3), which can complicate the obtention of the desired compound [4,11].
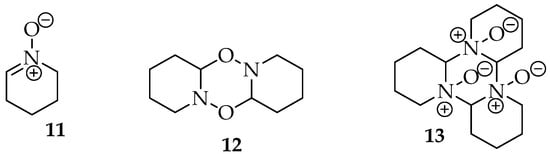
Figure 3.
Dimer 12 and trimer 13 of 2,3,4,5 tetrahydropyridine oxide 11, through C-O and C-N bonding, respectively [4].
2.1. Oxidations
One approach involves the oxidation of nitrogenous precursors, such as secondary amines, hydroxylamines, and imines (Scheme 2), with the use of hydroxylamines being the least used method due to the use of highly toxic reagents, such as mercuric oxide [3]. Nevertheless, a more environmentally friendly approach using sodium hypochlorite was successfully tested [13]. In the early days, lead and mercury oxides and potassium permanganate were commonly used as oxidizing agents, but an urge to lower the ecological footprint has led to the development of reactions using hydroperoxides or air’s oxygen, sometimes in the presence of catalysts [4].
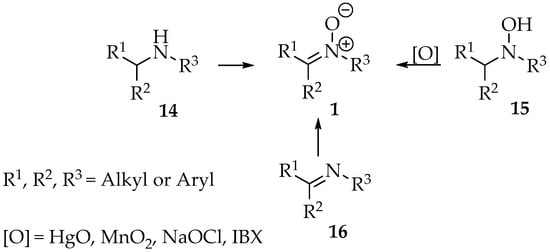
Scheme 2.
Nitrones 1 formation by oxidation of secondary amines 14, N,N dialkyl hydroxylamines 15 and imines 16.
In the case of hydroxylamine derivatives 15, various oxidants have been reported in the literature. Examples include t-butyl hydroperoxide, m-CPBA (3-chlorobenzene-1-carboperoxoic acid), airflow (catalyzed by Cu2+), and both palladium and mercury(II) oxide (Scheme 2) [3,12,13,14,15]. Although this method is not widely used, Goti and colleagues have developed several approaches that operate under mild conditions, primarily utilizing less hazardous catalytic systems [13,16,17,18,19]. It is worth highlighting the efficient use of poly(4-vinylpyridine)/MTO compounds or microencapsulated polystyrene/MTO systems as catalysts, which could be recovered and used with hydrogen peroxide, making this procedure an environmentally friendly one (Scheme 2) [19].
To better elucidate the redox chemistry phenomena in these reactions, Scheme 3A shows the process’s main steps. Although they are not exclusive, as the reactions depend on the oxidant reagents used, they are the most ubiquitous for hydroxylamine derivatives 15. In the case of the hydroxylamine derivatives 15, they lose the N-hydroxyl hydrogen, forming the nitroxide radical. The simplicity of this step determines the hydroxylamines’ anti-oxidative properties in their applications. Then, depending on the substituents’ hindrance, they may either form a dimer or proceed readily to their disproportionation. As a result, the nitrone product 1 is obtained from each of the two nitroxide radicals, and the hydroxylamine reagent 15 can be recovered (Scheme 3A) [20]. An illustrative example is the oxidation of isoxazolidine 17 (Scheme 3B) [21,22], whereas the isoxazolidine’s behavior is similar to that of hydroxylamine.
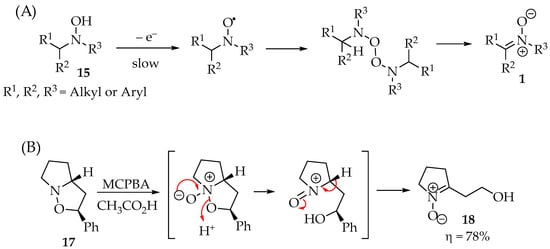
Scheme 3.
(A) Hydroxylamine 15 oxidation to nitrone 1; (B) isoxazolidine 17 oxidation into nitrone 18 and the respective mechanism.
Although isoxazolidine are heterocyclic structures commonly synthesized from nitrones, they can form new nitrones as well. The N-O bond can be cleaved with oxidants, producing γ-hydroxynitrones 18 (Scheme 3B). The reaction starts with the nucleophilic attack of the nitrogen to the oxidant species, such as m-CPBA, resulting in the isoxazolidine N-oxide. Then, the ring opens, forming a nitrosonium ion derivative that is released from an α-hydron, restores the molecular neutrality, and forms, selectively, the nitrone 18 in very good yields (Scheme 3B).
For amines and imines, DMD (dimethyldioxirane) and Oxone® have also been used [3,12]. Hydrogen peroxide is another option in an aqueous solution or a solid complex with urea. However, a catalyst is required to promote the organic compound oxidation instead of the peroxide disproportionation [23]. Sodium tungstate seems to provide the best results [23]. To guarantee the highest conversion, the oxidant is often present in significant excess, having to be removed/reduced at the reaction end. The photocatalytic oxidation of aldimines to nitrones by the O2/TiO2 system achieved good yields (>80%) in one study [24]. An efficient metal-free procedure, using only Oxone® as oxidant and a biphasic basic medium composed of acetonitrile and THF (4:1) and an aqueous of EDTA and NaHCO3 solution, was used to obtain nitrones from amines [25]. The yields were very good, and most importantly, other functional groups or existing stereogenic centers were tolerant to the conditions used.
The amines 14 are usually oxidized by losing one of the electrons in the electron pair, forming a radical cation (Scheme 4A). Then, it loses the α-hydrogen (or a hydron and an electron, depending on whether the mechanism is concerted or not) to produce the imine 16 [26,27]. In molecules with other α-nucleophiles, such as the carboxylic group in α-amino acids, they might be lost instead [9].
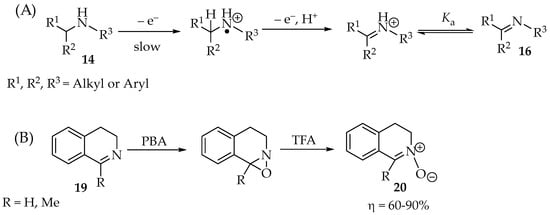
Scheme 4.
(A) Amine 14 oxidation to imine 16, and (B) example with further oxidation to nitrone 20.
The imines, such as 19 (Scheme 4B), are more acidic than the corresponding amines. Therefore, the following deionization happens to a good extent. Primary amines require stronger oxidants than secondary or tertiary amines. Their anodic (oxidation) potentials against Ag/0.1 M Ag+ (MeCN) are between 1.2 and 2.0 V for primary amines, between 0.82 and 1.08 V for secondary amines, and 0.71 V for the N,N-dimethylbenzylamine [26,28]. The imine 19 might be further oxidized by the same oxidant, given enough time and quantity of the PBA reagent, to an oxaziridine derivative (Scheme 4B). By acidification of the mixture, for example, with trifluoroacetic acid (TFA), the nitrone 20 is formed in moderate to excellent yields, depending on the solvent used (Scheme 4B). Sometimes, the intermediate compound converts readily to the nitrone without further processing [3,29,30].
2.2. Nucleophilic Substitutions
Nucleophilic substitutions of carbonyl compounds with N-alkyl/aryl hydroxylamines were one of the first methodologies implemented for nitrones syntheses and are still one of the most commonly applied nowadays. Oximes are also capable of nucleophilic substitutions with compounds with good leaving groups (Scheme 5).
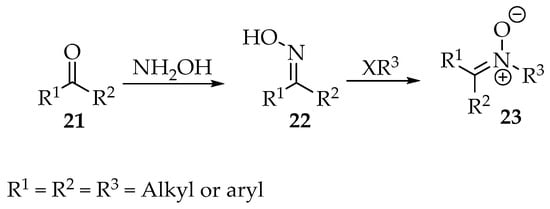
Scheme 5.
General synthesis of nitrones 23, through nucleophilic substitutions involving oximes 22.
Starting with N-alkyl/aryl hydroxylamines or simply hydroxylamine, as shown in Scheme 5, countless examples exist in the literature for their condensation with aldehydes and ketones. The hydroxylamine moiety can be prepared in situ from nitro compounds, the most sensitive compounds, to avoid degradation in air. As an example, the 3-(benzo[d][1,3]dioxol-5-yl)-4-nitro-1-phenylbutan-1-one 24 was reduced by zinc dust in acidic media, affording the terminal hydroxylamine 25, which readily condensed, intramolecularly, with the carbonyl to produce the cyclic 3,4-dihydro-2H-pyrrole-1-oxide derivative 26 (Scheme 6) [11].
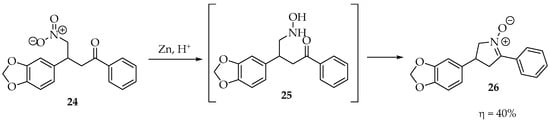
Scheme 6.
Example of an intramolecular cyclization between a ketone and a hydroxylamine formed in situ.
In the hope of finding a more sustainable methodology, Cisneros et al. tried a different, one-pot approach, using nanocatalysts for the selective hydrogenation of nitro groups, followed by the formation of hydroxylamines and cyclization into nitrones (Scheme 7) [31]. Excellent results were achieved with 0.2 wt% Pt/C at 308 K, using 5 bar hydrogen pressure as a reagent, even in the presence of alkenes and alkynes. Unfortunately, this system only shows selectivity in the case of nitroaryl compounds, having poor yields in the case of nitroalkyl compounds, such as in the cases of nitrones 30 and 31 (Scheme 7) [31].
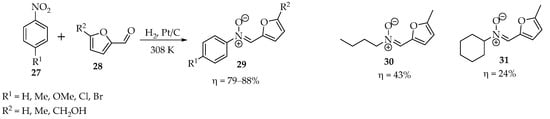
Scheme 7.
Example of a one-pot transformation of nitro compounds into nitrones.
The direct reaction between hydroxylamines and aldehydes using ball-milling was performed without solvent, with better results than microwave irradiation [32]. The frequencies were optimized to 30 Hz and hydrogen carbonate was used as a base to increase hydroxylamines’ nucleophilicity, because commercial hydroxylamines are acidified with hydrogen chloride to avoid evaporation [32]. From the three hydroxylamines used, the N-methylhydroxylamine needed to be recrystallized twice with AcOEt/EtOH at −20 °C prior to usage, which depicts the low stability of hydroxylamines. In the conventional methodology, the MgSO4 can be used as a desiccant, and the reaction is performed at room temperature in dichloromethane for some hours [33].
The scope of this reaction has been extended to imines, nitriles, and cyanohydrins—in the latter case, they must be converted to imines first (Scheme 8) [34]. That reduction is performed by diisobutylaluminium hydride (DIBAL) at −10 °C, followed by protonation with methanol. Finally, after adding a hydroxylamine derivative, the transamination takes about 4 h at room temperature.
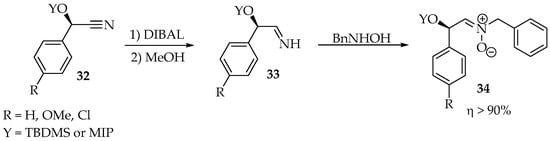
Scheme 8.
Cyanohydrins’ 32 reduction to imines 33, followed by transamination with N-benzylhydroxylamine to produce N-benzylnitrones 34.
Nucleophilic substitutions with oximes are less frequent, the main reason being the lower reaction yields. This functional group presents ambidexterity, attacking electrophilic species with nitrogen or oxygen atoms. Furthermore, it forms diastereoisomeric products [35].
The oxime structure 22 is a tautomeric equilibrium with the nitrone form 35, though the first is thermodynamically more stable (Scheme 9) [36]. As nitrogen is considerably more acidic in oximes than in hydroxylamines, the strength of the nucleophilic attack by this atom (nitrogen) should be inferior. Increasing the mixture’s pH will further improve the nucleophilicity of the oxygen [2]. In relation to the oxime, the E-oxime also favors substitution on the oxygen, when compared with the Z-conformer, mainly due to steric reasons [37]. The same reasoning explains the formation of oxime O-ether when ketoximes are used instead of aldoximes [36].
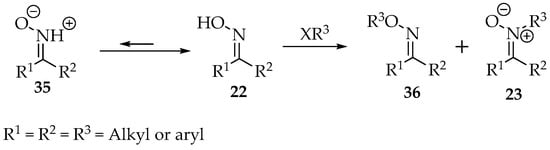
Scheme 9.
Oxime alkylation produces a mixture of oxime O-ether 36 and nitrone 23.
Furthermore, in the case of aryl oximes, the presence of electron-releasing groups yields more ether 36, whereas the electron-withdrawing groups produce more nitrone 23 (Scheme 9) [4]. Finally, the electrophile’s nature has another impact on the product ratio. Using iodide reagents makes the reaction faster and improves the nitrones’ yield. The bromine and chlorine substituents prefer the formation of ethers’ [35]. Fortunately, their separation seems easy, as they recrystallize the nitrone from an ether–alkane solvent mixture [37].
A particular system in which nitrones are obtained in good yields is the case of activated hydroxyl substituents (as leaving groups from the electrophile) (Scheme 10). This has been achieved by mesylating aldose oximes with methanesulfonyl chloride (MsCl) in pyridine [38]. An alternative is using Mitsunobu conditions: adding triphenylphosphine and diisopropyl azodicarboxylate reagents to the alcohol, to activate the hydroxyl group [35].
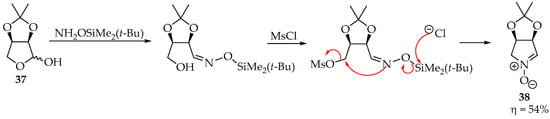
Scheme 10.
Example of a one-pot nitrone synthesis using NH2OSiMe2(t-Bu).
Although it is not a true nucleophilic substitution, nitrones have also been produced from oximes by Chan–Lam coupling with vinyl and aryl boronic acids [39]. The reaction showed good yields using copper(II) acetate as a catalyst when pyridine and 1,2-dichloroethane were used as solvents.
Finally, no interconversion between the oxime O-ether and the nitrone was observed when the reaction was left for a longer reaction time, suggesting good stability of both products when formed [35]. However, there is a special case in which it was achieved with a copper(I) catalyst [40].
2.3. Additions to Multiple Bonds
Besides substitutions, the nucleophilic hydroxylamines 8 and oximes 22 can be added to alkynes 39, allenes 41, or alkenes 43 to produce nitrone derivatives (Scheme 11).
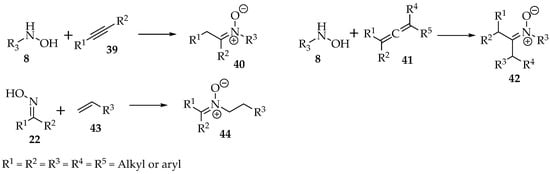
Scheme 11.
General examples of addition reactions of hydroxylamines 8 and oximes 22.
The first reaction using oximes addition to alkenes became famous with the studies of Grigg, hence receiving his name [3,41]. However, in Grigg’s synthesis, many side reactions may occur, like in the nucleophilic substitutions. The ambidexterity of oximes results in nitrones and oxime O-ethers, depending on the reaction conditions and the nature of the reagents. Furthermore, both the obtained nitrone derivatives can react with alkenes through 1,3-dipolar cycloadditions, producing heterocyclic compounds. Many examples showing this reactivity of nitrones can be found in the literature [42]. The examples provided by Grigg’s synthesis show that it is difficult to obtain the isolated nitrone. For instance, oxime 45 undergoes an intramolecular cyclisation to produce nitrone 46 in low yield and oxazine 47 in good yield (61%) (Scheme 12) [43].
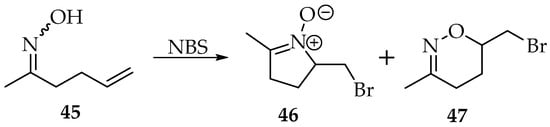
Scheme 12.
Example of Grigg’s intramolecular synthesis.
From Grigg’s studies, it became clear that the alkene should contain an electron-withdrawing group or be activated by an electrophile or a Lewis acid [41,43]. For example, in the fortunate experiment represented in Scheme 13, iodine was used as the electrophile, and the nitrone 48 was obtained as the sole product, although isolated in its salt form. This was lucky because, as explained in the previous section, the iodine tends to form nitrone products, and the simple oxime does not cause substantial steric hindrance. In a more recent study, it was calculated that the transition state’s energy could be halved when bases are added, such as potassium tert-butoxide [44]. The base’s cation has a high impact on stabilizing the transition state’s negative charges.
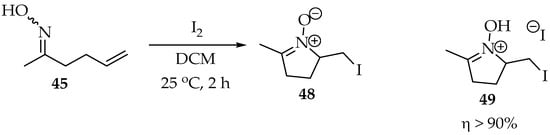
Scheme 13.
Intramolecular Grigg’s synthesis, using iodine as an electrophile to activate the double bond.
Finding suitable Lewis’s acids for this reaction can be arduous. In another study, in which several aldoximes were reacted with α,β-unsaturated carbonyl compounds, the best combination of Lewis acids was the proportion of 1:1 ZnI2/BF3•OEt2 (Scheme 14) [45]. When only zinc iodide was tried, a mixture with the oxime O-ether was formed, whereas the reaction was not completed with only the second component. According to Roca-López et al. [36], the nitrone is formed when there is an orthogonal disposition of the oxime with the alkene moiety.
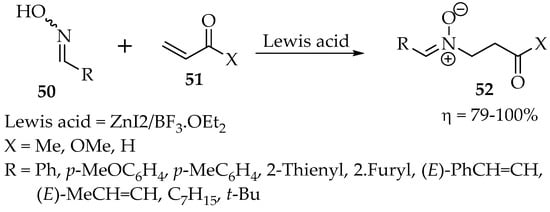
Scheme 14.
Nitrones synthesis through the reaction of oximes with α,β-unsaturated carbonyl compounds.
One final discovery was presented in the reaction of ketoximes with cyclopropenes, which had high enantioselectivities and no ring-opening side products. A CuCl Lewis acid with different bulky ligands was used at 5 mol%, along with sodium tert-butoxide (30 mol%) under an inert atmosphere [46].
Depending on the reagents used, the addition mechanism can be either a nucleophilic attack or radical formation. The second case is rarer, however, successfully achieved for the first time by Han et al. [47]. The radical initiator was diethyl diazenedicarboxylate, and 5-exo-trig selectivity was evident. Additions through both nitrogen and oxygen are possible, with the product favored by the previous selectivity. For the intramolecular reaction of hydroxylamine derivatives with alkynes, cyclic nitrones 53 are produced, and their ring members are between five and seven (Scheme 15). In the case where the hydroxylamine derivatives with alkynes are silylated, that is, X = Si(CH3)3, the substituent is lost and nitrones 55 (Scheme 15) [48] are obtained. Between the three substituents presented in Scheme 15, the methyl group has the worst results for not polarizing the triple bond. Therefore, a slower reaction and worse yields are expected. This feature is generally common in Cope eliminations [49].
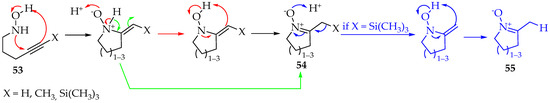
Scheme 15.
Intramolecular reaction between hydroxylamines and alkynes producing five to seven-membered cyclic nitrones.
Mechanistically, this synthesis is concerted in the first step, with no radicals being formed (Scheme 15) [48,49,50]. Then, the hydron is transferred from nitrogen to oxygen in a bimolecular process, requiring another molecule with labile hydrons, which can be obtained from the solvent (Scheme 15—red step). Beauchemin et al. [50] have calculated the energy involved, and found this to be more favorable than direct intramolecular hydron transfer between the two atoms. Finally, the last step towards forming the nitrone 54 is similar to the first step, with a concerted mechanism. Fox et al. [48] proposed that the hydron is transferred directly from nitrogen to the α carbon, due to tautomerization (Scheme 15—green step). Regardless of the mechanism, the reaction was tried successfully between a hydroxylamine derivative and an alkyne from two distinct molecules. The solvent used was propanol, at 110 °C for 14 h. The yield was good (52%) [50]. Excellent yields were obtained in another study for intramolecular reactions, using 5 mol% AgOTf in 1,2-dichloroethane, at room temperature, to a maximum of 4 h [51].
A comparison in reactivity of hydroxylamines with alkenes or alkynes has also been highlighted, using the hydroxylamine 56, which contains the three moieties (Scheme 16). The preferred product of the hydroxylamine 56 containing the three moieties depends on the ring size and the alkene/alkyne substituents [48,49].
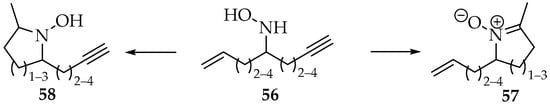
Scheme 16.
Competitive intramolecular reactions of hydroxylamines with alkenes and alkynes.
As a rule, the nitrone 57 is formed when the product’s ring is six-membered. If not, the hydroxylamines 58 are preferred. However, as mentioned previously, the presence of alkyl substituents increases the activation energy. Hence, when the alkene is substituted and the alkyne is terminal (or silylated), the nitrone is always favored over the hydroxylamine [48,49].
In a new gold catalysis reaction, the intramolecular cyclization of the hydroxylamine 59 with its alkyne moiety, with a concomitant transfer of a sulfonyl group from N- to α-position, has been achieved (Scheme 17). In addition to the Au catalyst, methanol at 60 °C and molecular sieves were used. The resulting nitrone 60 was obtained with yield ranging between 39% and 88% [52].
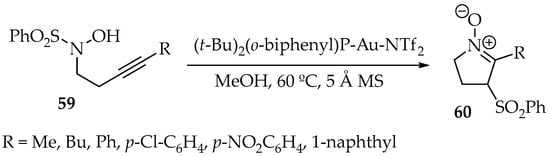
Scheme 17.
Intramolecular reaction between a hydroxylamine and an alkyne, with concomitant 1,3-tranfer of a sulfonyl substituent.
In another study by Beauchemin et al., the hydroxylamines were also reacted with allenes. The temperatures ranged between 90 and 140 °C in tert-butanol for 18 h. The yields were very good in most cases (Scheme 18). Mechanistically, the reaction is similar to the one with alkynes [53].
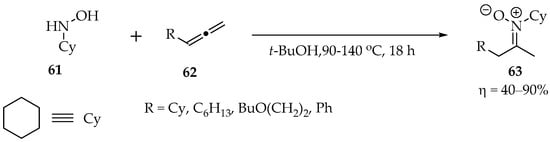
Scheme 18.
Example of a hydroxylamine reaction with allenes.
2.4. From Nitroso Reagents
Nitrones can be prepared from nitroso compounds 64 (Scheme 19) by reacting with structures resembling methylene 65. For example, the diazo compounds fulfill that role. In essence, the reactive carbon must be substituted by a group that can leave with the bond electrons (nucleophile Nu−) and another group leaving positively charged (electrophile E+). In the case of the azo compounds, that substituent executes both functions. A possible mechanism is represented in Scheme 19.
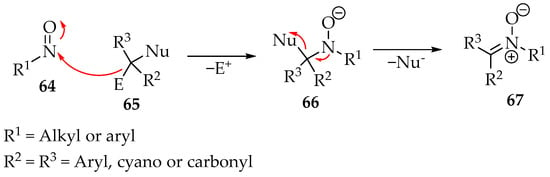
Scheme 19.
Proposed mechanism for the reaction between nitroso compounds 64 and active methylene species 65 to produce nitrones 67.
The nitroso compound 64 is in a high-oxidation state, so acting primarily as an electrophile is natural. As a result, a hydroxylamine’s conjugated base forms 66 as an intermediate. Then, the nucleophile is expelled from the molecule by the nitrogen’s electron pair (Scheme 19). This theory has already been supported in forming imines when the nucleophile group is absent by eliminating a water molecule from the intermediate 66 [3,54]. Nevertheless, more conclusive studies are required.
Many examples of this reaction type were reviewed by Hamer and Macaluso [4]. The construction of species 65 usually consists of aryl, cyano or carbonyl substituents in positions R2 and R3 (Scheme 19). Hence, the hydron or another electrophile is eliminated more easily from the molecule, for example, by adding a base. Several compounds with halogens, diazo, or nitro groups (Figure 4) have been reported for the nucleophile. The nitro compounds with α-hydrogens 73 are excellent; the nitro substituent facilitates hydron removal through structure stabilization by resonance and acting as the nucleophile leaving group [3]. Additionally, pyridinium salts 69 and sulfur ylides 70 (Figure 4) are also very good nucleophiles [3,4].
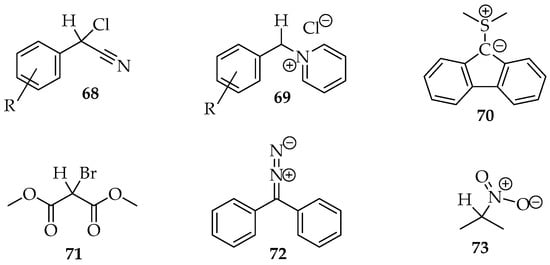
Figure 4.
Some examples of active methylene compounds.
The nitroso reagents can also react with multiple bonds, which has led to the discovery of new nitrone syntheses in the last two decades. Instead of having an electrophile in the structure, the π bond provides the electrons to the nitroso group. Then, there is either a nucleophile, as in compound 75, or another π bond, as in alkyne 71, to set up the final nitrone (Scheme 20).
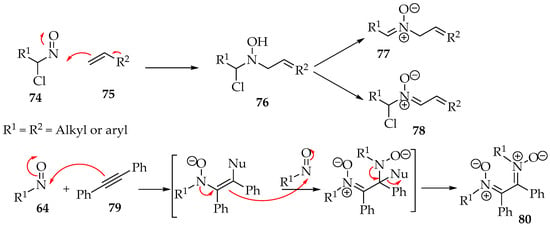
Scheme 20.
Electrophilic addition of nitroso compounds to alkenes or alkynes.
The preparation of a compound like 77 requires, for example, a geminal chloronitroso reagent, which has been synthesized by Bou-Moreno et al. [55] from secondary nitro substituents and oxalyl chloride. Then, Adam and Krebs explored how it reacts with the alkene [56]. First, an aziridine N-oxide is believed to be formed as an intermediate (Scheme 21). Then, in the transformation to the hydroxylamine derivative 82, the hydron captured by its oxygen belongs to the twix position (Scheme 21). Moreover, in the case of the formation of chiral derivatives, the carbon-nitrogen bond formation can be directed by adjacent hydroxyl groups to the threo-stereochemistry, as exemplified by compound 84 (Scheme 21) [56].
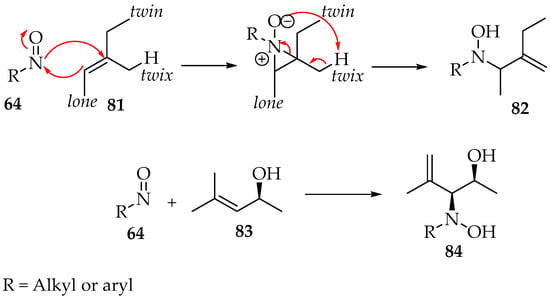
Scheme 21.
Regio- and stereoselectivity of the addition of nitroso compounds to alkenes.
One more excellent reaction is the formation of nitrones from 1,2-dicarbonyl compounds in the presence of N,N,N′,N′,N″,N″-hexamethylphosphanetriamine [P(NMe2)3 = PL3]. The phosphine derivative transforms the 1,2-dicarbonyl 85 into a dioxaphosphole derivative 86, which then reacts with the nitroso compound (Scheme 22) [57]. The mechanism has still not been verified experimentally. Still, the most accepted possibility is the formation of dioxazaphospholidine derivative 87, which, upon ring opening and elimination of the phosphine moiety, gives the desired nitrone 88 (Scheme 22) [57].
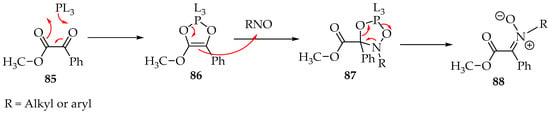
Scheme 22.
Possible mechanism for the reaction of nitroso compounds with 1,2-dicarbonyls, in the presence of a phosphine derivative.
The last illustrative reaction with alkenes combines an α,β-unsaturated nitroso 89 with an imidamide 90 (Scheme 23) [3]. It is an ingenious protocol to produce imidazoline oxides 91, and many other cyclic nitrones can be obtained. However, one must be aware that nucleophilic attack by the oxygen is now possible, leading to the formation of 5,6 dihydro-4H-1,2,5-oxadiazines 92 as by-products [3].
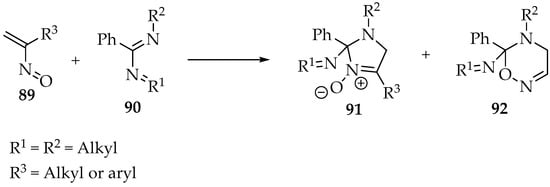
Scheme 23.
Reaction of an α,β-unsaturated nitroso 89 with an imidamide 90.
The reactions between nitroso compounds and alkynes are not explored well, at least to produce nitrones. The most common product is the previously presented dinitrone 80 (Scheme 20) [4]. Using a gold catalyst, one of the nitrone moieties is reduced to an imine [52]. Hamer and Macaluso have shown an unexpected product [4]. The reaction between 1-ethynyl-2-nitrobenzene 93 and nitrosobenzene 94 forms a nitrone with an indoline heterocycle 95 (Scheme 24) [4]. This results from reducing the nitro group to an amine and one nitrone moiety to an imine (Scheme 16), followed by an intramolecular cyclization.
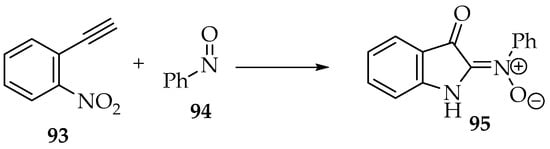
Scheme 24.
Reaction between 1-ethynyl-2-nitrobenzene 93 and nitrosobenzene 94, producing nitrone 95.
One last methodology through which nitroso compounds may form nitrones takes advantage of its reduction potential. For example, toluene and cycloheptatrienes react with one molecule of nitrosobenzene 94, furnishing hydroxylamines 97 (Scheme 25). Then, the hydrogen atom of the hydroxyl group transfers to another nitrosobenzene 94 molecule, oxidizing the hydroxylamine 97 to a nitrone 98 (Scheme 25) [4,58].
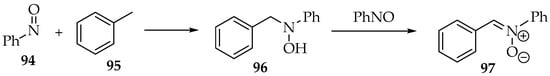
Scheme 25.
Reaction of nitrosobenzene with toluene, with a final oxidation step to nitrone.
2.5. From Nitro Compounds
Nitrones can be prepared by the alkylation of nitro groups. The nucleophilic carbons reported are usually from enolates 99 or Grignard reagents 103 (Scheme 26). Then, either the nitro compound already has an α-hydron 102 or the newly attached carbon must have it to form a double bond after condensation (Scheme 26) through water elimination.
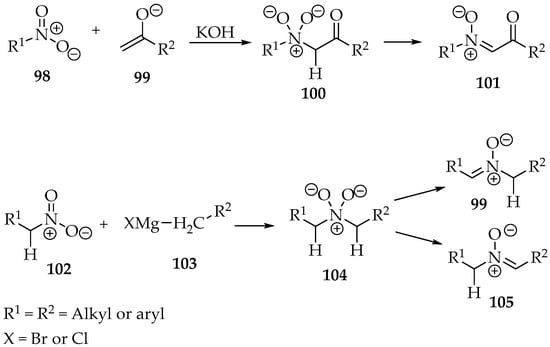
Scheme 26.
Synthesis of nitrones through condensation reaction with nitro compounds.
Nowadays, these reactions are rarely used; however, a more recent work aiming to study organocatalytic reactions on electron-deficient alkynes developed a new synthesis of bicyclic nitrones 108 using ethyl 2-(nitromethylene)-4-arylbut-3-ynoate 106 and diethyl 2-[(2-hydroxyben-zylidene)amino]malonates 107 (Scheme 27) [59]. The proposed mechanism is quite complex, but the authors could identify several intermediates by high-resolution mass spectrometry. Additionally, they suggest that the reaction starts with a conjugate addition of the deprotonated 2-[(2-hydroxyben-zylidene)amino]malonate to the enynoate 107 (Scheme 27) [59].
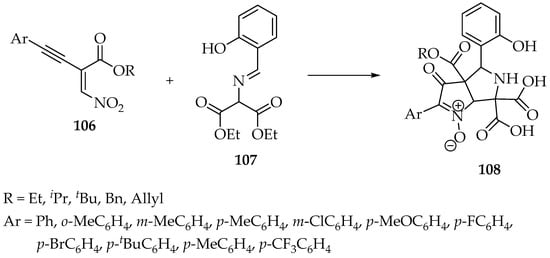
Scheme 27.
Synthesis of bicyclic nitrone derivatives 108 from a nitro enyne 106 and a 2-(benzylideneamino)malonate 107.
2.6. From N-Hydrxyamide Compounds
The preparation of nitrones from N-hydroxamides, also known as hydroxamic acids, is not a frequently used method. Two interesting examples deserve to be highlighted, both employing a similar strategy: the protection of oxygens, followed by reduction and deprotection.
In the first example, the 1,1,3,3-tetramethyldisiloxane [(Me2HSi)2O] was used as protecting group and reductor, concomitantly (Scheme 28) [60]. Nevertheless, the iridium catalyst was essential for obtaining the nitrone derivative 111, and the method was applied to a few substituents in the hydroxamic acid 109, demonstrating high chemoselectivity due to the bulky oxygen-protected atoms (Scheme 28, structure 110). Finally, the deprotection with PPTS or TBAF also promotes the elimination into nitrone 111 (Scheme 28).
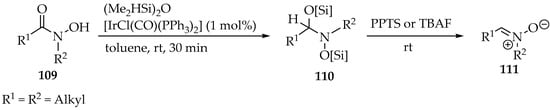
Scheme 28.
Partial reduction in O-protected N-hydroxyamides 109 with a siloxane producing nitrones 111.
In the second example, protection was achieved through cyclisation with acetals, resulting in dioxazole 113 (Scheme 29) [61]. Then, the reduction was disguised as a [3+2]-annulation with dimethyl 2-phenylcyclopropane-1,1-dicarboxylate 114 (Scheme 29) [62]. This was performed in the presence of molecular sieves (MSs) and an ytterbium Lewis acid, with the nitrone 115 being produced and purified by silica gel chromatography [62].
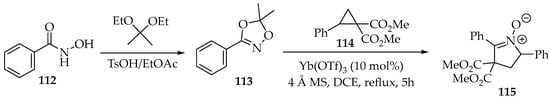
Scheme 29.
Partial reduction in O-protected N-hydroxyamide 113 through a [3+2]-annulation with a cyclopropane, producing nitrone 115.
2.7. Oximes Rearrangement
Although this rearrangement is rare, due to the oximes’ reported stability, it was achieved by Nakamura et al. [40]. Using a copper(I) halide catalyst, the authors successfully converted O-propargyl oximes 116 into stable cyclic nitrones 120 (Scheme 30) [40]. Only the E-oxime 119 could be transformed in this way, as the other diastereomer would undergo an alkylidene transfer, forming the oxazoline 118, which keeps the oxazoline ring from the intermediate 117 (Scheme 30).
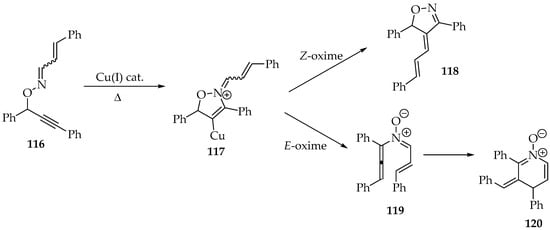
Scheme 30.
The O-propargyl oxime 116 transformation into cyclic nitrones 120, through a copper(I) halide catalysis.
Mechanistically, it was proposed that the copper catalyst would facilitate the 5-endo-dig cyclization, which would then be removed for another cycle. As a result, the N-allenyl imine oxide 119 is produced, which undergoes further rearrangement to the dihydropyridine oxide derivative 120 (Scheme 30).
3. Nitrones Transformations
Nitrone transformations are a significant topic because they allow for the synthesis of new compounds that can become critical bioactive compounds. However, an exhaustive description of the existing nitrones transformations will not be provided herein, as it is not the objective, the review size is limited, and, above all, because an excellent review was published in 2019, which presents and discusses the main transformations [9]. Nevertheless, a few vital transformations and/or more recent ones will be addressed.
3.1. Beckmann Rearrangement
Nitrones rearrangements are often related to the N-O bond liability, and they are quite useful for the preparation of lactams. The Beckmann rearrangement is an exceptional example; however, prolonged irradiation times favor amide formation [63].
The Beckmann rearrangement corresponds to the oxygen shift from the nitrogen to the nitrone’s carbon, so it can be seen as a subsequent transformation of oxaziridines, which were found as intermediaries in the production of amides and lactams [64]. Actually, one of the most important isomerization transformation of nitrones is its cyclization into a three-membered ring by those same three atoms, named oxaziridine. This transformation is usually directly related to the degradation of nitrone under poor storage conditions; however, it can also be induced photochemically. For example, it was found that the conversion into 2,3-dimethyl-1,2-oxaziridine 122 (Scheme 31) could be achieved upon irradiation of (Z)-N-methylethanimine oxide 121 with a mercury lamp whose wavelength is 313 nm [65,66]. The singlet electronic ground state’s electron S0 is excited to the first singlet excited state S1, and then the oxygen forms a single bond with the carbon.
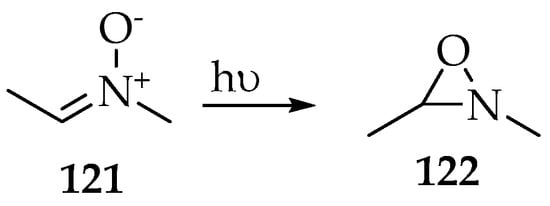
Scheme 31.
The isomerization of nitrone 121 to oxaziridine 122 after excitation to the S1 state by irradiation (λ 313 nm).
Indeed, the Beckmann rearrangement is a versatile method for transforming nitrones into other valuable organic compounds. For instance, irradiating the nitrone derivatives 123 at λ 313 nm will convert them into oxaziridine derivatives 124 (Scheme 32) [64]. After isomerization into oxaziridine, radiation also promotes homolytic N-O bond cleavage, producing a transient diradical structure 125, which, upon the anti-hydrogen shifting to the nitrogen, gives the final amide derivative 126 (Scheme 28) [64]. Accordingly, the authors believe that the dissociation of the C-H activation energy is very low, which explains the good selectivity. However, for bicyclic oxaziridine derivatives 127, both cycles are broken to form the biradical intermediate 128, which subsequently undergoes an intramolecular cyclisation to amide derivatives 129 (Scheme 32). In another example, with spirooxaziridine derivatives 130, a mixture of the two lactams 132 and 134 was obtained, although in a highly selective manner towards lactam 134, in a ratio of 95:5 (Scheme 32). The stereochemistry is governed by stereo hindrance rather than the lower energy state, so the reaction proceeds via the less stable radical 133 rather than through the more stable one, 131 [64].
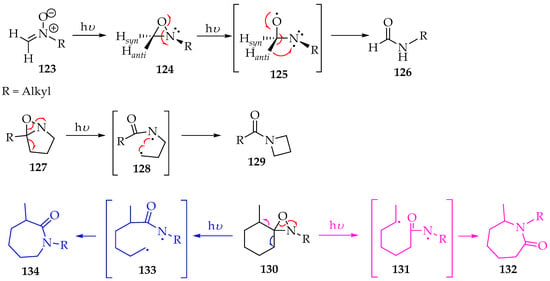
Scheme 32.
Nitrones photoisomerization, via oxaziridine intermediates.
Other strategies have been implemented for the second step of the reaction (transformation of the oxaziridine 135 into the amide), namely the addition of bases and Lewis acids (Scheme 33) [63,67,68]. In the case of acids such as iron(III) sulfate, the mechanism is purely ionic, as shown in Scheme 33 [63]. First, there is the formation of the iron complex 136, followed by the oxaziridine ring opening and the decomplexation into the amide 138. For the bases, it is advised to use photocatalysis in combination with a weak base [68]. The reason is that the hydron shift is the limiting step, and it is weakly acidic [68]. Strong base usage is possible, for example, with LDA or LTMP, but it also leads to the formation of by-products [67]. However, the addition of a photocatalyst, such as 9-mesityl-10-methylacrydinium tetrafluoroborate (Mes-Acr+), will oxidize the oxaziridine 135, making the hydron more acidic and reactive with weak bases.
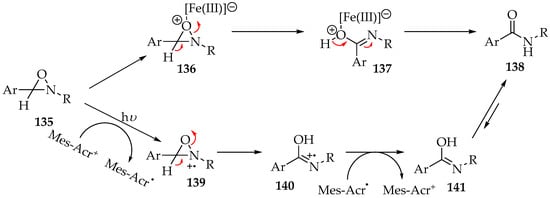
Scheme 33.
The transformation of oxaziridine 135 into amide 138, through the iron(III) complex, and by photoredox activation with 9-mesityl-10-methylacrydinium tetrafluoroborate (Mes Acr+).
3.2. Dipolar Additions
The dipolar additions to nitrones are a versatile method for obtaining new heterocyclic compounds. The most frequent system of dipolar additions is the [3+2]-cycloaddition, which produces oxazolidine analogous 142. The [3+1]-cycloadditions form 1,2-oxazetidine 143 type compounds, whereas the [3+3]-cycloadditions form 1,2-oxazinane 144 derivatives (Figure 5). Sometimes, other heteroatoms are incorporated into the rings, depending on the initial substrates, and intramolecular cycloadditions also occur, resulting in the formation of two new rings.
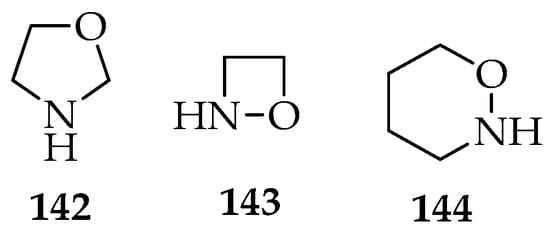
Figure 5.
Oxazolidine 142, 1,2-oxazetidine 143, and 1,2-oxazinane 144 structures.
Generally, dipolar addition involves the nitrone’s oxygen addition to one of the atoms involved in a π bond, and the other atom from the same π bond or another atom in a conjugated π system will form a new bond with the nitrone α-carbon and compensate the positively charged nitrogen. They are usually considered concerted mechanisms [69,70].
3.2.1. [3+1]-Cycloadditions
This reaction appears to be restricted to the addition of isonitriles 146 in the presence of BF3•OEt2 and triethylamine [71]. It is completed in two minutes at −40 °C, producing 1,2-oxazetidin-4-imines 147 (Scheme 34). Not many examples were reported, and the yield is dependent on the isonitrile substituent; good yields (>50%) were obtained with aliphatic carbons attached to the isonitrile 146, whereas with aromatic carbons, the yield was lower (η = 25%).
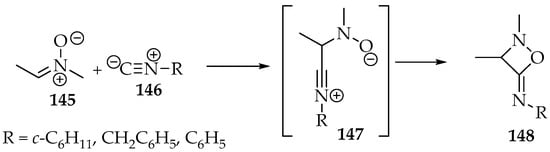
Scheme 34.
The [3+1]-cycloaddition of nitrone 145 to isonitriles 146, producing 1,2 oxazetidin-4-imine 147.
3.2.2. [3+3]-Cycloadditions
As mentioned before, the [3+3]-cycloaddition type often forms 1,2-oxazinane products 144 (Figure 4), although, depending on the reagents used, their final structure can present substituents, including N-alkyl groups (Scheme 35). The [3+3]-cycloadditions are, in general, less common [72], and their application using nitrones is also scarcer [73]. Among the ways to perform this reaction is the reaction of nitrones 149 with a three-membered ring derivative 150 (Scheme 35). By breaking one σ bond, the ring’s steric stress is released, and a three-atom bridge is added between the nitrone’s carbon and oxygen atoms. Nitrones’ reaction with cyclopropanes is relatively recent, with the debut dating back to the beginning of the twenty-first century, in a work of Young and Kerr [74]. Soon, it was found that the reaction followed a specific asymmetry, and a stepwise mechanism was suggested [75,76]. The nitrone’s oxygen first attacks one of the cyclopropane derivative’s more electrophilic carbons, forming a C-O bond and breaking the cyclopropane σ bond. This first step is the rate-limiting one and yields a high-energy zwitterionic intermediate with a negative charge on the other carbon atom of the broken σ bond and the positive charge on the nitrogen. Naturally, groups that stabilize the carbanion formed will contribute to lowering zwitterionic intermediate energy. The last step is the formation of the C-C bond to yield the 1,2-oxazinanes 151 [75,76].
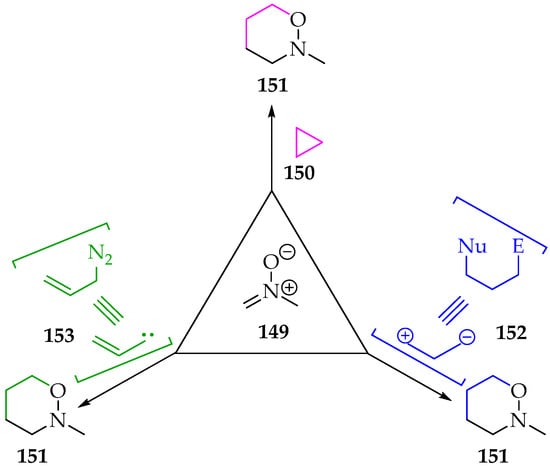
Scheme 35.
The possibilities for the [3+3]-cycloaddition of nitrone type 149.
The cyclopropane’s substituents and stereochemistry dictate the regioselectivity of the 1,2-oxazinane. A detailed example is the reaction of the nitrone 154 with each cyclopropane diastereoisomer 155 and 156 (Scheme 36) [76]. The orientation of the oxygen attack and the location of the new σ bond are anti to the broken σ bond, a feature also observed in other examples [77,78]. As a result, the stereochemistry of the cyclopropane carbon not involved in the broken σ bond is inverted, and the cis-2-methyl-3-phenylcyclopropane 155 leads to a 5,6-trans-1,2-oxazine 158, whereas the trans-2-methyl-3-phenylcyclopropane 156 forms the 5,6-cis-product 160 (Scheme 36). This reaction has only been tested with Z-nitrones, and the favored products that present the 3,6-cis-diastereoselectivity are depicted in Scheme 36 [76].
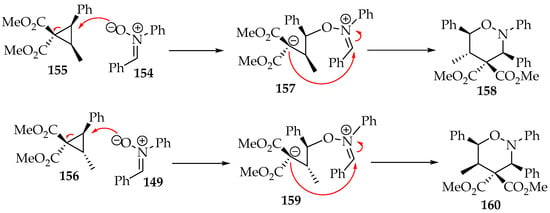
Scheme 36.
The mechanistic proposal for the [3+3]-cycloaddition of nitrones to diastereoisomeric cyclopropanes cis-155 and trans-156.
So far, no theoretical calculation has been performed on this system, and the rational mechanism depicted in Scheme 36 is based on the transition state proposed in Karadeolian and Kerr’s study [76]. Fundamentally, the carbanion binds to the nitrone’s carbon from the opposite side of the bulkiest group (phenyl). Nevertheless, other examples also establish this reaction’s high stereoselectivity [79].
The next [3+3] cycloaddition is the reaction of nitrones with open-chain dipoles prepared in situ, with an anionic/nucleophilic and a cationic/electrophilic part (Scheme 35, structure 152). The carbanions may be produced at the α-carbon of carboxylic acids or from silylated compounds. Alternative systems are enamines and heteroatoms. On the positively charged part, activated hydroxyl groups, for example, acetoxy, and halogens are the main strategies applied [7,9,80]. An interesting example is the 2-(1-hydroxyalkyl)indole 162, which has a nucleophilic character at the indole carbon C-3, and the 2-substituent acts as the electrophilic center (Scheme 37). The application of a chiral catalyst (phosphate derivative) resulted in good enantioselectivity of the tricyclic product 163. Additionally, the authors proposed a mechanism that explains the stereoselectivity and also evaluates the reaction scope of both indole and nitrone derivatives [81].
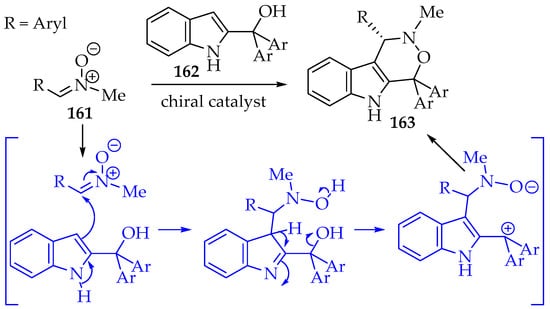
Scheme 37.
[3+3]-Cycloadditions between nitrones 161 and indole derivatives 162.
Another interesting example involves the use of silylated compounds, for instance, 1-phenyl-2-((trimethylsilyl)methyl)allyl acetate 164, which, upon palladium catalysis, can form in situ an open-chain dipolar intermediate that reacts with nitrones, giving 1,2-oxazines 165 in high yield (Scheme 38) [82]. Moreover, the reaction scope was investigated, and a high stereoselectivity towards the trans isomer was observed.
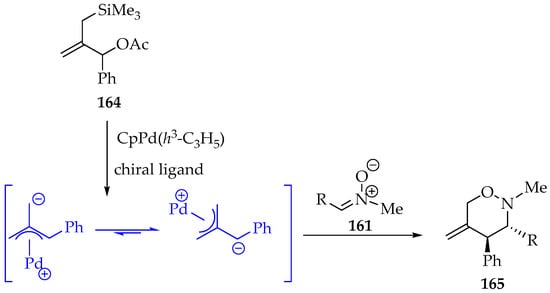
Scheme 38.
Another example of [3+3]-cycloadditions between nitrones 161 and open-chain dipolar compounds obtained in situ.
Regarding the vinyl carbene reagents, they have been prepared from vinyl diazo compounds 153 (Scheme 35) by coordinating it with a metal [7,9]. These metallo-vinylcarbenes of the Fischer type are generally electrophilic, due to the weak π-backdonation from the metal to the carbon. Considering the reaction of the vinyl diazo compound 166 as an example, there is in the first step the metal insertion, forming the metallo-vinylcarbene 167 (Scheme 39). Thus, as a result of the vinyl compound polarity inversion, the [3+3]-cycloaddition first goes through an addition of the nitrone’s oxygen at the β-unsaturated carbon, forming the intermediary 169. Then, the cyclization is completed, not due to the nucleophilic character of the carbon bound to the metal by itself, but due to the resonance of an electron releasing group at the α-carbon. The [3+3]-cycloaddition follows the syn-configuration and the 3,6-dihydro-1,2-oxazine 166 product is formed (Scheme 39) [83,84].
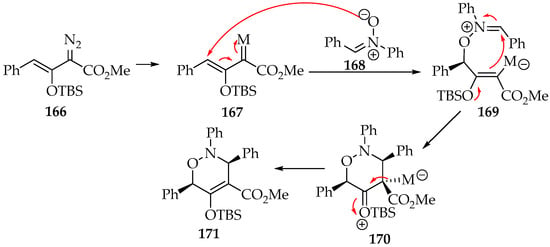
Scheme 39.
The [3+3]-cycloaddition of the nitrone 168 to the metallo-vinylcarbene 167.
The last chosen example is the reaction with the N-vinylindole derivatives 173 (Scheme 40). In this reaction, which requires a nickel(II) catalyst, to avoid a [3+2]-cycloaddition at the vinyl group, due to an interaction between the nitrone’s oxygen and the nickel ion [85], the mechanism is inverted, with the nitrone acting first as the electrophile and adding at the indole carbon C-2 (Scheme 36). It is followed by a 1,7-proton shift and the final cyclization to give the two possible isomers (Scheme 40). The obtained isomeric mixture showed a slight preference for the cis-diastereomer, though no chiral ligand was used [85].
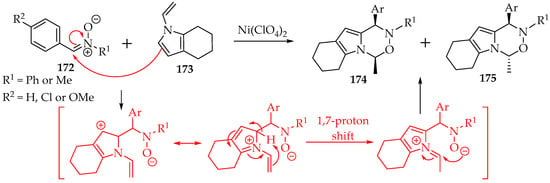
Scheme 40.
The reaction of the nitrone 172 with 1-vinyl-4,5,6,7-tetrahydro-1H-indole 173.
3.2.3. [3+2]-Cycloadditions
The [3+2]-cycloadditions involving nitrones have often been used to obtain new interesting compounds. This reaction is straightforward, yields high conversions, and can usually be performed in situ, immediately after the synthesis of the nitrone. It is also possible to carry out solid-supported reactions [86]. The reaction can be accelerated by increasing the pressure, raising the temperature, or using microwave irradiation. Additionally, a Lewis acid catalyst is frequently employed [3].
Nevertheless, this reaction type has two drawbacks: the variety of stereoisomers produced and its reversibility. Although protecting the nitrone might be helpful, the potential for changes in the stereoisomeric product ratio over time adds complexity. As a result, the products obtained under kinetic conditions may differ from those generated when the nitrone reacts for extended periods or at elevated temperatures (thermodynamic conditions). In such cases, one less favorable isomer may be transformed into a more stable one [87].
The most ubiquitous example is the reaction between a nitrone and an alkene, forming an isoxazolidine. The reaction can be either intramolecular or intermolecular. In the first case, bicyclic compounds are formed, and in the second case, the number of stereoisomers is larger (the alkene has more degrees of freedom).
There are five roots for the asymmetry: the purity of the nitrone’s diastereomer E/Z, the localization of the addition on the Re or Si nitrone’s face (Scheme 41), the orientation of the alkene, the alkene’s diastereoisomerism, and the endo/exo attack. Fortunately, the products can be predicted with some accuracy, and there is often a preference for one or two products, obtained in good yields.
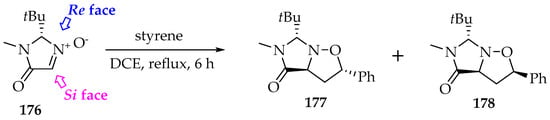
Scheme 41.
The [3+2]-cycloaddition of the nitrone 176 to styrene by the Re face, with the selective formation of the stereoisomers 177 (exo) and 178 (endo).
First, the reagents should be used as single stereoisomers. Then, the Re/Si face attack can be controlled by substituents on the nitrone or by appropriate chiral catalysts that crowd one face. In Scheme 41, the nitrone’s Si face 176 is hindered by the tert-butyl substituent (below the nitrone’s plane) [88]. In consequence, the styrene approaches through the Re face preferably (above the plane).
The alkenes’ substituents are the primary source of isomerism and are the most difficult to modify. The best is to understand their reaction models. Generally, the alkene’s orientation is such that its bulkier substituent is positioned at the C-2 of the isoxazolidine product (Scheme 41, products 177 and 178). The electron-withdrawing groups, namely through the heteroatoms’ presence, also favor that position, because it is easier for the nitrone’s oxygen nucleophilic attack [3]. Thus, both steric and electronic factors drive the reaction asymmetry.
The main exception is the reaction with α,β-unsaturated carbonyls [89]. There, the conjugated system’s resonance leads to the formation of 4-carbonyl isoxazolidine derivatives. In the same way, the α,β-unsaturated nitriles, nitro alkenes, and vinyl sulfones result in the same substitution pattern (at C-3) [90,91,92].
In terms of endo/exo selectivity, the two possibilities are depicted in Scheme 41 for a cyclic nitrone. In the case of an endo preference, a cis-isoxazolidine is obtained, whereas the exo configuration produces the trans-isomer. The same happens on linear E-nitrones. For the Z-nitrones, the products’ diastereoselectivity is the opposite. The alkenes’ substituents also influence the endo/exo preference, though with less impact than for the other kinds of isomerism. Hence, mixtures of trans- and cis-isoxazolidines are often observed.
In general, the exo approach is followed to minimize steric energy. However, due to the dipolarity of some groups and hydrogen bonding, the endo configuration is sometimes observed. This is the case of the α,β-unsaturated nitriles and aldehydes [93]. For α,β-unsaturated ketones and esters, the increased bulkiness often results in the exo configuration [89].
That dipolarity is also helpful for discerning between two competing asymmetries (Scheme 42) [94,95]. In this example, both the ester and the alkyl chain prefer the exo configuration. Since the ester exhibits some dipolarity at the carbonyl moiety, it is more readily accommodated by the endo approach, leaving the exo approach available for the alkyl and chlorine substituents. The same happens for the allocation of the C-2 and C-3 positions. As the ester is involved in the resonance, it ends up in the first option (C-3).
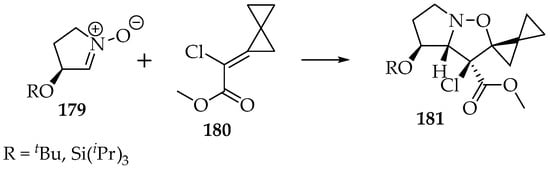
Scheme 42.
The [3+2]-cycloaddition of the nitrone 179 to the α,β-unsaturated ester 180 by the Re face, with the selective formation of the stereoisomer 181.
Although it is not sufficiently studied, the change in the solvent’s polarity can have an impact on the resulting product’s asymmetry, even changing the mechanism from a concerted to a stepwise route [93]. The diastereoisomerism arising from reactions with aldehydes can also be reversed by employing secondary amine catalysts [96]. As they attack the carbonyl before the nitrone, making it bulkier, the endo configuration is no longer available.
For the intramolecular cycloadditions, four types of bicyclic structures can be formed, which are represented in Scheme 43. All of them contain the isoxazolidine fragment. The other carbon ring is bound to different positions for each one. Thus, when the alkene is on the nitrone’s carbon side (Scheme 43, nitrone 182), its products have the ring bound to the 3,4-positions, compound 183, and 2,4-positions, compound 184. Whereas, for the alkene in the N-substituent (Scheme 43, nitrone 185), the products have the ring bound to the 3,5-positions, compound 186 or 2,5-positions 187.
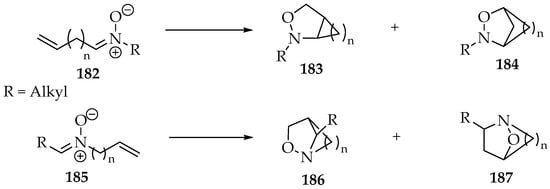
Scheme 43.
The possible bicyclic products of nitrones’ intramolecular cycloadditions.
The most frequent reactions involve the 182 nitrones, with n = 3 or 4 [3,97]. While the chain is small, the structure 183 is formed. For n = 4, there are already yields of compounds such as 180. For longer chains, it is expected that a larger ratio of 184/183 is required to maintain the same stereoselectivity observed in intermolecular cycloadditions. The case of compounds like 185 (Scheme 43) has not been studied to the same extent, and the products obtained are mostly of the type 187 [98,99]. It is likely that a chain on that side of the nitrone has more freedom than on the other side, since the carbon is in sp3 hybridization.
Alkynes are more reactive dipolarophiles than alkenes. Therefore, milder conditions are used, often by applying lower temperatures. Also, the heating and the employment of acid or base catalysis can accelerate the rearrangements of the 2,3-dihydroisoxazole product to other compounds [100]. Thus, it should be avoided. The cycloaddition follows a cis-configuration and occurs on the least-hindered nitrone’s face (Scheme 44) [101]. The choice of position per substituent in the 2,3-dihydroisoxazole product is similar to the reactions with alkenes [3].
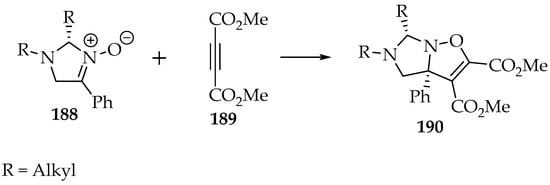
Scheme 44.
The asymmetric cycloaddition of nitrone 188 to the dimethyl but-2 ynedioate 189.
Contrary to the oxazolidines formed in the reaction with alkenes, the 2,3-dihydroisoxazoles 193 are seldom stable. This is even more significant when an aldonitrone 191 is used as a reagent. One possible rearrangement is the 1,3-H-shift, which forms the 2,5-dihydroisoxazole 195 (Scheme 45) [100]. This stabilizes the compound once a stronger conjugated π-system is established. It becomes more relevant in the presence of aromatic substituents.
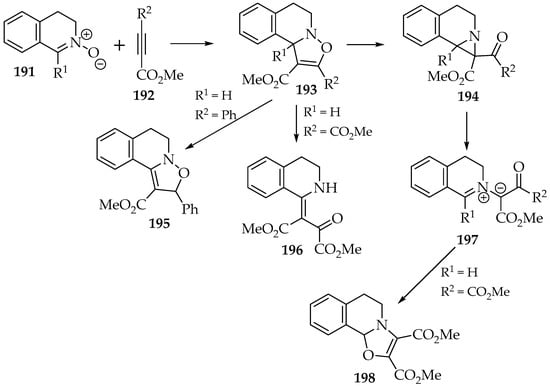
Scheme 45.
The reactivity of the 2,3 dihydroisoxazoles 193, resulting from the [3+2] cycloaddition between nitrone 191 and alkynes 192.
Apart from the hydrogen shift, the 2,3-dihydroisoxazole’s rearrangements follow two types of mechanism, with the formation of either the β-enamino-ketone 196 or the aziridine 194 (Scheme 45) [100]. They both result from the homolytic (when heated) or heterolytic (acid or base catalysis) cleavage of the N—O bond. However, the aziridine undergoes a second bond cleavage (C—C), yielding four possible products. The ylide intermediate 197 might be stable on its own [101]; it can rearrange into an oxazoline 198.
One last type of addition to terminal alkynes 200 is the Kinugasa reaction, which is catalyzed by copper(I) (Scheme 46) [9]. As the alkyne’s hydron is substituted by the Lewis acid (Scheme 46, complex 202), the resulting 2,3-dihydroisoxazole 205 rearranges to a ketene 210, and then to an azetidin-2-one 212 (β-lactam).
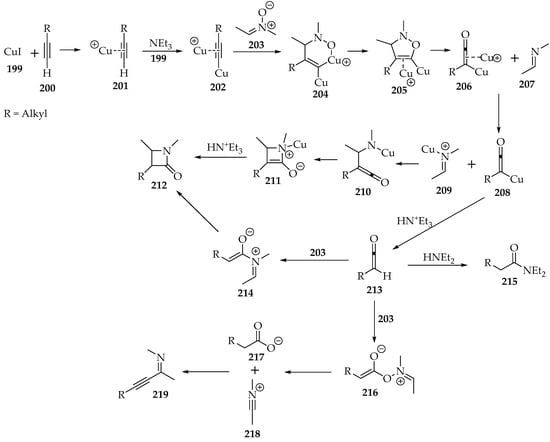
Scheme 46.
The Kinugasa reaction between a terminal alkyne 200 and the nitrone 199, catalyzed by copper(I) iodide.
Much discussion has revolved around this reaction’s mechanism. Kinetic studies have shown that the first cyclization to the 2,3-dihydroisoxazole 205 (Scheme 46) is the rate-determining step. It is of the first order on the nitrone and second order on the copper(I) cation, suggesting the dimetalate species [102]. It is supported by theoretical calculations [103]. The alkyne has a negative order of −1, as it consumes the copper required for the reaction catalysis. In essence, the alkyne’s excess forms the complex 201 (Scheme 46), and there is not enough copper(I) for the second complex 202. The base’s order was not analyzed.
The presence of the metal is detrimental to the cyclization asymmetry. The formation of complex 204 (Scheme 42) reduces the activation energy for that configuration, in contrast to the thermodynamically favored cycloaddition with the R group at position C-4 [103].
Then, it was found that the complete fragmentation of the 2,3-dihydroisoxazole 205 (Scheme 46) has a lower activation energy than the partial fragmentation of the N-O bond only [104]. Thus, the imine 209 and the ketenylcopper(I) 208 are produced, after a metal cation exchange. Afterwards, the C-C bond is formed, followed by the N-C bond. The β-lactam 212 is obtained after the final hydronation (Scheme 46).
Although it occurs to a lesser extent than the main pathway, ketenylcopper(I) 208 can be hydronated earlier, yielding ketene 213 (Scheme 46). It can still form the intended β-lactam 212, through a Staudinger ketene–imine cycloaddition (Scheme 46, intermediate 214) [105]. However, the ketene often adds to other nucleophiles. The use of nucleophilic amines should be avoided to reduce the amide side product 215 [102] significantly. In its absence, the nitrone 203 acts as the nucleophile and produces carboxylates 216 and alkynyl imines 219 (Scheme 46), but to a much lesser extent.
In terms of diastereoselectivity, the hydronation of the complex 211 (Scheme 46) happens at the least hindered site, favoring the kinetic cis-product [104]. Even if the Staudinger pathway is followed (through compound 214), and assuming an exo-attack of the ketene 213 to the E-imine 207, the same compound should be obtained (Scheme 46) [105]. Nevertheless, it isomerizes to the trans-β-lactam 212, which is thermodynamically more stable. The epimerization is considered the responsible mechanism, but the ring opening at the C—C bond should not be discarded (corresponds to the reverse transformation from 212 to 214) [106]. Moreover, it is more likely to occur when the N-substituent is smaller, or when there is an electron-releasing group at position four and an electron-withdrawing group at position 3 of the β-lactam 212 (Scheme 46) [105].
3.3. Transformations Fluorinated Nitrones
Nitrones have been identified as effective synthons for the synthesis of a variety of compounds, including heterocycles. While less studied, fluorinated nitrones are particularly noteworthy, as they are gaining recognition as an important synthon for future research and applications.
One of the first works using fluorinated nitrones was reported by Mitsuhashi et al. [107], who obtained the N-methyl-C-trifluoromethylnitrone 220 (Scheme 47) via a simple condensation between N-methylhydroxylamine hydrochloride and trifluoroacetaldehyde, followed by water removal. Upon obtaining the desired trifluoromethylnitrone 220, the authors studied its cycloaddition reaction with alkynes and obtained 3-trifluoromethyl-4-isoxazolines 222 (Scheme 47), in low-to-excellent yields, depending on the alkyne substituents (Scheme 47) [107]. The lower yield was obtained with oct-1-yne (26%), and the highest yield was obtained with ethynylbenzene (91%), suggesting that withdrawing groups in the alkyne favored the cycloaddition.
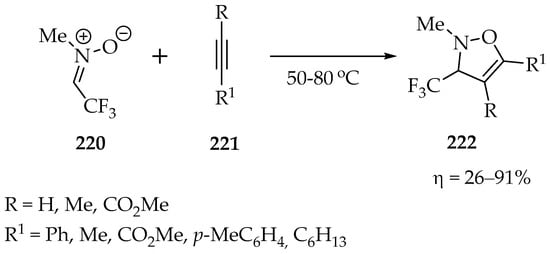
Scheme 47.
1,3-Dipolar cycloadditions of N-methyl-C-trifluoromethylnitrone 220.
Later on, other researchers used this type of fluoromethylnitrones, changing the N-alkyl group or the number of fluorine atoms [108,109,110], and in doing so they enlarged the scope of derivatives obtained. Moreover, the dipolarophiles were also changed, and alkenes [109,110] or even thioketones [108] were successfully employed. Particularly interesting was the use of N-vinyl nucleobases [109] to obtain, in good yields, isoxazolidinyl derivatives of nucleobases (Scheme 48). The yields depend on the nitrone substituent; for example, in the examples shown in Scheme 48, the methyl gives lower yields (38 to 47%), whereas the t-buthyl group gives higher yields (54–68%) [109]. Additionally, the methodology employed was proven to have some regio and stereo control, as the dipolarophile geometry is transferred to the cycloadduct. Again, the alkyl groups in the nitrone have some influence, since for the methyl group the major compounds are the endo-isomers 225 and 228, and for the t-buthyl group the exo-isomers 226 and 229 (Scheme 48) are the major compounds.
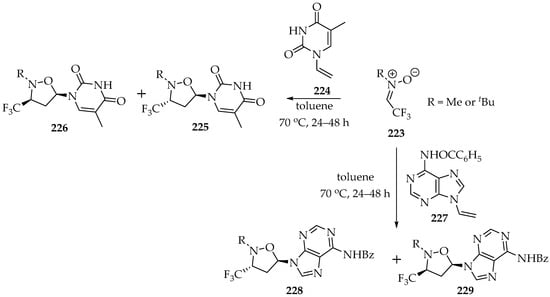
Scheme 48.
1,3-Dipolar cycloadditions of N-alkyl-C-trifluoromethylnitrone 223 to N-vinyl nucleobases 224 and 227.
In 2016, Heimgartner et al. published a fascinating study involving the reaction of fluorinated nitrones with alkynes [111]. They demonstrated that these compounds undergo a [3+2]-cycloaddition, followed by transformation into more stable β-lactams, which are obtained in good to very good yields (Scheme 48). While this process results in a diastereomeric mixture of the cis-isomer 232 and the trans-isomer 233 (Scheme 49), these isomers can be easily separated.
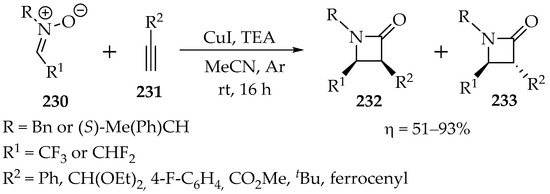
Scheme 49.
Kinugasa reaction with fluorinated nitrones 230 and acetylenes 231, leading to fluorinated β-lactams 232 and 233.
The Kinugasa reaction mechanism (Scheme 50) was recently reviewed [104]. This mechanism explains the production of both diastereomers and demonstrates that bulky substituents on the alkyne lead to higher amounts of the trans-isomer 233 (Scheme 49) [111]. Furthermore, the formation of intermediate 234 helps clarify why its cyclization to intermediate 235 may involve rotation around the single bond between carbons a and b (Scheme 50), yielding a mixture of diastereomers as the final product.
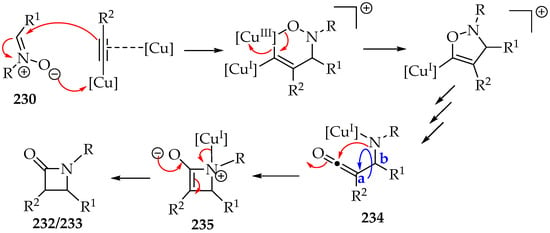
Scheme 50.
Kinugasa reaction mechanism.
Our last example is an interesting nucleophilic addition of organometallic reagents to trifluoromethyl nitrones 236 (Scheme 51), which were prepared in very good yields from the condensation of various hydroxylamines with fluoral [112]. The synthesis of trifluoroethyl N-hydroxylamines 237 was predominantly achieved using Grignard reagents, yielding results ranging from very good to exceptional (Scheme 51). This process not only underscores the remarkable versatility of trifluoromethyl nitrones but also showcases their potential.
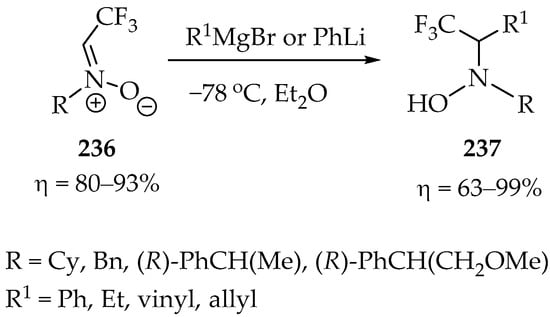
Scheme 51.
Synthesis of trifluoroethyl N-hydroxylamines.
In summary, it can be emphasized that these nitrones provide a valuable platform for a range of subsequent synthetic transformations. Due to their intrinsic stability, they present innovative opportunities for the synthesis of biologically active compounds.
4. Nitrones’ Applications
Nitrones are already in use or under investigation for future applications. For those purposes, it is helpful to have more insight into their physical–chemical properties.
Nitrones are usually stable at room temperature when kept in the dark. The smaller, aliphatic nitrones are yellowish oils that melt at room temperature, such as 5,5-dimethyl-1-pyrroline N-oxide. At the same time, most nitrones contain aromatic groups and have a solid appearance. Their melting points range from 80 to 100 °C for simpler structures, such as N,1-diphenylmethanimine oxide [113]. Additional electron-releasing groups or aromatic rings increase that temperature. For example, the N-methyl-1-(4-nitrophenyl)methanimine oxide and the N,1,1-triphenylmethanimine oxide both melt above 200 °C. However, one should always be concerned with the dimerization possibilities, namely by the [3+3]-cycloaddition mechanism.
The characterization of these compounds is performed by spectroscopic and spectrometric techniques. The 1H NMR signal of the proton bound to the sp2-carbon is shown around δ 7 ppm. On the N-substituent, the proton signal ranges from δ 3.77 ppm for the methyl to δ 5.89 ppm for the benzyl’s methylene. The 13C spectra show the sp2-carbon around δ 130 ppm, while the N-substituent’s carbon often ranges between δ 55 and 80 ppm. The effect of the solvent’s increasing polarity is down shield in the 1H spectra and slightly up shield in the 13C [114,115,116,117].
The UV-Vis spectrum varies for non-aromatic and aromatic nitrones. The first case shows a band with a maximum around λ 220–250 nm, which corresponds to the π-π* transition [118,119]. The second case shows two bands in the UV-Vis region. The least energetic is around λ 300–350 nm, and the most energetic is between λ 200 and 250 nm [113,117,120]. They were the subject of discussion in the 60 s [66,119]. The attribution to the nitrone’s π-π* transition and to the aromatic ring’s excitation, respectively, seems the most coherent. Moreover, the first band disappears during photoisomerization to the oxazirane. The spectra’s bands differ slightly between the diastereomers. They are more intense in the (E)-1-cyano-N,1-diphenylmethanimine oxide than in the cis isomer. There is also a hypsochromic shift in the first in comparison to the latter. The presence of substituents that hinder the planarity of the conjugated π-system often causes a reduction in the bands’ intensities [113]. The solvent’s polarity and hydrogen bonding also influence the light absorption. More polar solvents decrease the absorptivity, and the maximum wavelength shifts to shorter wavelengths. A hypothesis correlates the hypsochromic shift with the greater polarity of the nitrone relative to its excited state [119,121].
The nitrones’ IR spectra show two characteristic peaks. The one between λ 1650 and 1550 cm−1 is attributed to the C=N bond and the other, at λ 1150–1050 cm−1, to the N-O bond [66,114,116,122].
Another important piece of information is the redox potential of this family of compounds. McIntire et al. did cyclic voltammetry in water and acetonitrile, using a calomel reference electrode (SCE) [123]. In the first case, only the reduction potentials were determined, once water was oxidized at +1.2 V, before the nitrones’ oxidation. The reduction potential approximates to −1.8 V for simple nitrones, and the highest value obtained was −0.87 V for an N-alkylpyridinium derivative. In the organic solvent, the reduction potential was similar to or lower than that in water, with the difference in the measured potential not exceeding 0.7 V. The anodic potential was the lowest for the sulfonated phenyl nitrone, at 1.34 V. The maximum was obtained for the N-alkylpyridinium derivative, with 2.37 V.
Regarding lipophilicity, partition coefficients (KP) between 1-octanol and water are determined using HPLC or calculated with appropriate software [37,124,125,126,127]. There is still some variance between results, and the number of experimental works is limited to some nitrones’ derivatives. The two main references are the 5,5-dimethylpiperidine oxide and the N-tert-butyl-1-phenylmethanimine oxide, whose constants are around 0.1 and 15, respectively [37]. They confirm the good polarity of this organic family. The pyridinyl and phosphoryl substituents increase the hydrophilicity, whereas the bulkier aromatic moieties increase the hydrophobicity. This property is relevant to the toxicity of nitrones. The IC50 is often in the millimolar range, but it decreases for more apolar nitrones [124,128]. The related nitroso derivative, obtained upon the nitrone’s oxidative hydrolysis, is slightly more toxic (0.1 mM).
4.1. Spin Trap
Nitrones have been used as spin traps in electron paramagnetic resonance (EPR) experiments, also known as Electron Spin Resonance (ESR). Their goal is to trap radicals produced in specific environments, forming spin-active species (spin adducts), which are then analyzed in the resonance apparatus. Among the nitrone traps, the most ubiquitous are DMPO 238 (2,2-dimethyl-3,4-dihydro-2H-pyrrole 1-oxide), DEPMPO 239 (2-(diethoxyphosphoryl)-2-methyl-3,4-dihydro-2H-pyrrole 1-oxide), PBN 240 (N-t-butyl-1-phenylmethanimine oxide), and disufenton 241 (also known as NXY-059; 4-((t-butyloxidoazaneylidene)methyl)benzene-1,3-disulfonate), all depicted in Scheme 52 [124]. After the free radical trapping, the nitrone radical formed is stabilized by resonance between the two electronegative atoms (Scheme 52). Moreover, the EPR spectra are characteristic for each free radical with these trapping agents. The signals often show coupling frequencies between the unpaired electron with the nitrogen and the hydrogens of the adjacent carbon, which correlate to specific structures.
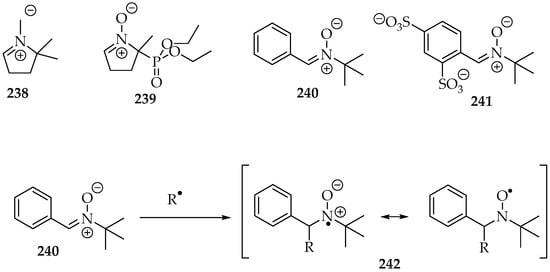
Scheme 52.
The most used nitrone spin traps: DMPO 238, DEPMPO 239, PBN 240, and disufenton 241; and the radical R• trapping, with the corresponding resonance stabilization.
However, before considering the trapping of other radicals, it is convenient to understand the radicals that can be produced by the nitrone on its own, especially under sonication conditions. One experiment identified the phenyl radicals from PBN 240 in inert-gas-saturated water solutions (Scheme 53) [129]. The spin adduct 243’s signal appeared as a double triplet, and its hyperfine coupling frequencies (hfcf) were aN = 1.60 mT and aHβ = 0.42 mT. The hydrogen radical from water was also identified as a triple triplet: aN = 1.67 mT and aHβ = 1.05 mT. The hydroxyl radical was too short-lived to be measured. The formation of organic radicals was suggested to occur at gas bubble interfaces, since the organic molecule’s high hydrophobicity favors its presence there. Hence, when the gas bubble bursts, the radicals are produced. That mechanical event is very energetic, far superior to the energy required for the C-C bond homolytic cleavage.
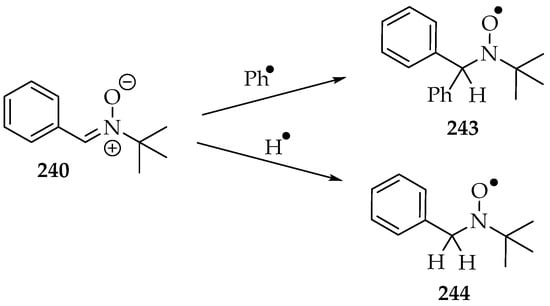
Scheme 53.
The auto spin adducts formation of water dissolved PBN 240, under sonication conditions.
Another concern in this spin-trapping activity is the formation of artefactual adducts, namely radicals produced by nucleophilic additions rather than by radical scavenging. Two main mechanisms have been described in the literature. They have already been covered before: nucleophile’s addition at the sp2-carbon, followed by the hydroxylamine oxidation to the nitroxide radical (either catalyzed by a metal that is reduced in the end, or not); and the initial nitrone’s oxidation, followed by the nucleophile’s attack at the same carbon [124]. The combination of these two pathways is known as the Forrester–Hepburn mechanism. Often, these reactions can be controlled by using chelating agents.
For the radicals to be trapped, not all of them have to have the same affinity for the nitrone. A qualitative scale might be HO• > H3C• > HS• > HO2• > O2•− > NO [124]. Some strategies can be envisioned to improve them, namely for the superoxide radical anion (O2•−). It can be protonated at lower pH, forming the slightly stronger HO2• radical. Including hydrogen-donating groups for hydrogen bonding, such as amides and cyclodextrins, is another possibility. For the weakest species of the list, nitric oxide (NO), other trapping agents are used, rather than the nitrones.
In practical experiments, the concentrations of nitrone spin traps range from 50 to 100 mM, so that the signal is detectable by ESR [115]. Considering biological applications, these high concentrations are a relevant drawback. Nevertheless, research has been conducted to improve the affinity of nitrones to the biological radicals in general, so that those concentrations can be lowered. For example, the combination of EMPO [2-(ethoxycarbonyl)-2-methyl-3,4-dihydro-2H-pyrrole-1-oxide] with gold nanoparticles enabled a 370-fold decrease in the concentration requirements, reducing from 10 mM for EMPO to 27 μM for Au@EMPO.
4.2. Therapeutic
One of the most important applications is the extensive study of nitrones for medical applications, as well as for the detection of biological radicals by ESR. Their own action against the reactive radicals, such as ROS and RNS (Reactive Oxygen and Nitrogen Species, respectively), by scavenging them, is already relevant in controlling the cells’ oxidative stress. Moreover, many of those adducts’ degradation products have beneficial properties. The degradation pathway accounts for the hydrogen on the sp2-carbon, which makes it much more reactive [124]. Furthermore, due to this degradation mechanism, the radical scavenging is irreversible.
The degradation of the nitrone-superoxide adduct results mainly in the hydrogen peroxide (H2O2), along with the nitric oxide (NO) and the hydroxamic acids (N-hydroxyamides) products. The peroxide production resembles the biological superoxide dismutase (SOD) enzyme and yields higher amounts than the other two products. Nitric oxide is a well-known signaling agent, a vasodilator, and a molecule involved in the immune response, by targeting iron–sulfur centers and damaging the DNA of parasites [130]. Nitrones are considered NO slow-releasers, as the superoxide disproportionation is faster than the former, which is even inhibited in alkaline media [131]. Finally, the hydroxamates are good metal ligands, especially for iron. Therefore, they can prevent iron-catalyzed Fenton reactions.
When trapping is performed with carbon radicals, the resulting nitroxide can be disproportionate to the nitrone and the hydroxylamine (Scheme 3A). That hydroxylamine further enhances the redox activity of such therapeutic agents [124,132].
Research into nitrones as pharmaceuticals began in 1986 with the work of Novelli et al. [133]. The study focused on the prevention capacity of PBN in rats that were subjected to a lethal body trauma, using a rotating drum. It was reported to be highly effective [133]. Since then, nitrones have been applied in the treatment of many diseases, such as cancer, stroke, Parkinson’s, Alzheimer’s, and aging, and have been recognized as potent anti-inflammatories [133]. The most promising compound so far, which entered phase 3 clinical trials for stroke under the AstraZeneca brand, is NXY-059 241 (Scheme 52) [134]. Unfortunately, no significant improvement in the patients’ treatment was observed in the last trial, in 2006. One reason might be the reported non-specificity for intracellular targets [124].
Nitrones’ partition coefficient varies with the bound groups, being 30 for PBN 240 (Scheme 52) and below 1 for more hydrophilic ones [124]. A solution to improve the substance’s retention within cells is to esterify it with the acetoxymethyl group. Other alternatives, such as binding the nitrone to specific compounds and receptors, have also been tested. One such example is the triphenylphosphonium lipophilic cation derivative, which was shown to concentrate the nitrone inside mitochondria. Other examples are amphiphilic, glycosylated, and peptidic molecules. Commonly used linkers are amides, esters, and phosphoryls. The IC50 for this family of organic molecules is usually in the millimolar range, with the more lipophilic ones having lower values [124]. For example, DMPO 238 (Scheme 52) is less toxic (IC50 140 mM) than PBN 240 (IC50 9 mM) in bovine aortic endothelial cells.
Despite the critical nature of this application and the emphasis on its importance, considerable effort is still needed to fully explore the therapeutic potential of nitrones. The journey ahead holds great promise and invites further investigation.
5. Conclusions
In summary, this comprehensive review highlights the critical role of nitrones in organic chemistry. It covers their synthetic methodologies, diverse applications in organic synthesis, physicochemical properties, potential therapeutic relevance, and opportunities for spin trapping.
The versatility of reactions involving nitrones highlights the need for further exploration of their synthesis. Some known methodologies for preparing nitrones have been thoroughly elucidated and explained, utilizing the best available knowledge. However, some mechanisms still require confirmation, and the conversion rates of specific reactions are not always complete. So, in-depth mechanistic studies can provide valuable insights into the fundamental principles governing these reactions and pave the way for the development of more efficient and selective processes. Additionally, the emergence of new metal-catalyzed reactions has opened up exciting possibilities in chemical synthesis and ensures that there remains ample room for improvement and discoveries.
Nitrones’ ability to scavenge free radicals and reduce oxidative stress in biological systems presents promising opportunities for the development of new therapeutic agents. Investigating nitrones as potential treatments for various diseases, including neurodegenerative disorders and cardiovascular conditions, is a promising area for future research.
In industry, nitrones can be explored in radical polymerization, enabling the production of advanced polymeric materials with new molecular structures that can be efficiently designed. The extent of their impact on fields such as nanotechnology, optics, and supramolecular chemistry remains to be fully explored.
Ultimately, this review of nitrones aims to reveal their chemical properties, stimulate the development of novel reactions, facilitate the discovery of potential therapeutics, and broaden their applications across various fields.
Author Contributions
Conceptualization, A.M.S.S. and D.C.G.A.P.; methodology, R.A.L.S.S.; writing—original draft preparation, R.A.L.S.S. and D.C.G.A.P.; writing—review and editing, D.C.G.A.P. and A.M.S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors are grateful to the University of Aveiro, to the FCT (Fundação para a Ciência e a Ténologia) and the MCTES (Ministério da Ciência, Técnologia e Ensino Superior) for their support, through the project LAQV-REQUIMTE (UID/50006/2025). R.A.L.S. Santos is also thankful to the FCT and to the ESF (European Social Fund) through the NORTE 2020 program (Northern Region Operational Program) for funding his PhD scholarship ref. UI/BD/151268/2021.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Ac | Acetyl |
| AIBN | 2,2′-Azobis(2-methylpropionitrile) |
| Ar | Aryl |
| BINAP | ([1,1′-Binaphthalene]-2,2′ diyl)bis(diphenylphosphane) |
| Bn | Benzyl |
| BPO | Benzoyl peroxide |
| bpy | 2,2’-Bipyridine |
| Bu | Butyl |
| Cp* | 1,2,3,4,5-Pentamethylcyclopentadienyl |
| DCE | 1,2-Dichloroethane |
| DCM | Dichloromethane |
| DEPMPO | 2-(Diethoxyphosphoryl)-2-methyl-3,4-dihydro-2H-pyrrole-1-oxide |
| DIBAL | Diisobutylaluminium hydride |
| DIPEA | N-Ethyl-N-(propan-2-yl)propan-2-amine |
| DMD | Dimethyldioxirane |
| DMPO | 2,2-Dimethyl-3,4-dihydro-2H-pyrrole 1-oxide |
| DNA | Deoxyribonucleic acid |
| E/E+ | Electrophile |
| EMPO | 2-(Ethoxycarbonyl)-2-methyl-3,4-dihydro-2H-pyrrole-1-oxide |
| ESR | Electron spin resonance |
| EPR | Electron paramagnetic resonance |
| Et | Ethyl |
| Hfcf | Hyperfine coupling frequency |
| HPLC | High performance liquid chromatography |
| IBX | o-Iodoxybenzoic acid |
| IPr | 1,3-Bis(2,5-diisopropylphenyl)imidazol-2-ylidene |
| IR | Infrared |
| IUPAC | International Union of Pure and Applied Chemistry |
| LDA | Lithium diisopropylamide |
| LTMP | Lithium tetramethylpiperidide |
| m-CPBA | 3-Chlorobenzene-1-carboperoxoic acid |
| Me | Methyl |
| Mes-Acr+ | 9-Mesityl-10-methylacrydinium |
| MIP | 2-Methoxyisopropyl |
| MTO | Methylrhenium trioxide |
| Ms | Mesyl/methylsulfonyl |
| MS | Molecular sieves |
| NBS | 1-Bromopyrrolidine-2,5-dione |
| NMR | Nuclear magnetic resonance |
| Nu/Nu− | Nucleophile |
| Oxone® | Potassium peroxysulfate |
| PBA | Perbenzoic acid |
| PBN | N-t-Butyl-1-phenylmethanimine oxide |
| Ph | Phenyl |
| Phen | 1,10-Phenanthroline |
| PIN | Preferred IUPAC Name |
| PPTS | Pyridinium p-toluenesulfonate |
| ROS | Reactive Oxygen Species |
| RNS | Reactive Nitrogen Species |
| SCE | Saturated calomel electrode (0.241 V vs. SHE, 25 °C) |
| SHE | Standard hydrogen electrode |
| SOD | Superoxide dismutase |
| TBAF | Tetrabutylammonium fluoride |
| TBDMS | tert-Butyldimethylsilyl |
| TBDPS | tert-Butyldiphenylsilyl |
| TEA | Triethylamine |
| Tf | Triflyl |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TIPS-EBX | 1-[(Triisopropylsilyl)ethynyl]-1,2-benziodoxol-3-(1H)-one |
| TMS | Trimethylsilyl |
| Tol | Tolyl/(methylphenyl) |
| Ts | Tosyl/((4-methylphenyl)sulfonyl) |
References
- Beckmann, E. Zur kenntniss der aldoxime. VII, Ber. Dtsch. Chem. Ges. 1890, 23, 3331–3341. [Google Scholar] [CrossRef]
- Smith, L.I. Aliphatic diazo compounds, nitrones, and structurally analogous compounds. systems capable of undergoing 1,3-additions. Chem. Rev. 1938, 23, 193–285. [Google Scholar] [CrossRef]
- Feuer, H. Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis: Novel Strategies in Synthesis, 2nd ed.; John Wiley & Sons: New Jersey, NY, USA, 2008; pp. 129–434. [Google Scholar] [CrossRef]
- Hamer, J.; Macaluso, A. Nitrones. Chem. Rev. 1964, 64, 473–495. [Google Scholar] [CrossRef]
- Favre, H.A.; Powell, W.H. Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names 2013; Royal Society of Chemistry: London, UK, 2013. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent advances of catalytic asymmetric 1,3-dipolar cycloadditions. Chem. Rev. 2015, 115, 5366–5412. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L. Diverse applications of nitrones for the synthesis of heterocyclic compounds. Asian J. Org. Chem. 2016, 5, 9–30. [Google Scholar] [CrossRef]
- Shi, W.-M.; Ma, X.-P.; Su, G.-F.; Mo, D.-L. New developments of ketonitrones in organic synthesis. Org. Chem. Front. 2016, 3, 116–130. [Google Scholar] [CrossRef]
- Murahashi, S.-I.; Imada, Y. Synthesis and transformations of nitrones for organic synthesis. Chem. Rev. 2019, 119, 4684–4716. [Google Scholar] [CrossRef]
- Tamura, O. Exploration and development of nitrone chemistry. Chem. Pharm. Bull. 2024, 72, 731–746. [Google Scholar] [CrossRef]
- Bonnett, R.; Brown, R.F.C.; Clark, V.M.; Sutherland, I.O.; Todd, A. 423. Experiments towards the synthesis of corrins. Part II. The preparation and reactions of Δ1-pyrroline 1 oxides. J. Chem. Soc. 1959, 2094–2102. [Google Scholar] [CrossRef]
- De La Mare, H.E.; Coppinger, G.M. Oxidation of N,N-dialkyl hydroxylamines with t-butyl hydroperoxide. A new synthesis for nitrones. J. Org. Chem. 1963, 28, 1068–1070. [Google Scholar] [CrossRef]
- Cicchi, S.; Corsi, M.; Goti, A. Inexpensive and environmentally friendly oxidation of hydroxylamines to nitrones with bleach. J. Org. Chem. 1999, 64, 7243–7245. [Google Scholar] [CrossRef]
- Murahashi, S.-I.; Mitsui, H.; Watanabe, T.; Zenki, S.-I. The reaction of N-mono an N,N-disubstituted hydroxylamines with palladium catalyst. Tetrahedron Lett. 1983, 24, 1049–1052. [Google Scholar] [CrossRef]
- Ali, S.A.; Hashmi, S.M.A.; Siddiqui, M.N.; Wazeer, M.I.M. Regiochemistry of mercury(II) oxide oxidation of unsymmetrical N,N-disubstituted hydroxylamines. Tetrahedron 1996, 52, 14917–14928. [Google Scholar] [CrossRef]
- Goti, A.; De Sarlo, F.; Romani, M. Highly efficient and mild synthesis of nitrones by catalytic oxidation of hydroxylamines with tetra-n-propylammonium perruthenate. Tetrahedron Lett. 1994, 35, 6571–6574. [Google Scholar] [CrossRef]
- Cicchi, S.; Cardona, F.; Brandi, A.; Corsi, M.; Goti, A. Oxidation of hydroxylamines to nitrones catalyzed by (Salen)Mn(III) complexes. Enantioselective synthesis of a protected cis-dihydroxypyrroline N-oxide with Jacobsen catalyst. Tetrahedron Lett. 1999, 40, 1989–1992. [Google Scholar] [CrossRef]
- Cicchi, S.; Marradi, M.; Goti, A.; Brandi, A. Manganese dioxide oxidation of hydroxylamines to nitrones. Tetrahedron Lett. 2001, 42, 6503–6505. [Google Scholar] [CrossRef]
- Saladino, R.; Neri, V.; Cardona, F.; Goti, A. Oxidation of N,N-disubstituted hydroxylamines to nitrones with hydrogen peroxide catalyzed by polymer-supported methylrhenium trioxide systems. Adv. Synth. Catal. 2004, 346, 639–647. [Google Scholar] [CrossRef]
- Bowman, D.F.; Gillan, T.; Ingold, K.U. Kinetic applications of electron paramagnetic resonance spectroscopy. III. Self-reactions of dialkyl nitroxide radicals. J. Am. Chem. Soc. 1971, 93, 6555–6561. [Google Scholar] [CrossRef]
- Ali, S.A.; Wazeer, M.I.M. Peracid induced ring opening of isoxazolidines. A mechanistic study. Tetrahedron Lett. 1992, 33, 3219–3222. [Google Scholar] [CrossRef]
- Balagam, B.; Richardson, D.E. The mechanism of carbon dioxide catalysis in the hydrogen peroxide N-oxidation of amines. Inorg. Chem. 2008, 47, 1173–1178. [Google Scholar] [CrossRef]
- Marcantoni, E.; Petrini, M.; Polimanti, O. Oxidation of secondary amines to nitrones using urea-hydrogen peroxide complex (UHP) and metal catalysts. Tetrahedron Lett. 1995, 36, 3561–3562. [Google Scholar] [CrossRef]
- Somasundaram, N.; Srinivasan, C. Oxygenation of aldimines and deoxygenation of nitrones on irradiated TiO2. Tetrahedron Lett. 1998, 39, 3547–3550. [Google Scholar] [CrossRef]
- Gella, C.; Ferrer, È.; Alibés, R.; Busqué, F.; March, P.; Figueredo, M.; Font, J. A metal-free general procedure for oxidation of secondary amines to nitrones. J. Org. Chem. 2009, 74, 6365–6367. [Google Scholar] [CrossRef]
- Eckert, T.S.; Bruice, T.C. Chemical properties of phenanthrolinequinones and the mechanism of amine oxidation by o-quinones of medium redox potentials. J. Am. Chem. Soc. 1983, 105, 4431–4441. [Google Scholar] [CrossRef]
- Furukawa, S.; Ohno, Y.; Shishido, T.; Teramura, K.; Tanaka, T. Reaction mechanism of selective photooxidation of amines over niobium oxide: Visible-light-induced electron transfer between adsorbed amine and Nb2O5. J. Phys. Chem. C 2013, 117, 442–450. [Google Scholar] [CrossRef]
- Barnes, K.K.; Mann, C.K. Electrochemical oxidation of primary aliphatic amines. J. Org. Chem. 1967, 32, 1474–1479. [Google Scholar] [CrossRef]
- Lin, Y.-M.; Miller, M.J. Practical synthesis of hydroxamate-derived siderophore components by an indirect oxidation method and syntheses of a DIG−siderophore conjugate and a biotin−siderophore conjugate. J. Org. Chem. 1999, 64, 7451–7458. [Google Scholar] [CrossRef]
- Ogata, Y.; Sawaki, Y. Peracid oxidation of imines. Kinetics and mechanism of competitive formation of nitrones and oxaziranes from cyclic and acyclic imines. J. Am. Chem. Soc. 1973, 95, 4692–4698. [Google Scholar] [CrossRef]
- Cisneros, L.; Serna, P.; Corma, A. Selective reductive coupling of nitro compounds with aldehydes to nitrones in H2 using carbon-supported and -decorated platinum nanoparticles. Angew. Chem. 2014, 126, 9460–9464. [Google Scholar] [CrossRef]
- Colacino, E.; Nun, P.; Colacino, F.M.; Martinez, J.; Lamaty, F. Solvent-free synthesis of nitrones in a ball-mill. Tetrahedron 2008, 64, 5569–5576. [Google Scholar] [CrossRef]
- Diez-Martinez, A.; Gultekin, Z.; Delso, I.; Tejero, T.; Merino, P. Synthesis of N-(benzyloxyethyl)- and N-(alkoxycarbonylmethyl)nitrones. Synthesis 2010, 2010, 678–688. [Google Scholar] [CrossRef]
- Hulsbos, E.; Marcus, J.; Brussee, J.; van der Gen, A. Direct conversion of chiral cyanohydrins to chiral nitrones by transamination. Tetrahedron Asymmetry 1997, 8, 1061–1067. [Google Scholar] [CrossRef]
- Sin, N.; Venables, B.L.; Liu, X.; Huang, S.; Gao, Q.; Ng, A.; Dalterio, R.; Rajamani, R.; Meanwell, N.A. The alkylation of isatin-derived oximes: Spectroscopic and X-ray crystallographic structural characterization of oxime and nitrone products. J. Heterocycl. Chem. 2009, 46, 432–442. [Google Scholar] [CrossRef]
- Roca-López, D.; Darù, A.; Tejero, T.; Merino, P. Revisiting oxime–nitrone tautomerism. Evidence of nitrone tautomer participation in oxime nucleophilic addition reactions. RSC Adv. 2016, 6, 22161–22173. [Google Scholar] [CrossRef]
- Allouch, A.; Roubaud, V.; Lauricella, R.; Bouteiller, J.-C.; Tuccio, B. Preparation and use as spin trapping agents of new ester -nitrones. Org. Biomol. Chem. 2003, 1, 593–598. [Google Scholar] [CrossRef]
- Cicchi, S.; Marradi, M.; Vogel, P.; Goti, A. One-pot synthesis of cyclic nitrones and their conversion to pyrrolizidines: 7a-epi-crotanecine inhibits α-mannosidases. J. Org. Chem. 2006, 71, 1614–1619. [Google Scholar] [CrossRef]
- Kontokosta, D.; Mueller, D.S.; Mo, D.-L.; Pace, W.H.; Simpson, R.A.; Anderson, L.L. Copper-mediated synthesis of N-alkenyl-α,β-unsaturated nitrones and their conversion to tri- and tetrasubstituted pyridines. Beilstein J. Org. Chem. 2015, 11, 2097–2104. [Google Scholar] [CrossRef]
- Nakamura, I.; Zhang, D.; Terada, M. Copper-catalyzed tandem [2,3]-rearrangement and 6π-3-azatriene electrocyclization in (E)-O-propargylic α,β-unsaturated oximes. J. Am. Chem. Soc. 2010, 132, 7884–7886. [Google Scholar] [CrossRef] [PubMed]
- Grigg, R.; Hadjisoteriou, M.; Kennewell, P.; Markandu, J.; Thornton-Pett, M. Mercuric acetate induced formation of cyclic nitrones from alkenyl oximes. J. Chem. Soc., Chem. Commun. 1992, 19, 1388–1389. [Google Scholar] [CrossRef]
- Noguchi, M.; Okada, H.; Nishimura, S.; Yamagata, Y.; Takamura, S.; Tanaka, M.; Kakehi, A.; Yamamoto, H. A simple oxime–nitrone isomerisation and intramolecular nitrone-cycloaddition reaction of 3-(alk-2-enylamino)propionaldehyde oximes. J. Chem. Soc. Perkin Trans. 1 1999, 185–192. [Google Scholar] [CrossRef]
- Grigg, R.; Hadjisoteriou, M.; Kennewell, P.; Markandu, J.; Thornton-Pett, M. Halogen-induced formation of nitrones from oximes and alkenes. J. Chem. Soc., Chem. Commun. 1993, 1340–1342. [Google Scholar] [CrossRef]
- Peng, X.; Tong, B.M.K.; Hirao, H.; Chiba, S. Inorganic-base-mediated hydroamination of alkenyl oximes for the synthesis of cyclic nitrones. Angew. Chem. 2014, 126, 1990–1993. [Google Scholar] [CrossRef]
- Nakama, K.; Seki, S.; Kanemasa, S. A new synthetic access to N-alkylated nitrones through Lewis acid-catalyzed conjugate additions of aldoximes. Tetrahedron Lett. 2001, 42, 6719–6722. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, J.; Sun, B.; Zhou, T.; Liu, M.; Liu, S.; Zhang, M.; Zhang, Q. Asymmetric nitrone synthesis via ligand-enabled copper-catalyzed Cope-type hydroamination of cyclopropene with oxime. J. Am. Chem. Soc. 2017, 139, 11702–11705. [Google Scholar] [CrossRef]
- Han, B.; Yang, X.L.; Fang, R.; Yu, W.; Wang, C.; Duan, X.Y.; Liu, S. Oxime radical promoted dioxygenation, oxyamination, and diamination of alkenes: Synthesis of isoxazolines and cyclic nitrones. Angew. Chem. Int. Ed. 2012, 51, 8816–8820. [Google Scholar] [CrossRef]
- Fox, M.E.; Holmes, A.B.; Forbes, I.T.; Thompson, M. N-alkenyl nitrone dipolar cycloaddition routes to piperidines and indolizidines. Part 7. Hydroxylamine–alkyne cyclisations. Formation of cyclic nitrones and application to the synthesis of the proposed structure for (±)-acacialactam. J. Chem. Soc. Perkin Trans. 1 1994, 3379–3395. [Google Scholar] [CrossRef]
- Krenske, E.H.; Davidson, E.C.; Forbes, I.T.; Warner, J.A.; Smith, A.L.; Holmes, A.B.; Houk, K.N. Reverse Cope elimination of hydroxylamines and alkenes or alkynes: Theoretical investigation of tether length and substituent effects. J. Am. Chem. Soc. 2012, 134, 2434–2441. [Google Scholar] [CrossRef]
- Beauchemin, A.M.; Moran, J.; Lebrun, M.E.; Seguin, C.; Dimitrijevic, E.; Zhang, L.; Gorelsky, S.I. Intermolecular Cope-type hydroamination of alkenes and alkynes. Angew. Chem. Inter. Ed. 2008, 47, 1410–1413. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhang, L.; Yang, J.; Xu, B.; Xiao, Y.; Zhang, J. Pyrroles versus cyclic nitrones: Catalyst-controlled divergent cyclization of N-(2-perfluoroalkyl-3-alkynyl) hydroxylamines. Chem. Commun. 2014, 50, 4203–4206. [Google Scholar] [CrossRef]
- Huple, D.B.; Ghorpade, S.; Liu, R.-S. Recent advances in gold-catalyzed N- and O-functionalizations of alkynes with nitrones, nitroso, nitro and nitroxy species. Adv. Synth. Catal. 2016, 358, 1348–1367. [Google Scholar] [CrossRef]
- Moran, J.; Pfeiffer, J.Y.; Gorelsky, S.I.; Beauchemin, A.M. Ketonitrones via Cope-type hydroamination of allenes. Org. Lett. 2009, 11, 1895–1898. [Google Scholar] [CrossRef]
- Perrine, T.D.; Sargent, L.J. Studies in the acridine series. VI. The reaction of certain 9-formylacridines with 3-dibutylaminopropylmagnesium chloride. J. Org. Chem. 1949, 14, 583–592. [Google Scholar] [CrossRef]
- Bou-Moreno, R.; Luengo-Arratta, S.; Motherwell, W.B. Observations on the reaction of nitronate anions with oxalyl chloride: A new method for the preparation of geminal chloronitroso compounds. Tetrahedron Lett. 2011, 52, 2097–2099. [Google Scholar] [CrossRef]
- Adam, W.; Krebs, O. The nitroso ene reaction: A regioselective and stereoselective allylic nitrogen functionalization of mechanistic delight and synthetic potential. Chem. Rev. 2003, 103, 4131–4146. [Google Scholar] [CrossRef]
- Chavannavar, A.P.; Oliver, A.G.; Ashfeld, B.L. An umpolung approach toward N-aryl nitrone construction: A phosphine-mediated addition of 1,2-dicarbonyls to nitroso electrophiles. Chem. Commun. 2014, 50, 10853–10856. [Google Scholar] [CrossRef]
- More, S.A.; Kardile, R.D.; Kuo, T.-C.; Cheng, M.-J.; Liu, R.-S. Gold catalysts can generate nitrone intermediates from a nitrosoarene/alkene mixture, enabling two distinct catalytic reactions: A nitroso-activated cycloheptatriene/benzylidene rearrangement. Org. Lett. 2021, 23, 5506–5511. [Google Scholar] [CrossRef]
- Huang, W.-Y.; Gurubrahamam, R.; Chen, K. An unprecedented organocascade synthesis of functionalized bicyclic nitrones from 2-aminomalonate derived nucleophiles and 1-nitro-1,3-enynes via allenes formation and subsequent rearrangement. Adv. Synth. Cat. 2019, 361, 170–175. [Google Scholar] [CrossRef]
- Katahara, S.; Kobayashi, S.; Fujita, K.; Matsumoto, T.; Sato, T.; Chida, N. An iridium-catalyzed reductive approach to nitrones from N-hydroxyamides. J. Am. Chem. Soc. 2016, 138, 5246–5249. [Google Scholar] [CrossRef]
- Couturier, M.; Tucker, J.L.; Proulx, C.; Boucher, G.; Dubé, P.; Andresen, B.M.; Ghosh, A. 5,5-Dimethyl-1,4,2-dioxazoles as versatile aprotic hydroxamic acid protecting groups. J. Org. Chem. 2002, 67, 4833–4838. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Zhang, H.-H.; Xu, P.-F.; Luo, Y.-C. Synthesis of five-membered cyclic nitrones based on the Lewis acid-catalysed [3+2]-annulation reaction of donor–acceptor cyclopropanes with 1,4,2-dioxazoles. Chem. Commun. 2018, 54, 10128–10131. [Google Scholar] [CrossRef]
- Kraïem, J.; Ollevier, T. Atom economical synthesis of N-alkylbenzamides via the iron(III) sulfate catalyzed rearrangement of 2-alkyl-3-aryloxaziridines in water and in the presence of a surfactant. Green Chem. 2017, 19, 1263–1267. [Google Scholar] [CrossRef]
- Oliveros, E.; Riviere, M.; Malrieu, J.P.; Teichteil, C. Theoretical exploration of the photochemical rearrangement of oxaziridines. J. Am. Chem. Soc. 1979, 101, 318–322. [Google Scholar] [CrossRef]
- Shinzawa, K.; Tanaka, I. The photochemical isomerization of α,N-diphenylnitrone. J. Phys. Chem. 1964, 68, 1205–1213. [Google Scholar] [CrossRef]
- Koyano, K.; Tanaka, I. The photochemical and thermal isomerization of trans- and cis-α-cyano-α-phenyl-N-phenylnitrones. J. Phys. Chem. 1965, 69, 2545–2550. [Google Scholar] [CrossRef]
- Newcomb, M.; Reeder, R.A. Reactions of trans-2-tert-butyl-3-phenyloxaziridine with lithium amide bases. J. Org. Chem. 1980, 45, 1489–1493. [Google Scholar] [CrossRef]
- Park, J.; Park, S.; Jang, G.S.; Kim, R.H.; Jung, J.; Woo, S.K. Weak base-promoted selective rearrangement of oxaziridines to amides via visible-light photoredox catalysis. Chem. Commun. 2021, 57, 9995–9998. [Google Scholar] [CrossRef]
- Darù, A.; Roca-López, D.; Tejero, T.; Merino, P. Revealing stepwise mechanisms in dipolar cycloaddition reactions: Computational study of the reaction between nitrones and isocyanates. J. Org. Chem. 2016, 81, 673–680. [Google Scholar] [CrossRef]
- Merino, P.; Tejero, T.; Mannucci, V. Experimental and theoretical evidences of 2-aza-Cope rearrangement of nitrones. Tetrahedron Lett. 2007, 48, 3385–3388. [Google Scholar] [CrossRef]
- Moderhack, D.; Lorke, M. Stable 4-imino-1,2-oxazetidines from N-neopentylidene-tert-butylamine N-oxide and isocyanides. Angew. Chem. Int. Ed. Eng. 1980, 19, 45–46. [Google Scholar] [CrossRef]
- Xu, X.; Doyle, M.P. The [3 + 3]-cycloaddition alternative for heterocycle syntheses: Catalytically generated metalloenolcarbenes as dipolar adducts. Acc. Chem. Res. 2014, 47, 1396–1405. [Google Scholar] [CrossRef]
- Wang, X.; Xu, X.; Zavalij, P.Y.; Doyle, M.P. Asymmetric formal [3 + 3]-cycloaddition reactions of nitrones with electrophilic vinylcarbene intermediates. J. Am. Chem. Soc. 2011, 133, 16402–16405. [Google Scholar] [CrossRef]
- Young, I.S.; Kerr, M.A. A homo [3+2] dipolar cycloaddition: The reaction of nitrones with cyclopropanes. Angew. Chem. 2003, 115, 3131–3134. [Google Scholar] [CrossRef]
- Wanapun, D.; Van Gorp, K.A.; Mosey, N.J.; Kerr, M.A.; Woo, T.K. The mechanism of 1,3-dipolar cycloaddition reactions of cyclopropanes and nitrones—A theoretical study. Can. J. Chem. 2005, 83, 1752–1767. [Google Scholar] [CrossRef]
- Karadeolian, A.; Kerr, M.A. Examination of homo-[3 + 2]-dipolar cycloaddition: Mechanistic insight into regio- and diastereoselectivity. J. Org. Chem. 2007, 72, 10251–10253. [Google Scholar] [CrossRef]
- Xu, P.-W.; Liu, J.K.; Shen, L.; Cao, Z.Y.; Zhao, X.L.; Yan, J.; Zhou, J. Diastereo- and enantioselective [3 + 3] cycloaddition of spirocyclopropyl oxindoles using both aldonitrones and ketonitrones. Nat. Commun. 2017, 8, 1619. [Google Scholar] [CrossRef]
- Schneider, T.F.; Kaschel, J.; Werz, D.B. A new golden age for donor–acceptor cyclopropanes. Angew. Chem. Int. Ed. 2014, 53, 5504–5523. [Google Scholar] [CrossRef]
- Kang, Y.-B.; Sun, X.-L.; Tang, Y. Highly enantioselective and diastereoselective cycloaddition of cyclopropanes with nitrones and its application in the kinetic resolution of 2-substituted cyclopropane-1,1-dicarboxylates. Angew. Chem. 2007, 119, 3992–3995. [Google Scholar] [CrossRef]
- Thakur, S.; Das, A.; Das, T. 1,3-Dipolar cycloaddition of nitrones: Synthesis of multisubstituted, diverse range of heterocyclic compounds. New J. Chem. 2021, 45, 11420–11456. [Google Scholar] [CrossRef]
- Li, T.-Z.; Liu, S.-J.; Sun, Y.-W.; Deng, S.; Tan, W.; Jiao, Y.; Zhang, Y.-C.; Shi, F. Regio- and enantioselective [3+3]-cycloaddition of nitrones with 2-indolylmethanols enabled by cooperative organocatalysis. Angew. Chem. Int. Ed. 2021, 60, 2355–2363. [Google Scholar] [CrossRef]
- Shintani, R.; Park, S.; Duan, W.-L.; Hayash, T. Palladium-catalyzed asymmetric [3+3]-cycloaddition of trimethylenemethane derivatives with nitrones. Angew. Chem. Int. Ed. 2007, 46, 5901–5903. [Google Scholar] [CrossRef]
- Yang, J. Recent developments in nitrone chemistry: Some novel transformations. Synlett 2012, 23, 2293–2297. [Google Scholar] [CrossRef]
- Zheng, H.; Faghihi, I.; Doyle, M.P. Copper(I)-catalyzed highly enantioselective [3+3]-cycloaddition of β-aryl/alkyl vinyl diazoacetates with nitrones. Helv. Chim. Acta 2021, 104, e2100081. [Google Scholar] [CrossRef]
- Efremova, M.M.; Kostikov, R.R.; Stepakov, A.V.; Panikorovsky, T.L.; Shcherbakova, V.S.; Ivanov, A.V.; Molchanov, A.P. Unusual Lewis-acid catalyzed formal [3+3]-cycloaddition of azomethine imines and nitrones to N-vinylpyrroles. Tetrahedron 2017, 73, 671–680. [Google Scholar] [CrossRef]
- Rück-Braun, K.; Freysoldt, T.H.E.; Wierschem, F. 1,3-Dipolar cycloaddition on solid supports: Nitrone approach towards isoxazolidines and isoxazolines and subsequent transformations. Chem. Soc. Rev. 2005, 34, 507–516. [Google Scholar] [CrossRef] [PubMed]
- García Ruano, J.L.; Fraile, A.; Martín Castro, A.M.; Martín, M.R. The role of the sulfinyl group on the course of the reactions of 3-p-tolylsulfinylfuran-2(5H)-ones with nitrones. Synthetic uses of cycloreversion processes. J. Org. Chem. 2005, 70, 8825–8834. [Google Scholar] [CrossRef]
- Baldwin, S.W.; Long, A. 2-tert-Butyl-3-methyl-2,3-dihydroimidazol-4-one-N-oxide: A new nitrone-based chiral glycine equivalent. Org. Lett. 2004, 6, 1653–1656. [Google Scholar] [CrossRef]
- Kanemasa, S.; Uemura, T.; Wada, E. Lewis acid-catalyzed nitrone cycloadditions to bidentate and tridentate α,β-unsaturated ketones. High rate acceleration, absolutely endo-selective and regioselective reactions. Tetrahedron Lett. 1992, 33, 7889–7892. [Google Scholar] [CrossRef]
- Carmona, D.; Lamata, M.P.; Viguri, F.; Rodríguez, R.; Lahoz, F.J.; Fabra, M.J.; Oro, L.A. Asymmetric 1,3-dipolar cycloaddition reaction of α,β-unsaturated nitriles with nitrones catalyzed by chiral-at-metal rhodium or iridium complexes. Tetrahedron Asymmetry 2009, 20, 1197–1205. [Google Scholar] [CrossRef]
- Baranski, A. Synthesis and properties of azoles and their derivatives. XLIII. Regio-and stereoselectivity of [2+3] cycloaddition reaction of E-beta-nitrostyrene to Z-C-aryl-N-phenylnitrones. Pol. J. Chem. 2000, 74, 767–775. [Google Scholar]
- Zhang, H.; Chan, W.H.; Lee, A.W.; Wong, W.Y. 1,3-Dipolar cycloadditions of prop-1-ene-1,3-sultone with nitrile oxides/nitrones. Tetrahedron Lett. 2003, 44, 395–397. [Google Scholar] [CrossRef]
- Merino, P.; Tejero, T.; Delso, I.; Matute, R. New mechanistic interpretations for nitrone reactivity. Org. Biomol. Chem. 2017, 15, 3364–3375. [Google Scholar] [CrossRef]
- Revuelta, J.; Cicchi, S.; Faggi, C.; Kozhushkov, S.I.; de Meijere, A.; Brandi, A. Synthesis of enantiopure indolizinones by cascade ring enlargements of 4‘-chlorospiro[cyclopropane-1,5‘-isoxazolidines]. J. Org. Chem. 2006, 71, 2417–2423. [Google Scholar] [CrossRef]
- Alibés, R.; Busqué, F.; de March, P.; Figueredo, M.; Font, J.; Parella, T. On the regioselectivity in nitrone cycloadditions to γ-oxo α,β-unsaturated esters. Tetrahedron 1998, 54, 10857–10878. [Google Scholar] [CrossRef]
- Karlsson, S.; Högberg, H.-E. Catalytic enantioselective 1,3-dipolar cycloaddition of nitrones to cyclopent-1-enecarbaldehyde. Tetrahedron Asymmetry 2002, 13, 923–926. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Wong, A.W.F.; Ikeno, T.; Yamada, T. Experimental and theoretical studies on stereo- and regioselectivity in intramolecular nitrone-alkene cycloaddition of hept-6-enoses derived from carbohydrates. J. Org. Chem. 2006, 71, 3253–3263. [Google Scholar] [CrossRef] [PubMed]
- Gębarowski, P.; Sas, W. Asymmetric synthesis of novel polyhydroxylated derivatives of indolizidine and quinolizidine by intramolecular 1,3-dipolar cycloaddition of N-(3-alkenyl)nitrones. Chem. Commun. 2001, 915–916. [Google Scholar] [CrossRef]
- Aurich, H.G.; Geiger, M.; Sievers, U. Regio- and diastereoselectivity in the reaction sequence from secondary 3-oxa-5-hexen-1-ylamines to bicyclic compounds via nitrones. Synlett 1997, 1997, 1004–1006. [Google Scholar] [CrossRef]
- Black, D.S.C.; Crozier, R.F.; Davis, V.C. 1,3-Dipolar cycloaddition reactions of nitrones. Synthesis 1975, 1975, 205–221. [Google Scholar] [CrossRef]
- Coşkun, N.; Tirli Tat, F.; Özel Güven, Ö. Synthesis of di- and cis-triaryl-3a,4,5,6-tetrahydroimidazo [1,5-b]isoxazoles and their ring-opening reactions. Tetrahedron 2001, 57, 3413–3417. [Google Scholar] [CrossRef]
- Malig, T.C.; Yu, D.; Hein, J.E. A revised mechanism for the Kinugasa reaction. J. Am. Chem. Soc. 2018, 140, 9167–9173. [Google Scholar] [CrossRef]
- Santoro, S.; Liao, R.-Z.; Marcelli, T.; Hammar, P.; Himo, F. Theoretical study of mechanism and stereoselectivity of catalytic Kinugasa reaction. J. Org. Chem. 2015, 80, 2649–2660. [Google Scholar] [CrossRef]
- Santoro, S.; Himo, F. Mechanism of the Kinugasa reaction revisited. J. Org. Chem. 2021, 86, 10665–10671. [Google Scholar] [CrossRef]
- Jiao, L.; Liang, Y.; Xu, J. Origin of the relative stereoselectivity of the β-lactam formation in the Staudinger reaction. J. Am. Chem. Soc. 2006, 128, 6060–6069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hsung, R.P.; Li, H.; Zhang, Y.; Johnson, W.L.; Figueroa, R. A highly stereoselective synthesis of chiral α-amino-β-lactams via the Kinugasa reaction employing ynamides. Org. Lett. 2008, 10, 3477–3479. [Google Scholar] [CrossRef]
- Tanaka, K.; Ohsuga, M.; Sugimoto, Y.; Okafuji, Y.; Mitsuhashi, K. Applications of the fluorinated 1,3-dipolar compounds as the building blocks of the heterocycles with fluorine groups. Part XII. Synthesis of trifluoromethylisoxazolines and their rearrangement into trifluoromethylaziridines. J. Fluorine Chem. 1988, 39, 39–45. [Google Scholar] [CrossRef]
- Mlostoń, G.; Obijalska, E.; Celeda, M.; Mittermeier, V.; Linden, A.; Heimgartner, H. 1,3-Dipolar cycloadditions of fluorinated nitrones with thioketones. J. Fluorine Chem. 2014, 165, 27–32. [Google Scholar] [CrossRef]
- Kuprianowicz, M.; Kaźmierczak, M.; Muszyńska, E.; Bzdęga, K.; Wójtowicz-Rajchel, H. 1,3-Dipolar cycloaddition in the synthesis of trifluoromethyl-substituted isoxazolidinyl derivatives of nucleobases. J. Fluorine Chem. 2018, 212, 112–121. [Google Scholar] [CrossRef]
- Wójtowicz-Rajchel, H.; Kaźmierczak, M. Chemo-, regio-, and stereoselectivity in 1,3-dipolar cycloaddition of piperine with nitrones. A cycloadditive route to aminoalcohols. New J. Chem. 2020, 44, 6015–6025. [Google Scholar] [CrossRef]
- Kowalski, M.K.; Mloston, G.; Obijalska, E.; Linden, A.; Heimgartner, H. First application of fluorinated nitrones for the synthesis of fluoroalkylated β-lactams via the Kinugasa reaction. Tetrahedron 2016, 72, 5305–5313. [Google Scholar] [CrossRef]
- Milcent, T.; Hinks, N.; Bonnet-Delpon, D.; Crousse, B. Trifluoromethyl nitrones: From fluoral to optically active hydroxylamines. Org. Biomol. Chem. 2010, 8, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Yamakawa, M.; Mori, Y. The electronic spectra of nitrones and the solvent effect on them. Bull. Chem. Soc. Jpn. 1963, 36, 1552–1563. [Google Scholar] [CrossRef]
- Kim, S.-U.; Liu, Y.; Nash, K.M.; Zweier, J.L.; Rockenbauer, A.; Villamena, F.A. Fast reactivity of a cyclic nitrone−calix [4]pyrrole conjugate with superoxide radical anion: Theoretical and experimental studies. J. Am. Chem. Soc. 2010, 132, 17157–17173. [Google Scholar] [CrossRef]
- Du, L.; Huang, S.; Zhuang, Q.; Jia, H.; Rockenbauer, A.; Liu, Y.; Liu, K.J.; Liu, Y. Highly sensitive free radical detection by nitrone-functionalized gold nanoparticles. Nanoscale 2014, 6, 1646–1652. [Google Scholar] [CrossRef]
- Lasri, J.; Eltayeb, N.E.; Haukka, M.; Alghamdi, Y. Crystal and molecular structure studies of (Z)-N-methyl-C-4-substituted phenyl nitrones by XRD, DFT, FTIR and NMR methods. J. Mol. Struct. 2017, 1128, 70–78. [Google Scholar] [CrossRef]
- Lo Celso, F.; Barone, G.; Maiuolo, L.; Algieri, V.; Cretu, C.; Calandra, P. Dissolution of nitrones in alkylphosphates: A structural study. J. Mol. Liq. 2022, 367, 120517. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, S.; Gao, Y.; Sun, F. Reducing volumetric shrinkage of photopolymerizable materials using reversible disulfide-bond reactions. J. Mater. Sci. 2018, 53, 16169–16181. [Google Scholar] [CrossRef]
- Kaminsky, L.S.; Lamchen, M. Nitrones. Part VIII. The ultraviolet absorption of the 1-pyrroline 1-oxides. J. Chem. Soc. B: Phys. Org. 1968, 1085–1087. [Google Scholar] [CrossRef]
- Husmann, R.; Wertz, S.; Daniliuc, C.G.; Schäfer, S.W.; McArdle, C.B.; Studer, A. UV–Vis monitoring of radical polymerizations by spin trapping with chromophoric nitrones. Macromolecules 2014, 47, 993–1000. [Google Scholar] [CrossRef]
- Khoee, S.; Memarian, H.R. Fluorescence self-quenching of substituted N,α-diphenylnitrones in various solvents. J. Photochem. Photobiol. A Chem. 2006, 177, 276–285. [Google Scholar] [CrossRef]
- Mutlaq, D.Z.; Hassan, Q.M.A.; Sultan, H.A.; Emshary, C.A. The optical nonlinear properties of a new synthesized azo-nitrone compound. Opt. Mater. 2021, 113, 110815. [Google Scholar] [CrossRef]
- McIntire, G.L.; Blount, H.N.; Stronks, H.J.; Shetty, R.V.; Janzen, E.G. Spin trapping in electrochemistry. 2. Aqueous and nonaqueous electrochemical characterizations of spin traps. J. Phys. Chem. 1980, 84, 916–921. [Google Scholar] [CrossRef]
- Villamena, F.A.; Das, A.; Nash, K.M. Potential implication of the chemical properties and bioactivity of nitrone spin traps for therapeutics. Future Med. Chem. 2012, 4, 1171–1207. [Google Scholar] [CrossRef]
- Kim, S.; Guilherme, V.D.; Bouajila, J.; Dias, A.G.; Cyrino, F.Z.; Bouskela, E.; Costa, P.R.; Nepveu, F. α-Phenyl-N-tert-butyl nitrone (PBN) derivatives: Synthesis and protective action against microvascular damages induced by ischemia/reperfusion. Bioorganic Med. Chem. 2007, 15, 3572–3578. [Google Scholar] [CrossRef]
- Rizzi, C.; Marque, S.; Belin, F.; Bouteiller, J.C.; Lauricella, R.; Tuccio, B.; Cerri, V.; Tordo, P. PPN-type nitrones: Preparation and use of a new series of β-phosphorylated spin-trapping agents. J. Chem. Soc. Perkin Trans. 2 1997, 2513–2518. [Google Scholar] [CrossRef]
- Dhainaut, A.; Tizot, A.; Raimbaud, E.; Lockhart, B.; Lestage, P.; Goldstein, S. Synthesis, structure, and neuroprotective properties of novel imidazolyl nitrones. J. Med. Chem. 2000, 43, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ohlweiler, D.F.; Carr, A.A.; Nieduzak, T.R.; Hay, D.A.; Adams, G.; Vaz, R.; Bernotas, R.C. Characterization of the radical trapping Activity of a novel series of cyclic nitrone spin traps. J. Biol. Chem. 1996, 271, 3097–3104. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Riesz, P. Sonochemistry of nitrone spin traps in aqueous solutions. Evidence for pyrolysis radicals from spin traps. Free Radic. Biol. Med. 1989, 7, 259–268. [Google Scholar] [CrossRef]
- Wikiwand—Biological Functions of Nitric Oxide. Available online: https://www.wikiwand.com/en/Biological_functions_of_nitric_oxide (accessed on 17 November 2025).
- Locigno, E.J.; Zweier, J.L.; Villamena, F.A. Nitric oxide release from the unimolecular decomposition of the superoxide radical anion adduct of cyclic nitrones in aqueous medium. Org. Biomol. Chem. 2005, 3, 3220–3227. [Google Scholar] [CrossRef]
- Floyd, R.A.; Kopke, R.D.; Choi, C.-H.; Foster, S.B.; Doblas, S.; Towner, R.A. Nitrones as therapeutics. Free Radic. Biol. Med. 2008, 45, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.P.; Angiolini, P.; Tani, R.; Consales, G.; Bordi, L. Phenyl-t-butyl-nitrone is active against traumatic shock in rats. Free Radic. Res. Commun. 1986, 1, 321–327. [Google Scholar] [CrossRef]
- Antonic, A.; Dottori, M.; Macleod, M.R.; Donnan, G.A.; Howells, D.W. NXY-059, a failed stroke neuroprotectant, offers no protection to stem cell-derived human neurons. J. Stroke Cerebrovasc. Dis. 2018, 27, 2158–2165. [Google Scholar] [CrossRef] [PubMed]


























































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.