
Hydrogen Bonding of Trialkyl-Substituted Urea in Organic Environment

 ,
,  and
and
Abstract
1. Introduction
Characterization of Urea Species by IR
2. Results and Discussion
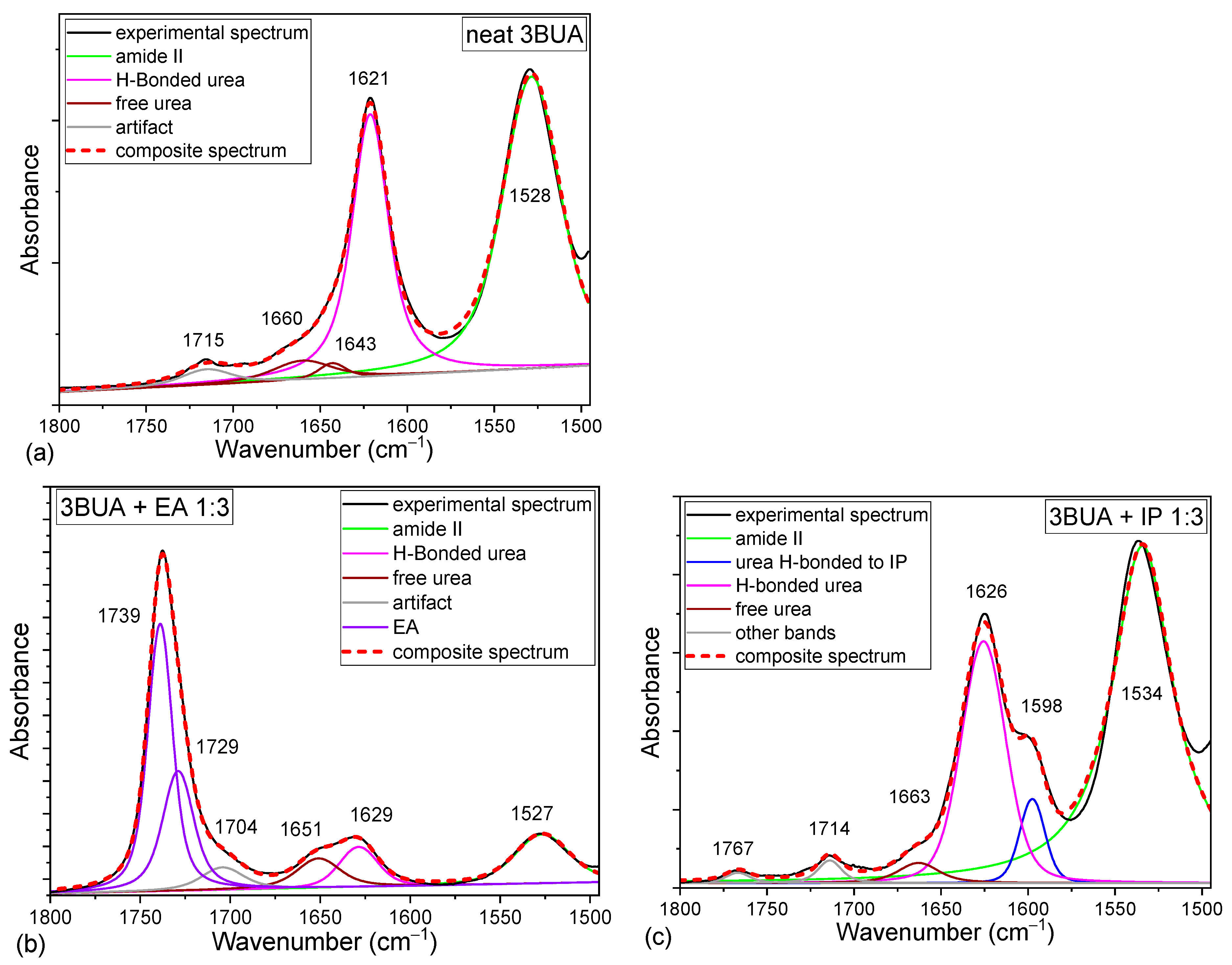
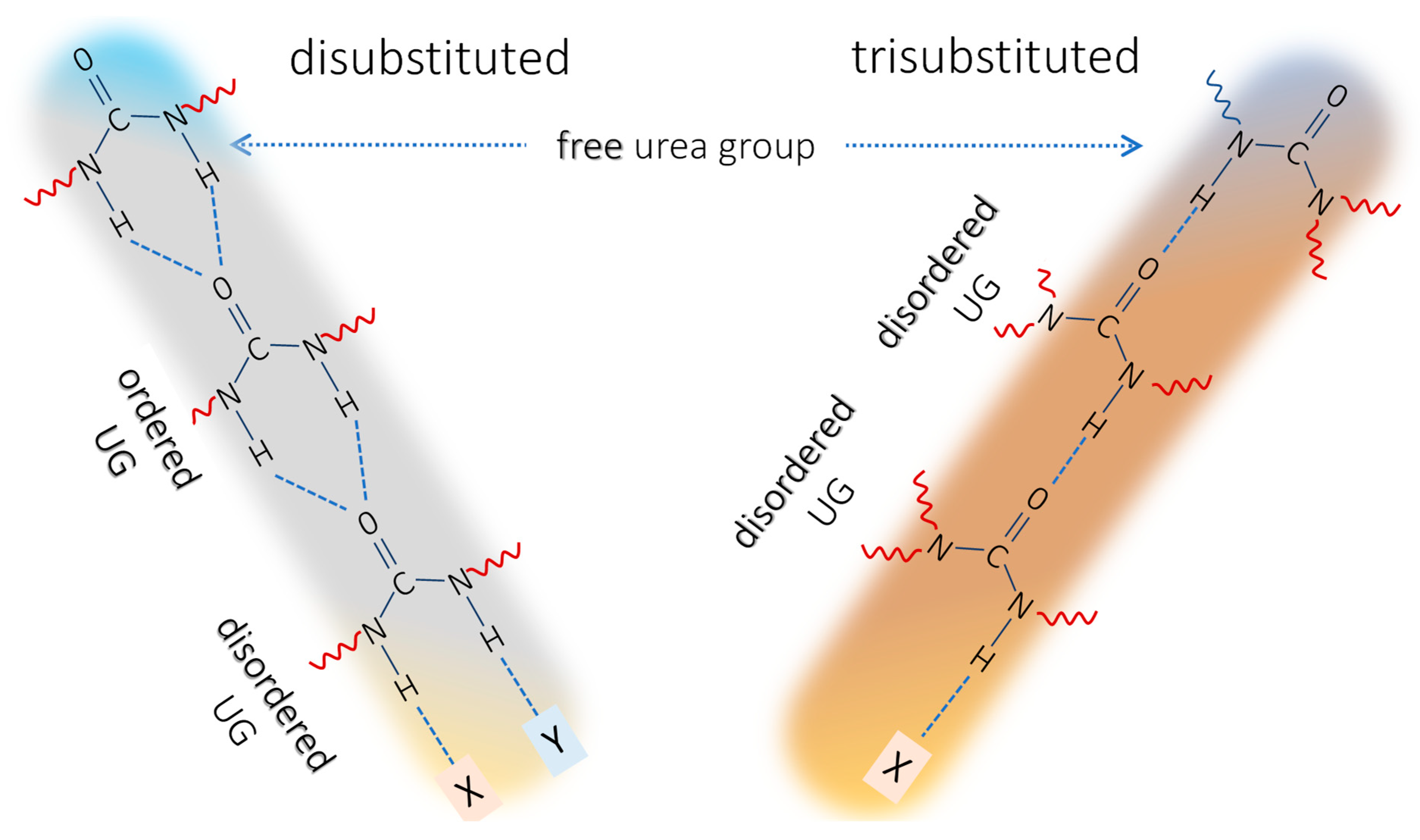
2.1. Background on the Urea IR Spectroscopy
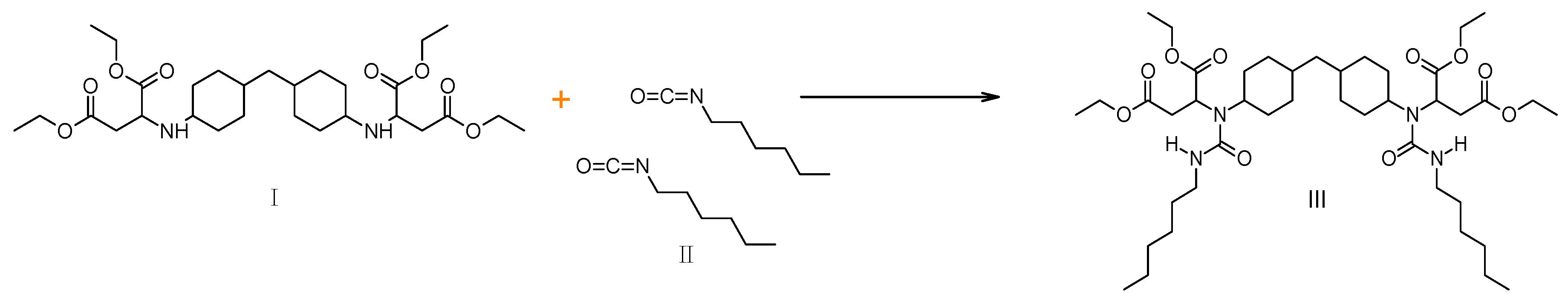
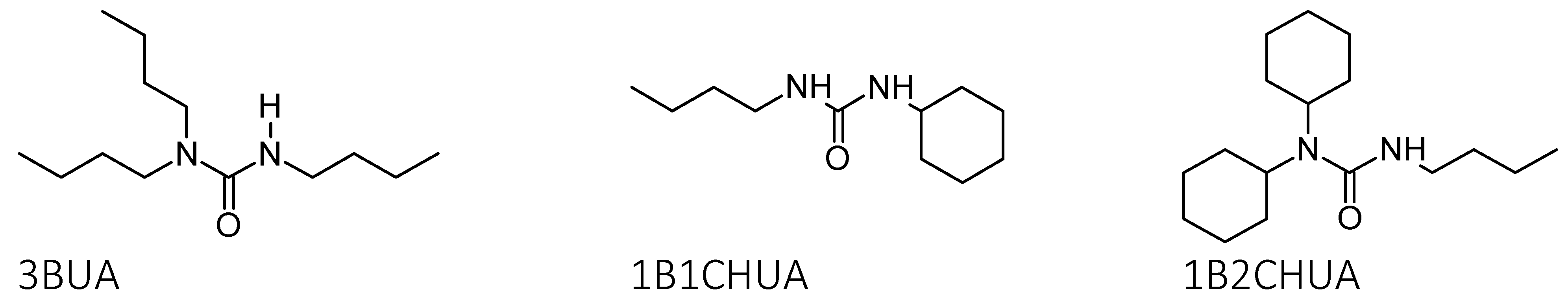
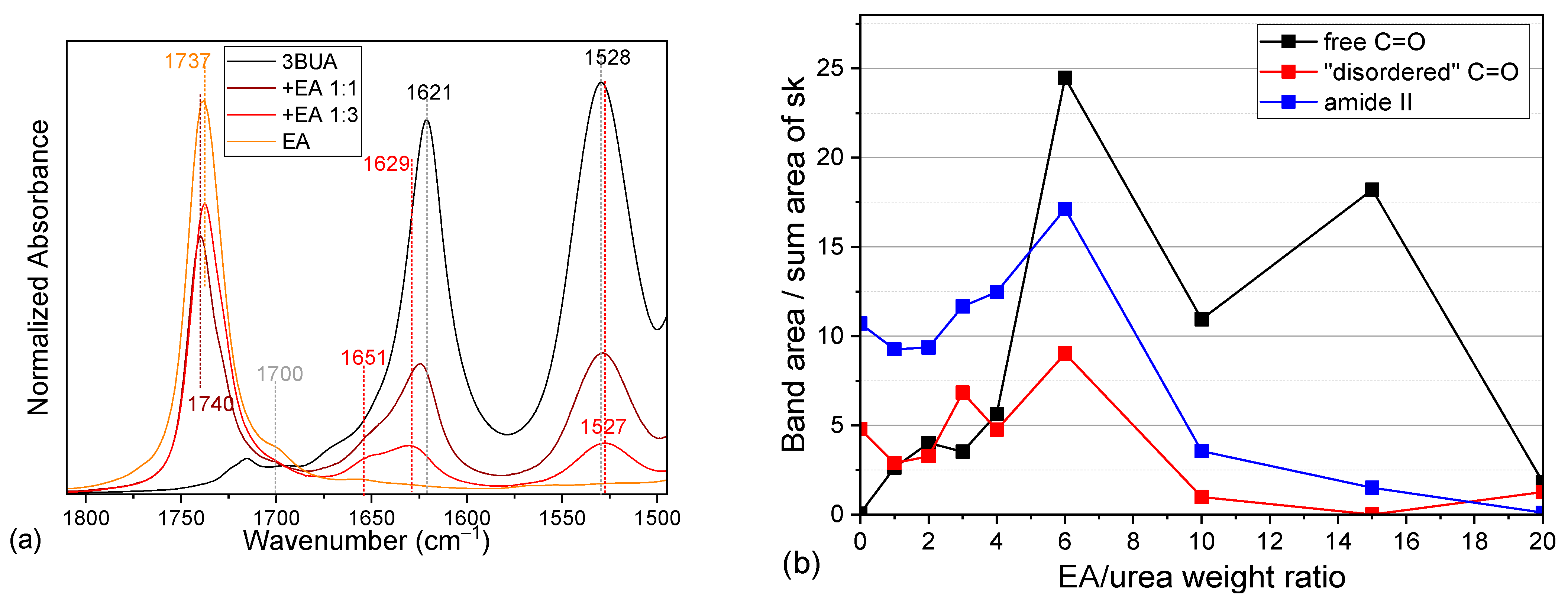
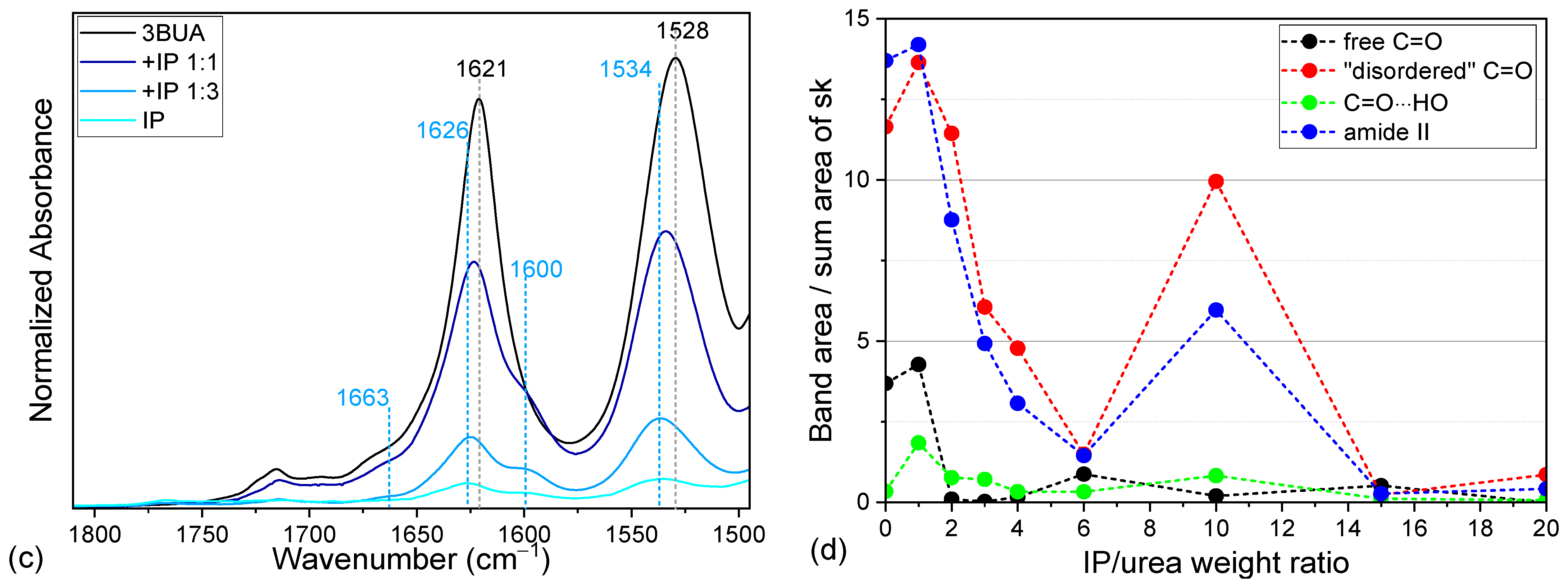
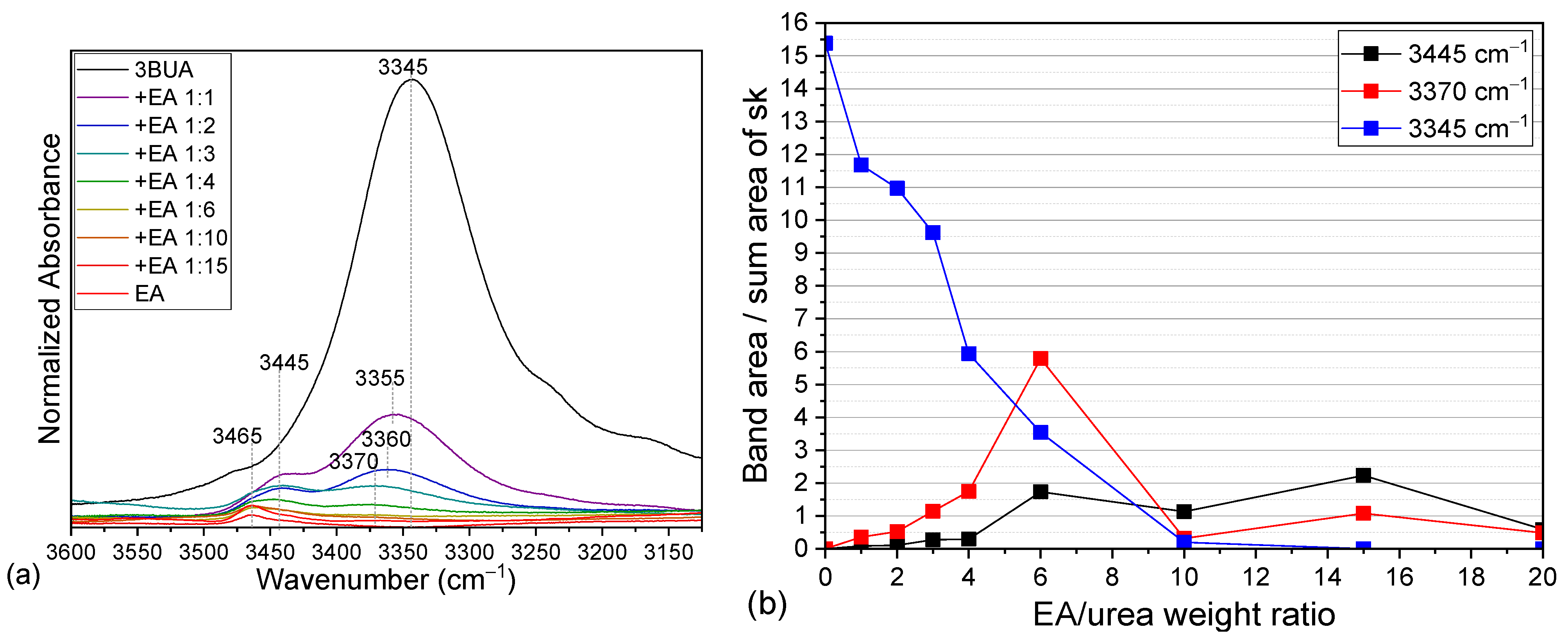
2.2. Tributyl Urea as a Model Molecule
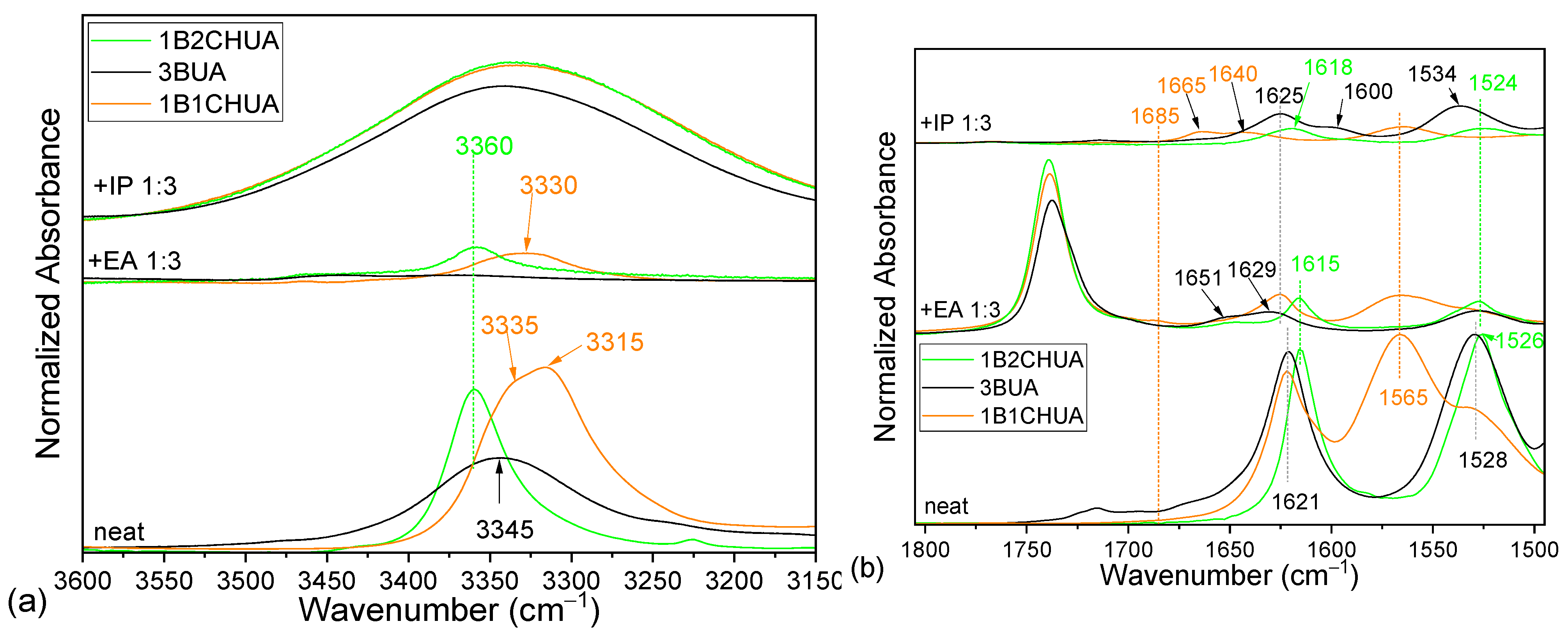
2.3. Comparison of Model Molecules
3. Experimental Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steed, J.W. Anion-tuned supramolecular gels: A natural evolution from urea supramolecular chemistry. Chem. Soc. Rev. 2010, 39, 3686. [Google Scholar] [PubMed]
- Mattia, J.; Painter, P. A comparison of hydrogen bonding and order in a polyurethane and poly(urethane-urea) and their blends with poly(ethylene glycol). Macromolecules 2007, 40, 1546–1554. [Google Scholar] [CrossRef]
- Platzer, N. Encyclopedia of polymer science and engineering, 2nd ed., by Mark Bikales Overberger Menges, Wiley-Interscience, New York, 1987, 840 pp. Price: $200.00. J. Polym. Sci. Polym. Lett. Ed. 1988, 26, 169–170. [Google Scholar] [CrossRef]
- Stoye, D.; Freitag, W.; Beuschel, G. Resins for Coatings: Chemistry, Properties, and Applications; Hanser Publishers: Munich, Germany, 1996. [Google Scholar]
- Ionescu, M. Chemistry and Technology of Polyols for Polyurethanes; iSmithers Rapra Publishing: Akron, OH, USA, 2016. [Google Scholar]
- Skleničková, K.; Abbrent, S.; Halecký, M.; Kočí, V.; Beneš, H. Biodegradability and ecotoxicity of polyurethane foams: A review. Crit. Rev. Environ. Sci. Technol. 2022, 52, 157–202. [Google Scholar]
- Wicks, D.A.; Yeske, P.E. Amine chemistries for isocyanate-based coatings. Prog. Org. Coat. 1997, 30, 265–270. [Google Scholar]
- Hutchby, M.; Houlden, C.E.; Ford, J.G.; Tyler, S.N.G.; Gagné, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Hindered Ureas as Masked Isocyanates: Facile Carbamoylation of Nucleophiles under Neutral Conditions. Angew. Chem. Int. Ed. 2009, 48, 8721–8724. [Google Scholar]
- Ying, H.; Zhang, Y.; Cheng, J. Dynamic urea bond for the design of reversible and self-healing polymers. Nat. Commun. 2014, 5, 3218. [Google Scholar]
- Natour, S.; Gajdošová, V.; Morávková, Z.; Šlouf, M.; Hodan, J.; Sharma, A.; Šomvársky, J.; Dušková-Smrčková, M. Aspartate-based polyurea coatings: Ambient cure process and inevitable transformation of urea groups into hydantoin cycles in polyurea networks and their impact on film properties. Prog. Org. Coat. 2024, 192, 108449. [Google Scholar]
- Williams, C.T.; Wicks, D.A.; Jarrett, W.L. Hydrogen bonding effects on aspartate ester reactions. J. Coat. Technol. Res. 2009, 6, 37–45. [Google Scholar]
- Liu, W.-X.; Yang, Z.; Qiao, Z.; Zhang, L.; Zhao, N.; Luo, S.; Xu, J. Dynamic multiphase semi-crystalline polymers based on thermally reversible pyrazole-urea bonds. Nat. Commun. 2019, 10, 4753. [Google Scholar]
- Jin, E.; Wang, X.; Liu, N.; Zhang, W. Self-assembled microspheres of glucose-containing polyaniline by alkali-guided method. Mater. Lett. 2007, 61, 4959–4962. [Google Scholar] [CrossRef]
- Marcos-Fernández, A.; Lozano, A.E.; González, L.; Rodríguez, A. Hydrogen bonding in copoly(ether-urea)s and its relationship with the physical properties. Macromolecules 1997, 30, 3584–3592. [Google Scholar]
- Harnagea, E.I.; Jagodzinski, P.W. Infrared spectra of cyclic and non-cyclic ureas in solution: Structures and interactions. Vib. Spectrosc. 1996, 10, 169–175. [Google Scholar]
- Luo, N.; Wang, D.N.; Ying, S.K. Hydrogen-bonding properties of segmented polyether poly(urethane urea) copolymer. Macromolecules 1997, 30, 4405–4409. [Google Scholar]
- Fischer, P.H.H.; McDowell, C.A. The Infrared Absorption Spectra of Urea–Hydrocarbon Adducts. Can. J. Chem. 1960, 38, 187–193. [Google Scholar]
- Kaválek, J.; Fiedler, P.; Papoušková, Z.; Exner, O. The confirmation of trisubstituted ureas and thioureas in solution. Collect. Czechoslov. Chem. Commun. 1982, 47, 35–44. [Google Scholar]
- Topalian, A.; Méchin, F.; Duchet-Rumeau, J.; Klucker, R.; Gérard, J.-F. Effect of the formation of hydantoin in aspartate-based polyurea networks. Prog. Org. Coat. 2025, 200, 109102. [Google Scholar]
- Ishihara, H.; Kimura, I.; Saito, K.; Ono, H. Infrared studies on segmented polyurethane-urea elastomers. J. Macromol. Sci. Part B 1974, 10, 591–618. [Google Scholar]
- Ramin, M.A.; Le Bourdon, G.; Heuzé, K.; Degueil, M.; Buffeteau, T.; Bennetau, B.; Vellutini, L. Epoxy-terminated self-assembled monolayers containing internal urea or amide groups. Langmuir 2015, 31, 2783–2789. [Google Scholar]
- Ramin, M.A.; Le Bourdon, G.; Daugey, N.; Bennetau, B.; Vellutini, L.; Buffeteau, T. PM-IRRAS investigation of self-assembled monolayers grafted onto SiO2/Au substrates. Langmuir 2011, 27, 6076–6084. [Google Scholar]
- Huang, W.B.; Lu, P.; Zhang, J.; Li, X.M. Study on morphology and mechanical properties of polyaspartic esters based polyureas. Adv. Mater. Res. 2011, 197–198, 1289–1293. [Google Scholar]
- Huang, Z.; Wang, C.; Li, H.; Ai, J.; Song, L.; Liu, B. Manual applied polyurethane-urea: High performance coating based on CO2-based polyol and polyaspartic ester. Prog. Org. Coat. 2023, 181, 107580. [Google Scholar]
- Lu, P.; Zhang, J.; Huang, W.; Liu, X. Hydrogen bond of polyaspartic ester polyurea coatings and its influence on aging behavior. Mater. Sci. Forum 2011, 689, 361–366. [Google Scholar]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies. Tables and Charts; John Wiley & Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Coleman, M.M.; Sobkowiak, M.; Pehlert, G.J.; Painter, P.C.; Iqbal, T. Infrared temperature studies of a simple polyurea. Macromol. Chem. Phys. 1997, 198, 117–136. [Google Scholar]
- Coleman, M.M.; Skrovanek, D.J.; Painter, P.C. Hydrogen bonding in polymers: III further infrared temperature studies of polyamides. Makromol. Chem. Macromol. Symp. 1986, 5, 21–33. [Google Scholar]
- Iqbal, N.; Tripathi, M.; Parthasarathy, S.; Kumar, D.; Roy, P.K. Aromatic versus Aliphatic: Hydrogen Bonding Pattern in Chain-Extended High-Performance Polyurea. ChemistrySelect 2018, 3, 1976–1982. [Google Scholar]
- Chan-Chan, L.H.; González-García, G.; Vargas-Coronado, R.F.; Cervantes-Uc, J.M.; Hernández-Sánchez, F.; Marcos-Fernandez, A.; Cauich-Rodríguez, J.V. Characterization of model compounds and poly(amide-urea) urethanes based on amino acids by FTIR, NMR and other analytical techniques. Eur. Polym. J. 2017, 92, 27–39. [Google Scholar]
- Born, L.; Hespe, H. On the physical crosslinking of amine-extended polyurethane urea elastomers: A crystallographic analysis of bis-urea from diphenyl methane-4-isocyanate and 1,4-butane diamine. Colloid. Polym. Sci. 1985, 263, 335–341. [Google Scholar]
- Le Questel, J.-Y.; Laurence, C.; Lachkar, A.; Helbert, M.; Berthelot, M. Hydrogen-bond basicity of secondary and tertiary amides, carbamates, ureas and lactams. J. Chem. Soc. Perkin Trans. 2 1992, 18, 2091. [Google Scholar]
- Besseau, F.; Laurence, C.; Berthelot, M. Hydrogen-bond basicity of esters, lactones and carbonates. J. Chem. Soc. Perkin Trans. 1994, 2, 485–489. [Google Scholar] [CrossRef]
- Li, Z.T.; Wu, L.Z. Hydrogen Bonded Supramolecular Structures; Springer: Berlin/Heidelberg, Germany, 2015; Volume 87. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disubstituted Ureas | Trisubstituted Ureas | ||||
|---|---|---|---|---|---|
| N,N′-sub. Urea i | 1B1CHUA | 3BUA | 1B2CHUA | PU-ASPE ii | State |
| (cm−1) | |||||
| ν(N–H) Amide A | |||||
| ~3300 | 3335, 3315 | 3345 | 3360 | ~3300 | neat |
| — | 3330 | 3370, 3445 | 3360 | — | in EA solution 1:3 |
| — | NO | NO | NO | — | in IP solution 1:3 |
| ν(C=O) Amide I “free” | |||||
| ~1690 | NO | 1660 | NO | ~1640 | neat |
| — | 1685 | 1651 | 1651 | — | in EA solution 1:3 |
| — | 1685 | 1663 | 1665 | — | in IP solution 1:3 |
| ν(C=O) Amide I “disordered” | |||||
| ~1660 | NO | 1621 | 1615 | ~1625 | neat |
| — | 1665 | 1629 | 1615 | — | in EA solution 1:3 |
| — | 1665, 1640 | 1626 | 1618 | — | in IP solution 1:3 |
| ν(C=O) Amide I “ordered” | |||||
| ~1635 | 1621 | NO | NO | NO | neat |
| — | 1625 | NO | NO | — | in EA solution 1:3 |
| — | 1625 | NO | NO | — | in IP solution 1:3 |
| ν(C–N) + δ(N–H) Amide II | |||||
| ~1530 | 1565, 1528 | 1528 | 1526 | ~1530 | neat |
| — | 1565 | 1527 | 1526 | — | in EA solution 1:3 |
| — | 1565 | 1534 | 1524 | — | in IP solution 1:3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morávková, Z.; Podešva, J.; Shabikova, V.; Abbrent, S.; Dušková-Smrčková, M. Hydrogen Bonding of Trialkyl-Substituted Urea in Organic Environment. Molecules 2025, 30, 1410. https://doi.org/10.3390/molecules30071410
Morávková Z, Podešva J, Shabikova V, Abbrent S, Dušková-Smrčková M. Hydrogen Bonding of Trialkyl-Substituted Urea in Organic Environment. Molecules. 2025; 30(7):1410. https://doi.org/10.3390/molecules30071410
Chicago/Turabian StyleMorávková, Zuzana, Jiří Podešva, Valeriia Shabikova, Sabina Abbrent, and Miroslava Dušková-Smrčková. 2025. "Hydrogen Bonding of Trialkyl-Substituted Urea in Organic Environment" Molecules 30, no. 7: 1410. https://doi.org/10.3390/molecules30071410
APA StyleMorávková, Z., Podešva, J., Shabikova, V., Abbrent, S., & Dušková-Smrčková, M. (2025). Hydrogen Bonding of Trialkyl-Substituted Urea in Organic Environment. Molecules, 30(7), 1410. https://doi.org/10.3390/molecules30071410