
The Conformations of Isolated Gallic Acid: A Laser-Ablation Rotational Study




Abstract
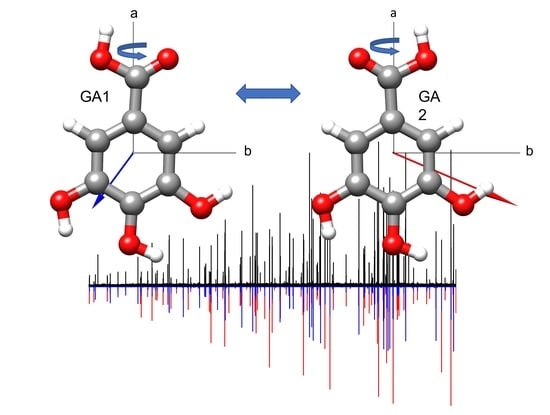
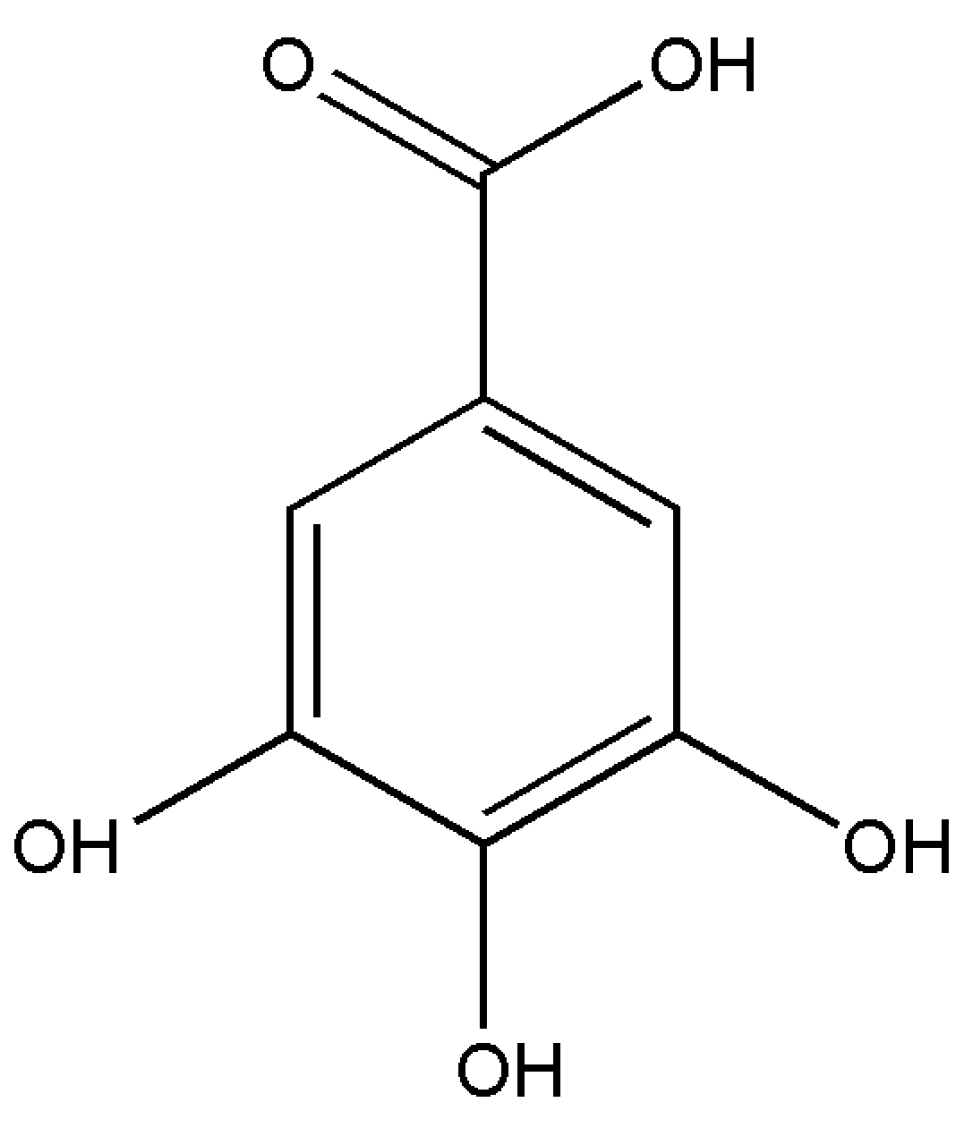
1. Introduction
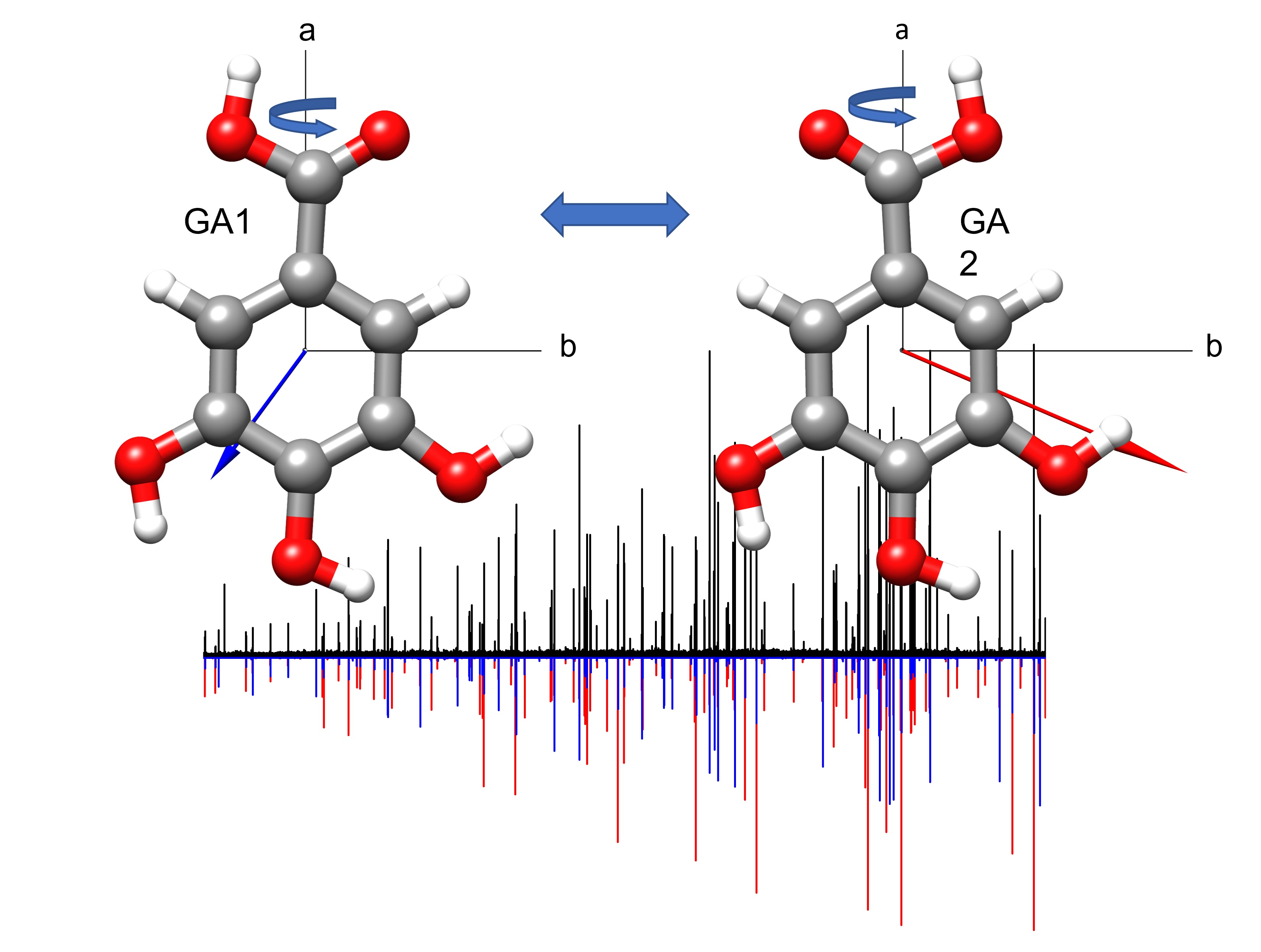
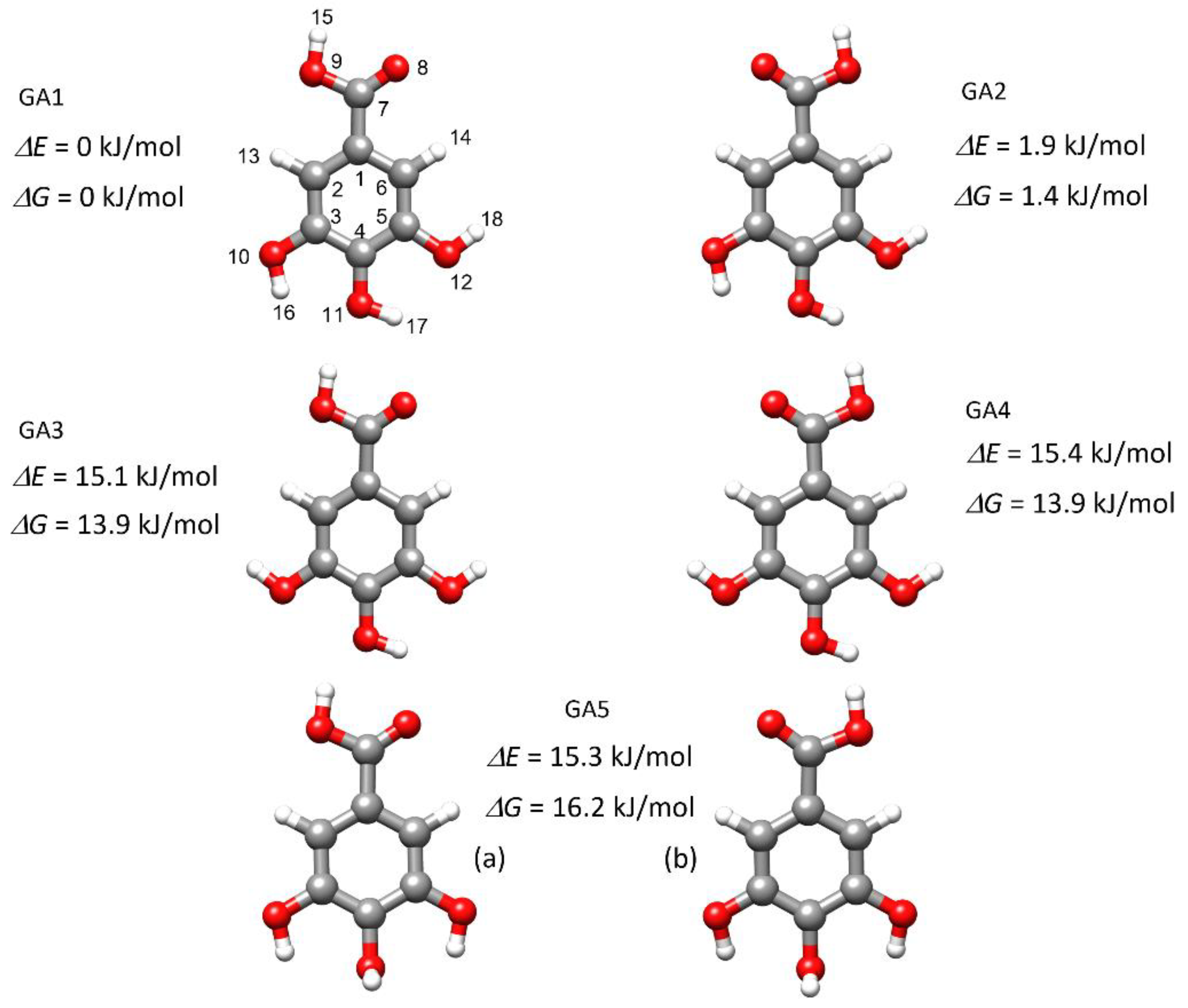
2. Results and Discussion
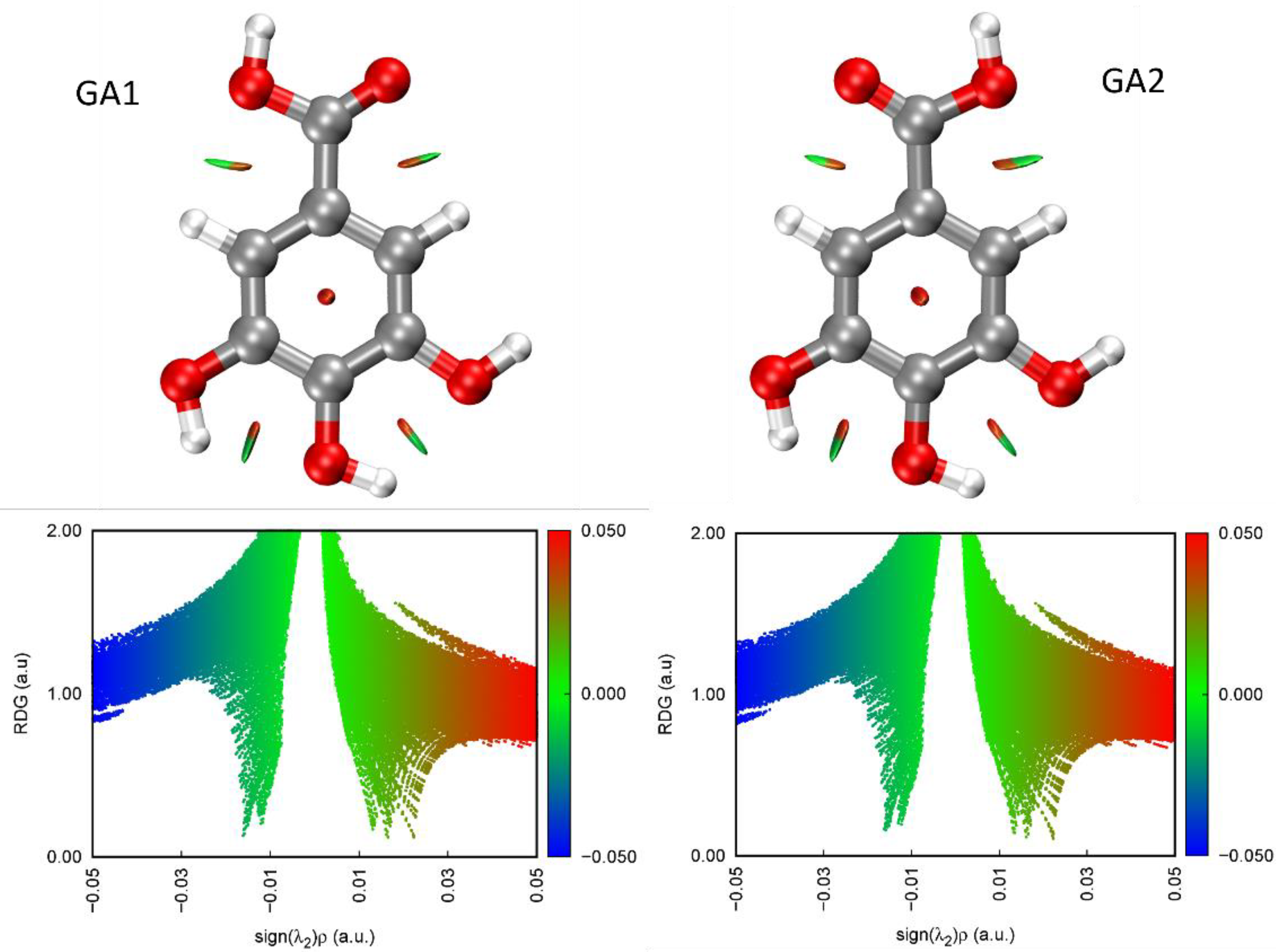
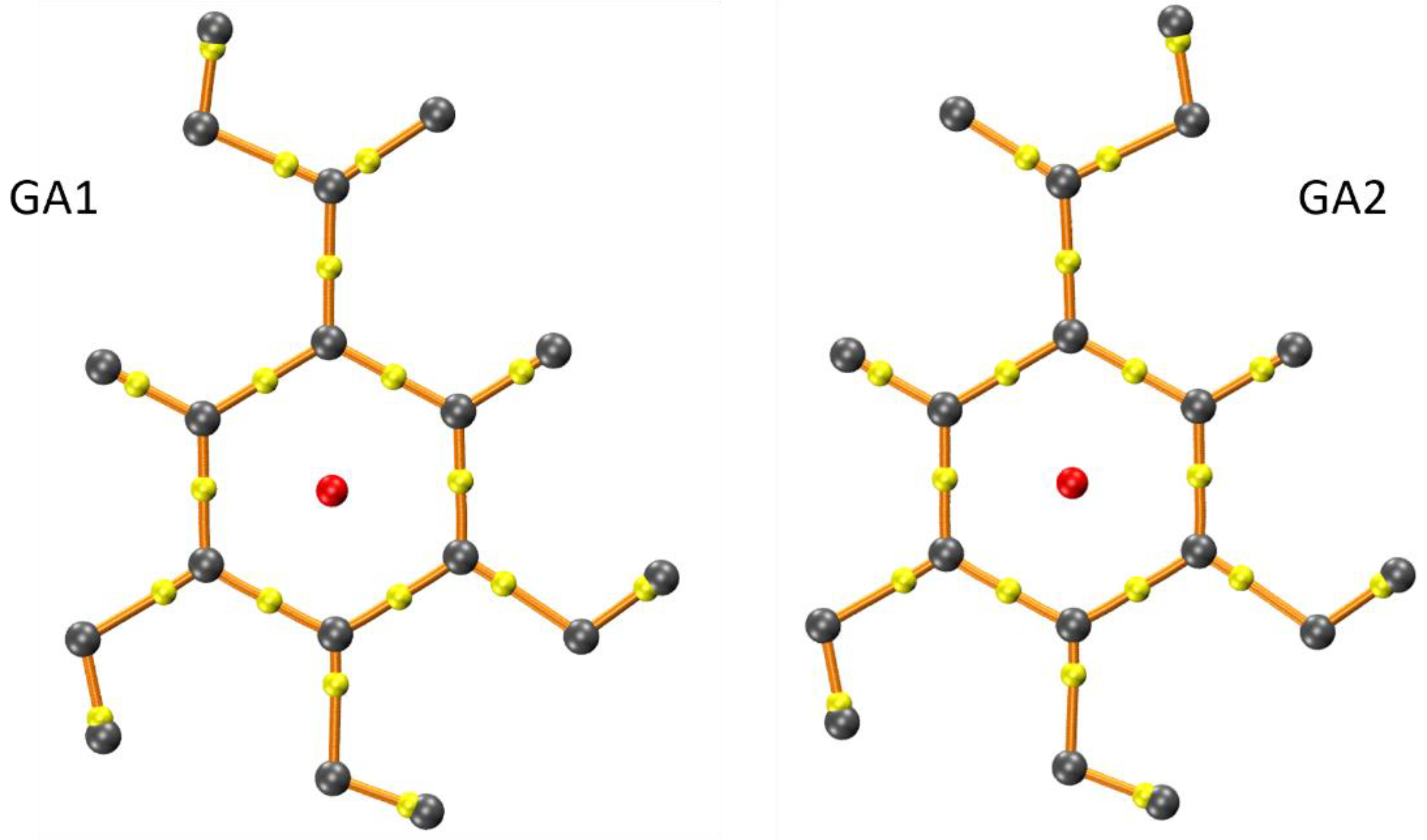
2.1. Conformational Search
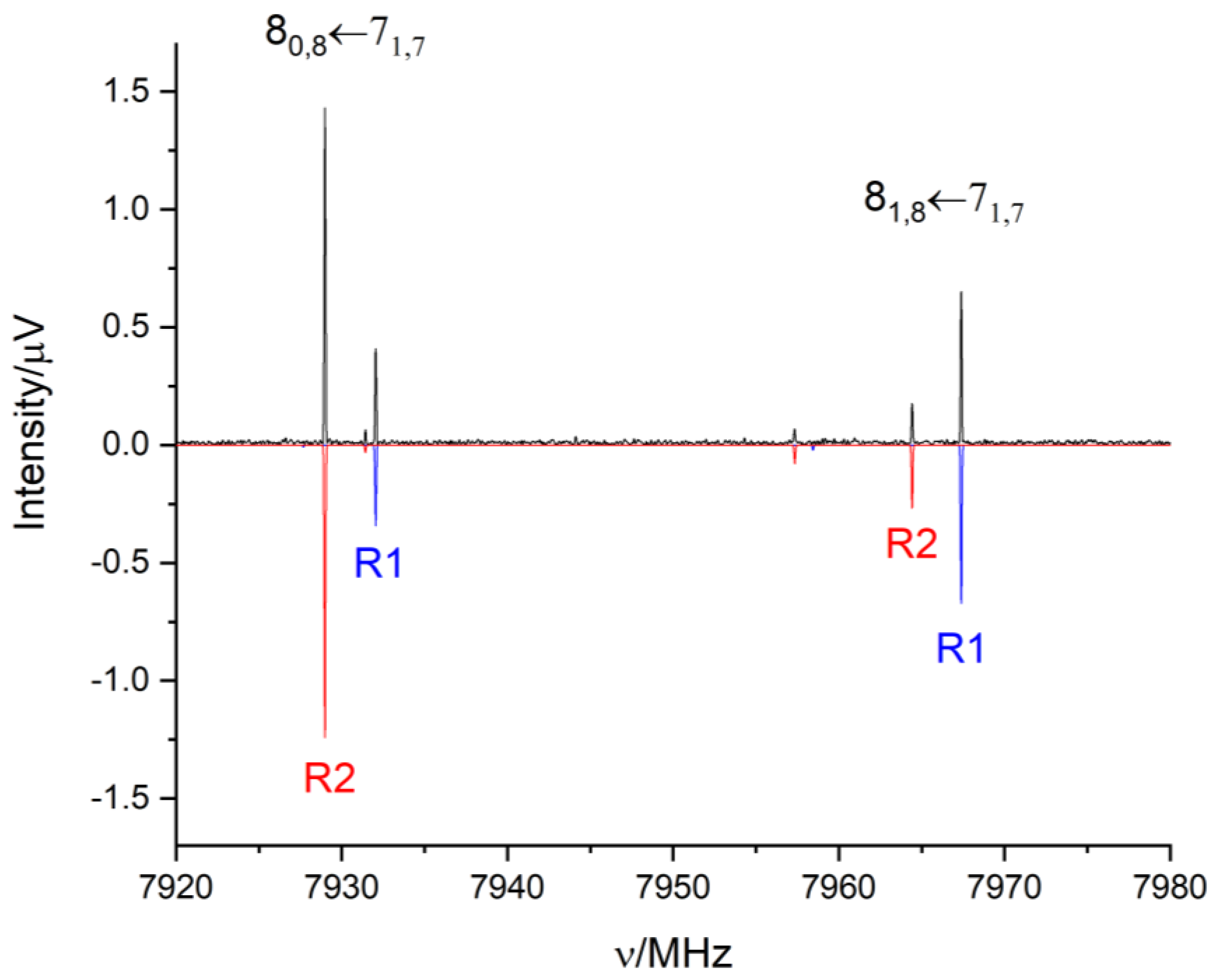
2.2. Rotational Spectrum
3. Discussion
4. Materials and Methods
4.1. Experimental
4.2. Theoretical
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Beckman, C.H. Phenolic-Storing Cells: Keys to Programmed Cell Death and Periderm Formation in Wilt Disease Resistance and in General Defence Responses in Plants? Physiol. Mol. Plant Pathol. 2000, 57, 101–110. [Google Scholar] [CrossRef]
- Hanuka Katz, I.; Eran Nagar, E.; Okun, Z.; Shpigelman, A. The Link between Polyphenol Structure, Antioxidant Capacity and Shelf-Life Stability in the Presence of Fructose and Ascorbic Acid. Molecules 2020, 25, 225. [Google Scholar] [CrossRef] [PubMed]
- Leopoldini, M.; Russo, N.; Toscano, M. The Molecular Basis of Working Mechanism of Natural Polyphenolic Antioxidants. Food Chem. 2011, 125, 288–306. [Google Scholar] [CrossRef]
- Vermerris, W.; Nicholson, R. Phenolic Compounds and Their Effects on Human Health. In Phenolic Compound Biochemistry; Springer: Dordrecht, The Netherlands, 2006; pp. 235–255. ISBN 9781402051630. [Google Scholar]
- Singla, R.K.; Dubey, A.K.; Garg, A.; Sharma, R.K.; Fiorino, M.; Ameen, S.M.; Haddad, M.A.; Al-Hiary, M. Natural Polyphenols: Chemical Classification, Definition of Classes, Subcategories, and Structures. J. AOAC Int. 2019, 102, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, F.; Janitha, P.K.; Wanasundara, P.D. Phenolic Antioxidants. Crit. Rev. Food Sci. Nutr. 2009, 32, 67–103. [Google Scholar] [CrossRef]
- Platzer, M.; Kiese, S.; Herfellner, T.; Schweiggert-Weisz, U.; Eisner, P. How Does the Phenol Structure Influence the Results of the Folin-Ciocalteu Assay? Antioxidants 2021, 10, 811. [Google Scholar] [CrossRef]
- Al-Mamary, M.A.; Moussa, Z.; Al-Mamary, M.A.; Moussa, Z. Antioxidant Activity: The Presence and Impact of Hydroxyl Groups in Small Molecules of Natural and Synthetic Origin. In Antioxidants—Benefits, Sources, Mechanisms of Action; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Bors, W.; Michel, C.; Stettmaier, K. Antioxidant Effects of Flavonoids. BioFactors 1997, 6, 399–402. [Google Scholar] [CrossRef]
- Bors, W.; Heller, W.; Michel, C.; Saran, M. Flavonoids as Antioxidants: Determination of Radical-Scavenging Efficiencies. Methods. Enzym. 1990, 186, 343–355. [Google Scholar] [CrossRef]
- Haslam, E. Gallic Acid and Its Metabolites. In Plant Polyphenols; Springer: Berlin/Heidelberg, Germany, 1992; pp. 169–194. [Google Scholar] [CrossRef]
- Delfanian, M.; Sahari, M.A.; Barzegar, M.; Ahmadi Gavlighi, H. Structure–Antioxidant Activity Relationships of Gallic Acid and Phloroglucinol. J. Food Meas. Charact. 2021, 15, 5036–5046. [Google Scholar] [CrossRef]
- Phonsatta, N.; Deetae, P.; Luangpituksa, P.; Grajeda-Iglesias, C.; Figueroa-Espinoza, M.C.; le Comte, J.; Villeneuve, P.; Decker, E.A.; Visessanguan, W.; Panya, A. Comparison of Antioxidant Evaluation Assays for Investigating Antioxidative Activity of Gallic Acid and Its Alkyl Esters in Different Food Matrices. J. Agric. Food Chem. 2017, 65, 7509–7518. [Google Scholar] [CrossRef]
- Velderrain-Rodríguez, G.R.; Torres-Moreno, H.; Villegas-Ochoa, M.A.; Ayala-Zavala, J.F.; Robles-Zepeda, R.E.; Wall-Medrano, A.; González-Aguilar, G.A. Gallic Acid Content and an Antioxidant Mechanism Are Responsible for the Antiproliferative Activity of ‘Ataulfo’ Mango Peel on LS180 Cells. Molecules 2018, 23, 695. [Google Scholar] [CrossRef] [PubMed]
- Badhani, B.; Sharma, N.; Kakkar, R. Gallic Acid: A Versatile Antioxidant with Promising Therapeutic and Industrial Applications. RSC Adv. 2015, 5, 27540–27557. [Google Scholar] [CrossRef]
- Gao, J.; Hu, J.; Hu, D.; Yang, X. A Role of Gallic Acid in Oxidative Damage Diseases: A Comprehensive Review. Nat. Prod. Commun. 2019, 14, 1934578X1987417. [Google Scholar] [CrossRef]
- Zanwar, A.A.; Badole, S.L.; Shende, P.S.; Hegde, M.V.; Bodhankar, S.L. Role of Gallic Acid in Cardiovascular Disorders. Polyphen. Hum. Health Dis. 2014, 2, 1045–1047. [Google Scholar] [CrossRef]
- Khan, B.A.; Mahmood, T.; Menaa, F.; Shahzad, Y.; Yousaf, A.M.; Hussain, T.; Ray, S.D. New Perspectives on the Efficacy of Gallic Acid in Cosmetics & Nanocosmeceuticals. Curr. Pharm. Des. 2019, 24, 5181–5187. [Google Scholar] [CrossRef]
- Giftson, J.S.; Jayanthi, S.; Nalini, N. Chemopreventive Efficacy of Gallic Acid, an Antioxidant and Anticarcinogenic Polyphenol, against 1,2-Dimethyl Hydrazine Induced Rat Colon Carcinogenesis. Invest. New. Drugs 2010, 28, 251–259. [Google Scholar] [CrossRef]
- Subramanian, A.P.; Jaganathan, S.K.; Mandal, M.; Supriyanto, E.; Muhamad, I.I. Gallic Acid Induced Apoptotic Events in HCT-15 Colon Cancer Cells. World J. Gastroenterol. 2016, 22, 3952–3961. [Google Scholar] [CrossRef]
- Hugo, P.C.; Gil-Chávez, J.; Sotelo-Mundo, R.R.; Namiesnik, J.; Gorinstein, S.; González-Aguilar, G.A. Antioxidant Interactions between Major Phenolic Compounds Found in ‘Ataulfo’ Mango Pulp: Chlorogenic, Gallic, Protocatechuic and Vanillic Acids. Molecules 2012, 17, 12657–12664. [Google Scholar] [CrossRef]
- You, B.R.; Kim, S.Z.; Kim, S.H.; Park, W.H. Gallic Acid-Induced Lung Cancer Cell Death Is Accompanied by ROS Increase and Glutathione Depletion. Mol. Cell Biochem. 2011, 357, 295–303. [Google Scholar] [CrossRef]
- Elango, S.; Balwas, R.; Padma, V.V.; Elango, S.; Balwas, R.; Padma, V.V. Gallic Acid Isolated from Pomegranate Peel Extract Induces Reactive Oxygen Species Mediated Apoptosis in A549 Cell Line. J. Cancer 2011, 2, 638–645. [Google Scholar] [CrossRef][Green Version]
- Sroka, Z.; Cisowski, W. Hydrogen Peroxide Scavenging, Antioxidant and Anti-Radical Activity of Some Phenolic Acids. Food. Chem. Toxicol. 2003, 41, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.W.; Ming, D.S.; But, P.P.H.; Mak, T.C.W. Gallic Acid Monohydrate. Acta. Cryst. C 2000, 56, 594–595. [Google Scholar] [CrossRef] [PubMed]
- Okabe, N.; Kyoyama, H.; Suzuki, M. Gallic Acid Monohydrate. Acta Cryst. Sect. E Struct. Rep. Online 2001, 57, o764–o766. [Google Scholar] [CrossRef]
- Billes, F.; Mohammed-Ziegler, I.; Bombicz, P. Vibrational Spectroscopic Study on the Quantum Chemical Model and the X-Ray Structure of Gallic Acid, Solvent Effect on the Structure and Spectra. Vib. Spectrosc. 2007, 43, 193–202. [Google Scholar] [CrossRef]
- Clarke, H.D.; Arora, K.K.; Wojtas, Ł.; Zaworotko, M.J. Polymorphism in Multiple Component Crystals: Forms III and IV of Gallic Acid Monohydrate. Cryst. Growth Des. 2011, 11, 964–966. [Google Scholar] [CrossRef]
- Hirun, N.; Saithong, S.; Pakawatchai, C.; Tantishaiyakul, V. 3,4,5-Trihydroxybenzoic Acid. Acta Cryst. Sect. E Struct. Rep. Online 2011, 67, o787. [Google Scholar] [CrossRef]
- Braun, D.E.; Bhardwaj, R.M.; Florence, A.J.; Tocher, D.A.; Price, S.L. Complex Polymorphic System of Gallic Acid—Five Monohydrates, Three Anhydrates, and over 20 Solvates. Cryst. Growth Des. 2013, 13, 19–23. [Google Scholar] [CrossRef]
- Thomas, L.H.; Wales, C.; Wilson, C.C. Selective Preparation of Elusive and Alternative Single Component Polymorphic Solid Forms through Multi-Component Crystallisation Routes. Chem. Commun. 2016, 52, 7372–7375. [Google Scholar] [CrossRef]
- Hoser, A.A.; Sovago, I.; Lanza, A.; Madsen, A. A Crystal Structure Prediction Enigma Solved: The Gallic Acid Monohydrate System—Surprises at 10 K. Chem. Commun. 2017, 53, 925–928. [Google Scholar] [CrossRef]
- Ponce, A.; Zavalij, P.Y.; Eichhorn, B.W. 3,4,5-Trihydroxybenzoic Acid Monohydrate. CSD Commun. 2014. [Google Scholar] [CrossRef]
- Zhao, J.; Khan, I.A.; Fronczek, F.R. Gallic Acid. Acta Cryst. Sect. E Struct. Rep. Online 2011, 67, o316–o317. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Wu, M.; Wang, Q.; Peng, X.; Wen, H.; McKinstry, W.J.; Chen, Q. Crystal Structure of Tannase from Lactobacillus Plantarum. J. Mol. Biol. 2013, 425, 2737–2751. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, E.; Stravodimos, G.A.; Kantsadi, A.L.; Chatzileontiadou, D.S.M.; Skamnaki, V.T.; Leonidas, D.D. Natural Flavonoids as Antidiabetic Agents. The Binding of Gallic and Ellagic Acids to Glycogen Phosphorylase b. FEBS Lett. 2015, 589, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, K.R.; Zatikyan, A.L. Ab Initio Studies of Complexation of Gallic Acid In Aqueous Solutions. J. Struct. Chem. 2018, 59, 1492–1499. [Google Scholar] [CrossRef]
- Kraus, J.; Kortus, J. A Theoretical Investigation into Gallic Acid Pyrolysis. J. Comput. Chem. 2022, 43, 1023–1032. [Google Scholar] [CrossRef]
- de Souza, G.L.C.; Peterson, K.A. Benchmarking Antioxidant-Related Properties for Gallic Acid through the Use of DFT, MP2, CCSD, and CCSD(T) Approaches. J. Phys. Chem. A 2021, 125, 198–208. [Google Scholar] [CrossRef]
- Lespade, L. Ab Initio Molecular Dynamics of Electron Transfer from Gallic Acid to Small Radicals: A Comparative Study between Hydroxyl and Nitrogen Dioxide Radicals. Comput. Chem. 2018, 1135, 6–10. [Google Scholar] [CrossRef]
- Badhani, B.; Kakkar, R. DFT Study of Structural and Electronic Properties of Gallic Acid and Its Anions in Gas Phase and in Aqueous Solution. Struct. Chem. 2017, 28, 1789–1802. [Google Scholar] [CrossRef]
- Rajan, V.K.; Muraleedharan, K. A Computational Investigation on the Structure, Global Parameters and Antioxidant Capacity of a Polyphenol, Gallic Acid. Food Chem. 2017, 220, 93–99. [Google Scholar] [CrossRef]
- Pardeshi, S.; Dhodapkar, R.; Kumar, A. Quantum Chemical Density Functional Theory Studies on the Molecular Structure and Vibrational Spectra of Gallic Acid Imprinted Polymers. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 116, 562–573. [Google Scholar] [CrossRef]
- Zaccaron, S.; Ganzerla, R.; Bortoluzzi, M. Iron Complexes with Gallic Acid: A Computational Study on Coordination Compounds of Interest for the Preservation of Cultural Heritage. J. Coord. Chem. 2013, 66, 1709–1719. [Google Scholar] [CrossRef]
- Pate, B.H. Taking the Pulse of Molecular Rotational Spectroscopy. Science 1979 2011, 333, 947–948. [Google Scholar] [CrossRef] [PubMed]
- Grabow, J.-U.; Caminati, W. Microwave Spectroscopy: Experimental Techniques. In Frontiers of Molecular Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2009; pp. 383–454. [Google Scholar] [CrossRef]
- Caminati, W.; Grabow, J.-U. Microwave Spectroscopy: Molecular Systems. In Frontiers of Molecular Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2009; pp. 455–552. [Google Scholar] [CrossRef]
- Dreizler, H.; Mata, S.; Lesarri, A.; Lopez, J.C.; Blanco, S.; Alonso, J.L. Rotational Spectra of Fluorodiacetylene and Fluorotriacetylene Produced by Electrical Discharge. Z Fur. Nat. 2002, 57a, 76–78. [Google Scholar]
- Alonso, J.L.; López, J.C. Microwave Spectroscopy of Biomolecular Building Blocks. Top Curr. Chem. 2015, 364, 335–401. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Macario, A.; García-Calvo, J.; Revilla-Cuesta, A.; Torroba, T.; López, J.C. Microwave Detection of Wet Triacetone Triperoxide (TATP): Non-Covalent Forces and Water Dynamics. Chem. –A Eur. J. 2021, 27, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Macario, A.; López, J.C. The Structure of Isolated Thalidomide as Reference for Its Chirality-Dependent Biological Activity: A Laser-Ablation Rotational Study. Phys. Chem. Chem. Phys. 2021, 23, 13705–13713. [Google Scholar] [CrossRef] [PubMed]
- López, J.C.; Macario, A.; Verde, A.; Perez, A.; Blanco, S. Decoding the Structure of Non-Proteinogenic Amino Acids: The Rotational Spectrum of Jet Cooled Laser Ablated Thioproline. Molecules 2021, 26, 7585. [Google Scholar] [CrossRef]
- Ray, B.S. Über Die Eigenwerte Des Asymmetrischen Kreisels. Zeitschrift. Für. Physik. 1932, 78, 74–91. [Google Scholar] [CrossRef]
- Plusquellic, D.F. Jb95 Spectral Fitting Program, NIST, Gaithersburg. Available online: https://www.nist.gov/services-resources/software/jb95-spectral-fitting-program (accessed on 24 December 2022).
- Kisiel, Z. Assignment and Analysis of Complex Rotational Spectra. In Spectroscopy from Space; Springer: Berlin/Heidelberg, Germany, 2001; pp. 91–106. [Google Scholar] [CrossRef]
- Kisiel, Z. PROSPE. Programs for Rotational Spectroscopy. Available online: http://www.ifpan.edu.pl/~kisiel/prospe.htm (accessed on 24 December 2022).
- Pickett, H.M. The Fitting and Prediction of Vibration-Rotation Spectra with Spin Interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- Watson, J.K.G. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure a Series of Advances; Durig, J.R., Ed.; Elsevier: New York, NY, USA, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Blanco, S.; Lesarri, A.; López, J.C.; Alonso, J.L. The Gas-Phase Structure of Alanine. J. Am. Chem. Soc. 2004, 126, 11675–11683. [Google Scholar] [CrossRef]
- Miller, D.R. Free Jet Sources. In Atomic and Molecular Beam Methods; Scoles, G., Ed.; Oxford University Press: Oxford, UK, 1998; Volume 1, pp. 14–53. ISBN 0-10-504280-8. [Google Scholar]
- Onda, M.; Asai, M.; Takise, K.; Kuwae, K.; Hayami, K.; Kuroe, A.; Mori, M.; Miyazaki, H.; Suzuki, N.; Yamaguchi, I. Microwave Spectrum of Benzoic Acid. J. Mol. Struct. 1999, 482–483, 301–303. [Google Scholar] [CrossRef]
- Caminati, W.; di Bernardo, S.; Schäfer, L.; Kulp-Newton, S.Q.; Siam, K. Investigation of the Molecular Structure of Catechol by Combined Microwave Spectroscopy and AB Initio Calculations. J. Mol. Struct. 1990, 240, 263–274. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Steiner, T. The Whole Palette of Hydrogen Bonds. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Becke, A. Density Functional Thermochemistry III The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Frisch, M.J. Gaussian 16, Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Param. a | Rotamer R1 | GA1 | Rotamer R2 | GA2 |
|---|---|---|---|---|
| A/MHz | 1602.84558(20) b | 1602.4 | 1602.69867(25) | 1602.3 |
| B/MHz | 663.46993(10) | 663.6 | 663.13549(10) | 663.3 |
| C/MHz | 469.477690(86) | 469.3 | 469.30622(10) | 469.1 |
| κ | −0.66 | −0.66 | −0.66 | −0.66 |
| Pa/uÅ2 | 761.44548(18) | 761.6 | 761.81977(20) | 762.0 |
| Pb/uÅ2 | 315.02548(18) | 315.4 | 315.04450(20) | 315.4 |
| Pc/uÅ2 | 0.27570(18) | 0.0 | 0.28558(20) | 0.0 |
| DJ /kHz | 0.00690(86) | 0.0054 | 0.00605(63) | 0.0054 |
| DJK /kHz | 0.0190(20) | 0.0427 | 0.0274(26) | 0.0428 |
| DK /kHz | 0.037(11) | 0.0202 | 0.039(13) | 0.0202 |
| d1 /kHz | 0.00209(23) | −0.0019 | 0.00253(33) | −0.0019 |
| d2 /kHz | 0.000510(88) | 0.0000 | 0.00025(11) | −0.0005 |
| σ/kHz | 3.1 | 3.7 | ||
| n | 133 | 116 | ||
| μa/D | moderate | 1.9 | moderate | 1.8 |
| μb/D | moderate | 1.4 | intense | 4.2 |
| μc/D | no | 0.0 | no | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verde, A.; Blanco, S.; López, J.C. The Conformations of Isolated Gallic Acid: A Laser-Ablation Rotational Study. Molecules 2023, 28, 159. https://doi.org/10.3390/molecules28010159
Verde A, Blanco S, López JC. The Conformations of Isolated Gallic Acid: A Laser-Ablation Rotational Study. Molecules. 2023; 28(1):159. https://doi.org/10.3390/molecules28010159
Chicago/Turabian StyleVerde, Andrés, Susana Blanco, and Juan Carlos López. 2023. "The Conformations of Isolated Gallic Acid: A Laser-Ablation Rotational Study" Molecules 28, no. 1: 159. https://doi.org/10.3390/molecules28010159
APA StyleVerde, A., Blanco, S., & López, J. C. (2023). The Conformations of Isolated Gallic Acid: A Laser-Ablation Rotational Study. Molecules, 28(1), 159. https://doi.org/10.3390/molecules28010159