




Biomolecules 2021, 11(11), 1622; https://doi.org/10.3390/biom11111622 - 2 Nov 2021
Cited by 5 | Viewed by 3279
Abstract
►
Show Figures
Traditionally, Cornelia de Lange Syndrome (CdLS) is considered a cohesinopathy caused by constitutive mutations in cohesin complex genes. Cohesin is a major regulator of chromatin architecture, including the formation of chromatin loops at the imprinted IGF2/H19 domain. We used 3C analysis
[...] Read more.
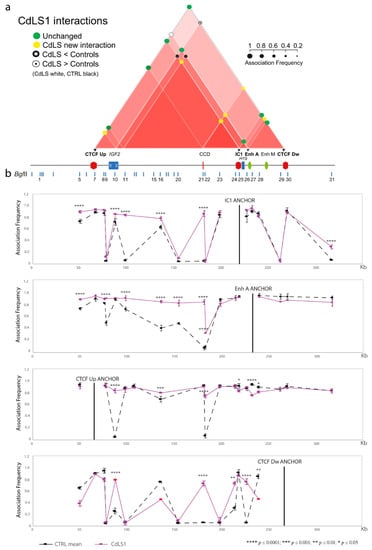
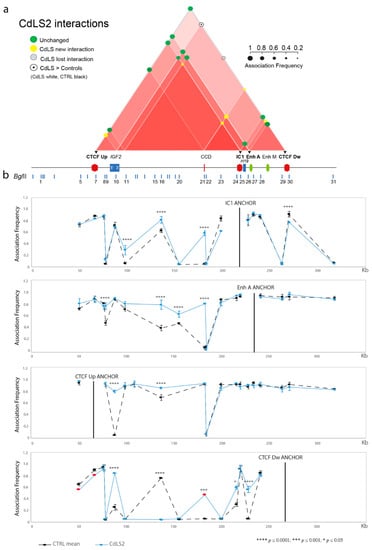
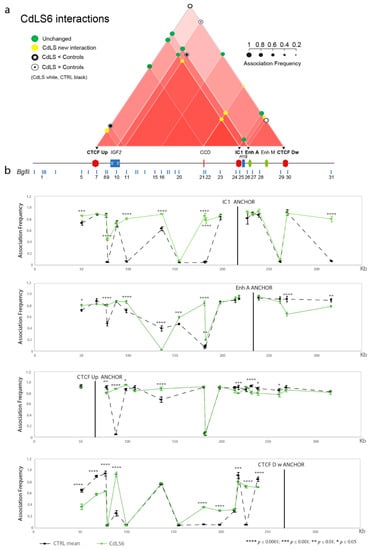
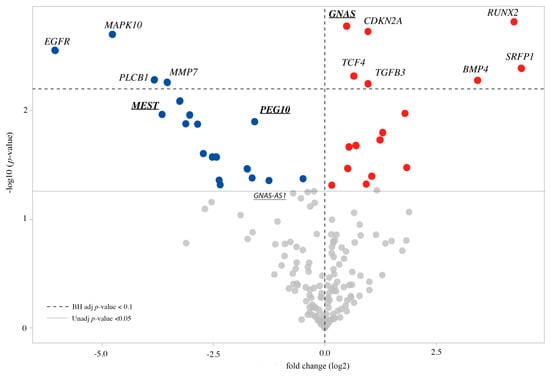
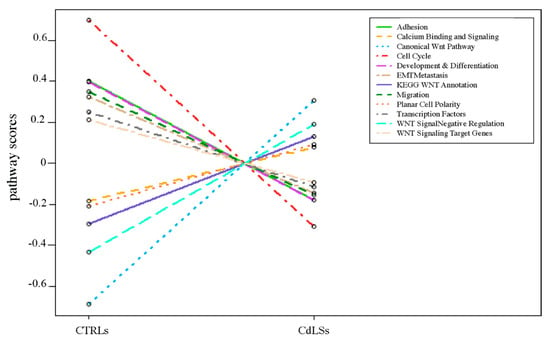
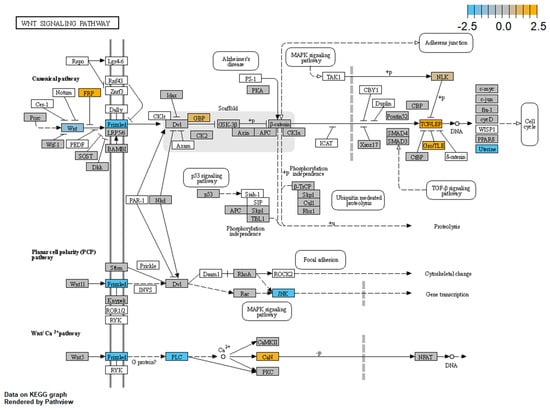
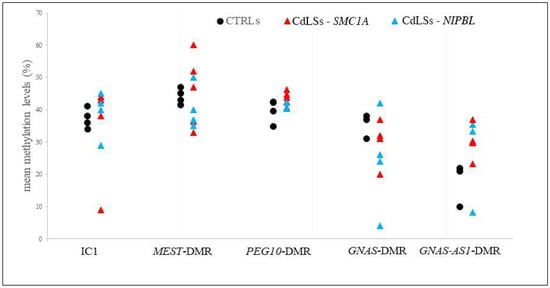
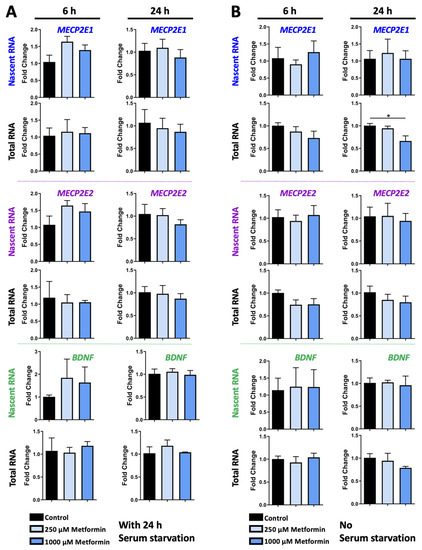
Traditionally, Cornelia de Lange Syndrome (CdLS) is considered a cohesinopathy caused by constitutive mutations in cohesin complex genes. Cohesin is a major regulator of chromatin architecture, including the formation of chromatin loops at the imprinted IGF2/H19 domain. We used 3C analysis on lymphoblastoid cells from CdLS patients carrying mutations in NIPBL and SMC1A genes to explore 3D chromatin structure of the IGF2/H19 locus and evaluate the influence of cohesin alterations in chromatin architecture. We also assessed quantitative expression of imprinted loci and WNT pathway genes, together with DMR methylation status of the imprinted genes. A general impairment of chromatin architecture and the emergence of new interactions were found. Moreover, imprinting alterations also involved the expression and methylation levels of imprinted genes, suggesting an association among cohesin genetic defects, chromatin architecture impairment, and imprinting network alteration. The WNT pathway resulted dysregulated: canonical WNT, cell cycle, and WNT signal negative regulation were the most significantly affected subpathways. Among the deregulated pathway nodes, the key node of the frizzled receptors was repressed. Our study provides new evidence that mutations in genes of the cohesin complex have effects on the chromatin architecture and epigenetic stability of genes commonly regulated by high order chromatin structure.
Full article
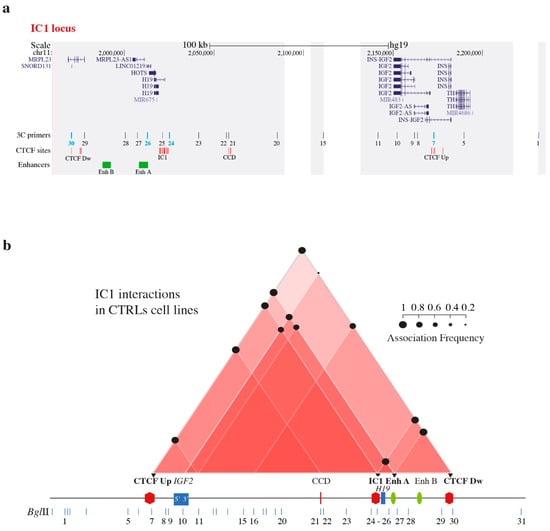
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}