Modeling DNA Methylation Profiles through a Dynamic Equilibrium between Methylation and Demethylation

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. DNA Extraction from Cells and Tissues
2.3. Sequence Handling
2.4. Computations and Statistical Analysis
3. Results
3.1. In-Depth DNA Methylation Profiling of DAO and DDO Genes
3.2. Mathematical Modeling of DNA Methylation
3.2.1. Model Assumptions and Rationale
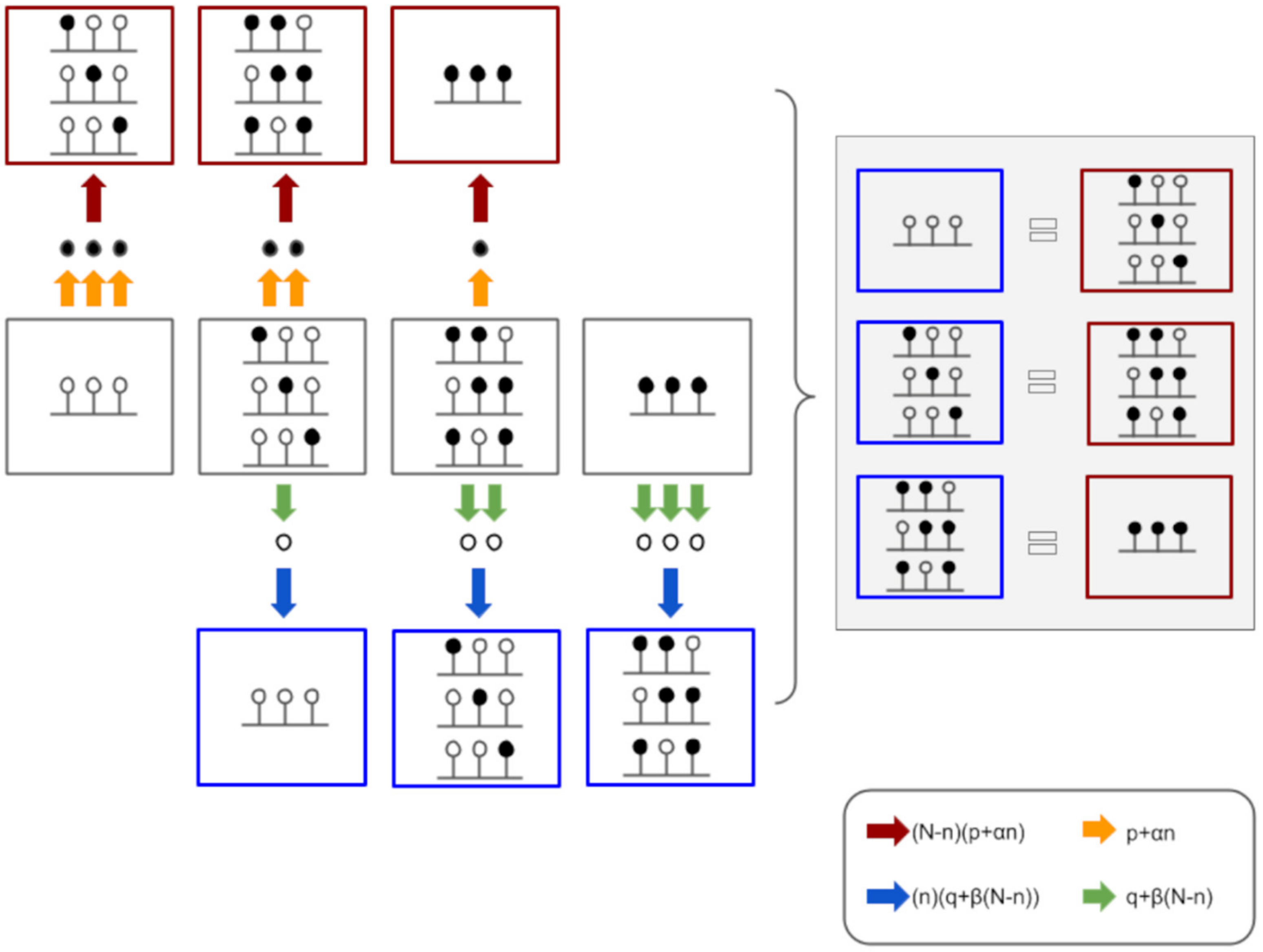
3.2.2. Model Implementation
3.2.3. Results Obtained by the Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Gowher, H.; Jeltsch, A. Mammalian DNA methyltransferases: New discoveries and open questions. Biochem. Soc. Trans. 2018, 46, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y. DNA Methylation in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA Methylation: Mechanisms, Genomics, and Biological Functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Saito, D.; Suyama, M. Linkage disequilibrium analysis of allelic heterogeneity in DNA methylation. Epigenetics 2015, 10, 1093–1098. [Google Scholar] [CrossRef][Green Version]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, J.; Tian, G.; Li, N.; Li, Q.; Ye, M.; Zheng, H.; Yu, J.; Wu, H.; Sun, J.; et al. The DNA Methylome of Human Peripheral Blood Mononuclear Cells. PLoS Biol. 2010, 8, e1000533. [Google Scholar] [CrossRef]
- Martin, T.C.; Yet, I.; Tsai, P.-C.; Bell, J.T. coMET: visualisation of regional epigenome-wide association scan results and DNA co-methylation patterns. BMC Bioinform. 2015, 16, 131. [Google Scholar] [CrossRef]
- Hu, K.; Ting, A.H.; Li, J. BSPAT: A fast online tool for DNA methylation co-occurrence pattern analysis based on high-throughput bisulfite sequencing data. BMC Bioinform. 2015, 16, 220. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Steigerwald, S.D.; Hansen, R.S.; Gartler, S.M.; Riggs, A.D. Polymerase chain reaction-aided genomic sequencing of an X chromosome-linked CpG island: Methylation patterns suggest clonal inheritance, CpG site autonomy, and an explanation of activity state stability. Proc. Natl. Acad. Sci. USA 1990, 87, 8252–8256. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Genome Res. 1975, 14, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Rausch, C.; Hastert, F.D.; Boneva, B.; Filatova, A.; Patil, S.J.; Nuber, U.A.; Gao, Y.; Zhao, X.; Cardoso, M.C. Methyl-CpG binding domain protein 1 regulates localization and activity of Tet1 in a CXXC3 domain-dependent manner. Nucleic Acids Res. 2017, 45, 7118–7136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xiong, J.; Zhang, Z.; Chen, J.; Huang, H.; Xu, Y.; Ding, X.; Zheng, Y.; Nishinakamura, R.; Xu, G.-L.; Wang, H.; et al. Cooperative Action between SALL4A and TET Proteins in Stepwise Oxidation of 5-Methylcytosine. Mol. Cell 2016, 64, 913–925. [Google Scholar] [CrossRef]
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. Tet Enzymes, Variants, and Differential Effects on Function. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Gu, T.; Lin, X.; Cullen, S.M.; Luo, M.; Jeong, M.; Estecio, M.; Shen, J.; Hardikar, S.; Sun, D.; Su, J.; et al. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 2018, 19, 88. [Google Scholar] [CrossRef]
- Rulands, S.; Lee, H.J.; Clark, S.J.; Angermueller, C.; Smallwood, S.A.; Krueger, F.; Mohammed, H.; Dean, W.; Nichols, J.; Rugg-Gunn, P.; et al. Genome-Scale Oscillations in DNA Methylation during Exit from Pluripotency. Cell Syst. 2018, 7, 63–76. [Google Scholar] [CrossRef]
- Gravina, S.; Dong, X.; Yu, B.; Vijg, J. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol. 2016, 17, 150. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.A.; Ponting, C.P.; Voet, T.; et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 2016, 13, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Tsukahara, T.; Nakamura, R.; Yurino, H.; Hashimoto, S.; Tsuji, S.; Takeda, H.; Morishita, S. Assessing Cell-to-Cell DNA Methylation Variability on Individual Long Reads. Sci. Rep. 2016, 6, 21317. [Google Scholar] [CrossRef] [PubMed]
- Landan, G.; Cohen, N.M.; Mukamel, Z.; Bar, A.; Molchadsky, A.; Brosh, R.; Horn-Saban, S.; Zalcenstein, D.A.; Goldfinger, N.; Zundelevich, A.; et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet. 2012, 44, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Florio, E.; Keller, S.; Coretti, L.; Affinito, O.; Scala, G.; Errico, F.; Fico, A.; Boscia, F.; Sisalli, M.J.; Reccia, M.G.; et al. Tracking the evolution of epialleles during neural differentiation and brain development: D-Aspartate oxidase as a model gene. Epigenetics 2017, 12, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Affinito, O.; Scala, G.; Palumbo, D.; Florio, E.; Monticelli, A.; Miele, G.; Avvedimento, V.E.; Usiello, A.; Chiariotti, L.; Cocozza, S. Modeling DNA methylation by analyzing the individual configurations of single molecules. Epigenetics 2016, 11, 881–888. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Scala, G.; Affinito, O.; Palumbo, D.; Florio, E.; Monticelli, A.; Miele, G.; Chiariotti, L.; Cocozza, S. ampliMethProfiler: A pipeline for the analysis of CpG methylation profiles of targeted deep bisulfite sequenced amplicons. BMC Bioinform. 2016, 17, 484. [Google Scholar] [CrossRef]
- Leitão, E.; Beygo, J.; Zeschnigk, M.; Klein-Hitpass, L.; Bargull, M.; Rahmann, S.; Horsthemke, B. Locus-Specific DNA Methylation Analysis by Targeted Deep Bisulfite Sequencing. Methods Mol. Biol. 2018, 1767, 351–366. [Google Scholar] [CrossRef]
- De Riso, G.; Cuomo, M.; Di Risi, T.; Della Monica, R.; Buonaiuto, M.; Costabile, D.; Pisani, A.; Cocozza, S.; Chiariotti, L. Ultra-Deep DNA Methylation Analysis of X-Linked Genes: GLA and AR as Model Genes. Genes (Basel) 2020, 11, 620. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Punzo, D.; Cuomo, M.; Affinito, O.; Coretti, L.; Sacchi, S.; Florio, E.; Lembo, F.; Carella, M.; Copetti, M.; et al. DNA methylation landscape of the genes regulating D-serine and D-aspartate metabolism in post-mortem brain from controls and subjects with schizophrenia. Sci. Rep. 2018, 8, 10163. [Google Scholar] [CrossRef] [PubMed]
- Affinito, O.; Palumbo, D.; Fierro, A.; Cuomo, M.; De Riso, G.; Monticelli, A.; Miele, G.; Chiariotti, L.; Cocozza, S. Nucleotide distance influences co-methylation between nearby CpG sites. Genomics 2020, 112, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Shipony, Z.; Mukamel, Z.; Cohen, N.M.; Landan, G.; Chomsky, E.; Zeliger, S.R.; Fried, Y.C.; Ainbinder, E.; Friedman, N.; Tanay, A. Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. Nature 2014, 513, 115–119. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Riso, G.; Fiorillo, D.F.G.; Fierro, A.; Cuomo, M.; Chiariotti, L.; Miele, G.; Cocozza, S. Modeling DNA Methylation Profiles through a Dynamic Equilibrium between Methylation and Demethylation. Biomolecules 2020, 10, 1271. https://doi.org/10.3390/biom10091271
De Riso G, Fiorillo DFG, Fierro A, Cuomo M, Chiariotti L, Miele G, Cocozza S. Modeling DNA Methylation Profiles through a Dynamic Equilibrium between Methylation and Demethylation. Biomolecules. 2020; 10(9):1271. https://doi.org/10.3390/biom10091271
Chicago/Turabian StyleDe Riso, Giulia, Damiano Francesco Giuseppe Fiorillo, Annalisa Fierro, Mariella Cuomo, Lorenzo Chiariotti, Gennaro Miele, and Sergio Cocozza. 2020. "Modeling DNA Methylation Profiles through a Dynamic Equilibrium between Methylation and Demethylation" Biomolecules 10, no. 9: 1271. https://doi.org/10.3390/biom10091271
APA StyleDe Riso, G., Fiorillo, D. F. G., Fierro, A., Cuomo, M., Chiariotti, L., Miele, G., & Cocozza, S. (2020). Modeling DNA Methylation Profiles through a Dynamic Equilibrium between Methylation and Demethylation. Biomolecules, 10(9), 1271. https://doi.org/10.3390/biom10091271