Abstract
Introduction: Radioresistance in prostate cancer (PCa) poses a major therapeutic challenge. Galectin-3 (Gal-3) is overexpressed in aggressive PCa and may contribute to resistance mechanisms. This study evaluated the role of Gal-3 in radioresistance and assessed the effect of its pharmacological inhibition using GB1107. Methods: Parental (22RV1-P) and radioresistant (22RV1-RR) PCa cell lines were treated with GB1107. Western blotting assessed Gal-3 and Protein Phosphatase 1 alpha (PP1α) expression. Cell viability (PrestoBlue™), migration (wound assay), and clonogenic survival post-irradiation were evaluated. Statistical significance was set at p < 0.05. Results: Gal-3 was significantly upregulated in 22RV1-RR cells (p = 0.0237). GB1107 reduced viability and impaired migration in both cell lines. Radiosensitisation was observed in 22RV1-P cells (p < 0.0001) but was not significant in 22RV1-RR cells (p = 0.1258). A non-significant increase in PP1α expression was detected in RR cells. Conclusion: Gal-3 contributes to radioresistance. Further studies are needed to clarify the role of PP1α and optimise Gal-3-targeted strategies.
1. Introduction
Prostate cancer (PCa) is the second most frequently diagnosed malignancy and the fifth leading cause of cancer-related deaths in men, with 1.5 million new cases and 397,000 deaths reported worldwide in 2022 [1]. Diagnosis relies on Digital Rectal Examination (DRE), Prostate-Specific Antigen (PSA), biopsy, and advanced imaging, though PSA’s limited specificity has driven the development of novel biomarkers [2,3,4].
Treatment depends on stage: active surveillance, surgery, external beam radiation therapy (EBRT), and brachytherapy are standard for localised disease, while advanced cases require androgen deprivation therapy (ADT), chemotherapy, new hormonal agents, immunotherapy, or radiopharmaceuticals [4]. Despite advances, castration-resistant PCa remains incurable, and radioresistance limits EBRT efficacy, with recurrence in up to 40% of patients [5,6]. Radioresistance is driven by enhanced DNA repair, apoptosis evasion, AR and PI3K/Akt signalling, checkpoint activation, Epithelial-to-Mesenchymal Transition (EMT), epigenetic alterations, and hypoxia [7,8,9,10,11,12,13,14,15,16,17,18]. Clinical strategies such as dose escalation, hypofractionation, and radiotherapy (RT)-ADT combinations improve outcomes but do not prevent progression to castration-resistant PCa stages. Thus, identifying biomarkers and new targets to overcome radioresistance remains a priority [15,19].
Galectin-3 (Gal-3), a β-galactoside-binding lectin encoded by LGALS3, is upregulated in PCa and correlates with Gleason score and tumour stage [20,21,22,23,24]. Its nuclear-to-cytoplasmic shift in malignant cells promotes invasion, apoptosis resistance, and poor outcomes. Gal-3 regulates proliferation, survival, and migration through PI3K/AKT, MAPK/ERK, and Bcl-2 signalling, while modulating the tumour microenvironment by suppressing T-cell activity and promoting M2 macrophage polarisation [8,17,25,26,27,28,29,30,31]. Pharmacological or RNAi-mediated Gal-3 inhibition reduces viability, induces apoptosis, and enhances sensitivity to chemo- and radiotherapy, though most studies remain preclinical. Emerging evidence also links Gal-3 overexpression to a PP1α–p53 regulatory axis in radioresistant cells (Figure 1) [32,33,34,35,36,37,38,39].
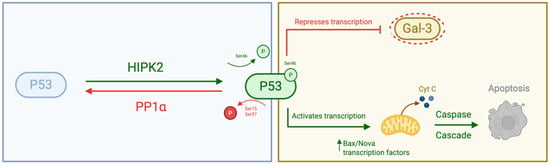
Figure 1.
Schematic representation of the molecular interactions between PP1α and p53, and between p53 and Galectin-3 (Gal-3). PP1α directly dephosphorylates p53 at key serine residues, modulating its transcriptional activity and stability. In turn, p53 transcriptionally regulates Gal-3 expression. Created with BioRender.com.
Currently, two pharmacological Gal-3 inhibitors are under investigation: GB1107, a small molecule, and belapectin, a carbohydrate-based compound [40,41]. GB1107 suppresses proliferation, migration, and invasion in PCa cell lines overexpressing Gal-3, while altering DNA, protein, and lipid profiles, including reducing monounsaturated fatty acids associated with invasiveness. It also affects glucose/lipid metabolism and inflammatory pathways, suggesting Gal-3-dependent metabolic reprogramming. Belapectin, a Galectin-3 inhibitor, showed limited antitumour activity as a single agent in preclinical models but demonstrated enhanced efficacy when combined with immunotherapeutic agents. Given its prior validation in PCa, GB1107 was selected for this study [41,42,43,44].
Based on these previously reported findings, we hypothesise that Gal-3 inhibition may overcome radioresistance by disrupting survival pathways (e.g., Bcl-2, PI3K/AKT, MAPK), enhancing radiation-induced cell death, and reducing pro-metastatic features. This study aims to (i) compare Gal-3 expression in radiosensitive and radioresistant PCa cells; (ii) evaluate the effects of GB1107 on viability, clonogenic survival, and migration; and (iii) investigate PP1α expression, given its potential interplay with Gal-3 in DNA damage response.
Therefore, this study serves as a proof-of-concept investigation. By employing a matched pair of parental and radioresistant cells, we aim to dissect the functional role of Gal-3 in radioresistance and evaluate the preliminary efficacy of GB1107 as a radiosensitizer in an aggressive prostate cancer context.
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
Two human prostate cancer (PCa) cell lines were used: 22RV1 parental (22RV1-P)—ATCC, Manassas, VA, USA—and a radioresistant derivative (22RV1-RR). The 22RV1 line was selected as the study model because it represents castration-resistant prostate cancer (CRPC), mimicking the aggressive, androgen-independent phenotype often encountered in the clinical setting where therapeutic resistance is most critical. The 22RV1-RR cell line was generated at IPO-Porto, as described in [45]. Briefly, 22RV1 parental cells were exposed to a cumulative radiation dose of 50 Gy [46], delivered as multifractionated irradiation (2.5 Gy per fraction, 5 fractions per week for 4 weeks), following an RT fractionation scheme, based on EAU guidelines for PCa treatment [4]. Cells were cultured in RPMI 1640 medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% foetal bovine serum (FBS), 100 IU/mL penicillin, and 100 μg/mL streptomycin and maintained at 37 °C in a humidified incubator with 5% CO2.
2.2. Gal-3 Inhibition
GB1107 [43] was used to pharmacologically inhibit Gal-3 activity in prostate cancer models. GB1107 was reconstituted in DMSO and diluted in culture medium to the final working concentration immediately before use. Based on previous in vitro studies demonstrating its efficacy, a concentration of 5 or 10 μM was selected for functional assays [47].
2.3. Protein Expression Analysis (Western Blot)
22RV1 parental and radioresistant cells were lysed with RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, Waltham, MA, USA) containing protease and phosphatase inhibitors, and protein concentration was quantified using the Pierce™ BCA Protein Assay Kit. Equal amounts of protein (25 µg) were separated by SDS-PAGE, transferred to PVDF membranes (Immobilon-P, Millipore, Burlington, MA, USA), and briefly stained with Ponceau S and scanned using the ChemiDoc™ system (Bio-Rad Laboratories, Hercules, CA, USA) to verify transfer efficiency and assess total protein loading. Membranes were blocked with 5% non-fat dry milk in TBS-T (Tris-buffered saline with 0.1% Tween-20); cells were incubated overnight with primary antibodies against Galectin-3 (mouse, 1:500; Santa Cruz Biotechnology, Dallas, TX, USA, Cat# sc-32790) and PP1α (rabbit, 1:100; Cell Signalling Technology, Danvers, MA, USA Cat# 2582) and then probed with IRDye®-conjugated secondary antibodies. Protein detection and quantification were performed with the Odyssey® Imaging System (LI-COR Biosciences, Lincoln, NE, USA), with band intensities normalised to tubulin. Densitometric analysis was conducted using ImageJ software (version 1.54g, National Institutes of Health, Bethesda, MD, USA).
2.4. Cell Viability Assay
Cell viability was measured using PrestoBlue™ (Thermo Fisher Scientific) following the manufacturer’s protocol. Briefly, 22RV1 parental (22RV1-P) and radioresistant (22RV1-RR) cells were seeded at 5000 cells/well in 96-well plates and incubated for 24 h at 37 °C, 5% CO2. After incubation, cells were treated with GB1107 (1, 5 and 10 µM) dissolved in DMSO or DMSO control for 48 h. PrestoBlue™ (10% v/v) was added and incubated for 1 h, and fluorescence was measured at wavelengths (excitation/emission) of 560 nm/590 nm. Background-corrected values were expressed relative to controls, considering the controls as 100% viability. Data represent at least three independent experiments with five technical replicates. Statistical analysis was performed using one-way ANOVA (p < 0.05).
2.5. Cell Wound Healing Assay
22RV1 parental (22RV1-P) and radioresistant (22RV1-RR) cells were seeded in 24-well plates (1 × 105 cells/well) and grown to confluence. A scratch was made using a sterile pipette tip, and cells were washed with PBS and incubated in serum-free medium with or without 10 µM GB1107 for 48 h. Images were captured at 0, 24, and 48 h using an EVOS™ M5000 system, and wound areas were quantified with ImageJ. Percentage wound closure was calculated relative to 0 h. Experiments were performed in triplicate.
2.6. Clonogenic Assay
Cells were treated with 5 µM GB1107 2 h prior to irradiation to allow for drug uptake. Cells were exposed to single doses of ionising radiation ranging from 0 to 8 Gy using a 6 MV photon beam generated by a TrueBeam linear accelerator. Irradiation was performed at room temperature with a dose rate of 600 MU/min and a field size of 25 × 25 cm2.
Twenty-four hours post-irradiation, cells were harvested and re-seeded into 6-well plates at optimised densities. Cultures were maintained for 7 days to allow for colony formation. Subsequently, colonies were fixed and stained, and those containing at least 50 cells were counted. The Surviving Fraction (SF) was calculated correcting for the Plating Efficiency (PE) of non-irradiated controls. Survival curves were fitted using the linear-quadratic (LQ) model [14].
2.7. Statistical Analysis
All experiments were conducted in triplicate, and results are expressed as mean ± standard deviation (SD). Data normality was tested with the Shapiro–Wilk test. Comparisons between two groups were performed using an unpaired Student’s t-test (parametric) or the Mann–Whitney U test (non-parametric). For more than two groups, one-way ANOVA with Tukey’s post hoc test (parametric) or the Kruskal–Wallis test with Dunn’s post hoc test (non-parametric) was applied. Survival curves from clonogenic assays were analysed by linear regression, with group differences evaluated by ANCOVA. Statistical significance was set at p < 0.05. Outliers were assessed using Grubbs’ test [48].
All statistical analyses were performed using GraphPad Prism version 10.0.0 for macOS (GraphPad Software, San Diego, CA, USA).
3. Results
3.1. Gal-3 Is Overexpressed in Radioresistant 22RV1 Cells Compared to 22RV1 Parental Cells
To investigate the expression of Gal-3 in radiotherapy-resistant prostate cancer (PCa) cells (22RV1-RR), a Western blot analysis was performed. The analysis revealed an increased expression of Gal-3 in the radioresistant PCa cell line (22RV1-RR) compared to the parental 22RV1-P (Figure 2), suggesting an association between Gal-3 upregulation and radioresistance.
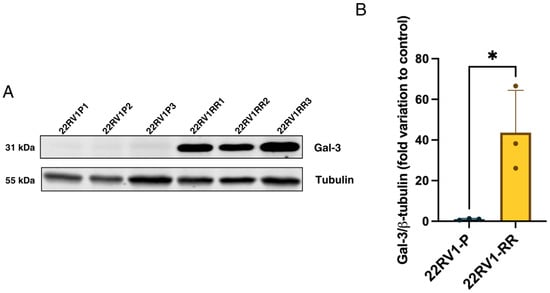
Figure 2.
(A) Western blot showing the variation in Gal-3 expression between parental cells and cells exposed to radiotherapy, including a tubulin blot for subsequent normalisation; (B) variation in Gal-3 expression in parental 22RV1 cells and radioresistant 22RV1 cells. Values were normalised to tubulin. An unpaired t-test was also performed. p-value = 0.0237; n = 3, * < 0.05.
3.2. GB1107 Reduces Cell Viability in Both Parental and Radioresistant 22RV1 Cells
Given the increased Gal-3 expression in radiotherapy-resistant cells, the functional role of Gal-3 in 22RV1 parental (22RV1-P) and radioresistant (22RV1-RR) cells was evaluated using the selective inhibitor GB1107. Both cell lines showed reduced viability upon Gal-3 inhibition after 24 h, with significant sensitivity observed at 5 μM and 10 μM (Figure 3).
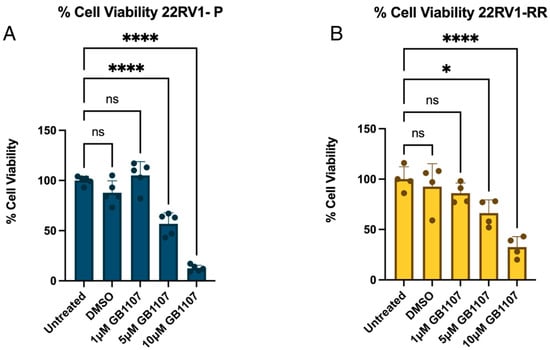
Figure 3.
22RV1-P and 22RV1-RR viability in the presence of Gal-3 inhibitor (GB1107). (A) 22RV1-P was incubated with different GB1107 concentrations for 24 h. (B) 22RV1-RR was incubated with different GB1107 concentrations for 24 h. An ANOVA test was performed (untreated was considered 100% viability), ns (non-significant), * < 0.05, **** < 0.0001; n = 3.
3.3. Gal-3 Inhibition Impairs Migration in Both 22RV1 Cell Lines
Following the viability assay, DMSO showed no effect on cell viability, while 10 μM GB1107 induced the strongest reduction. This concentration was therefore selected for a wound healing assay to assess the impact of Gal-3 inhibition on cell migration under four experimental conditions. In this assay, control 22RV1-P cells achieved near-complete wound closure within 24 h, whereas control 22RV1-RR cells displayed delayed migration, with significant closure only at 48 h (Figure 4A,B).
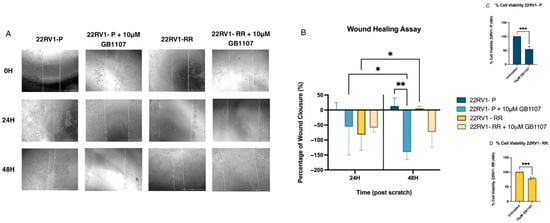
Figure 4.
Wound healing assay assessing the effect of GB1107 on prostate cancer cells. (A) Representative phase-contrast images of wound closure at 0, 24, and 48 h post-scratch in 22RV1 parental cells (22RV1-P), 22RV1-P treated with 10 µM GB1107, 22RV1 radioresistant cells (22RV1-RR), and 22RV1-RR treated with 10 µM GB1107. Images were acquired using the EVOS M5000 imaging system. (B) Quantification of wound closure at 24 and 48 h, expressed as the percentage of closure relative to 0 h. Data are presented as mean ± SD (n = 3). Statistical analysis was performed using two-way ANOVA followed by multiple comparisons; * < 0.05, ** < 0.01. 22RV1-P and 22RV1-RR viability in the presence of Gal-3 inhibitor (GB1107). (C) 22RV1-P was incubated with 10 µM GB1107 concentrations for 48 h. (D) 22RV1-RR was incubated with 10 µM GB1107 concentrations for 48 h. A t-test was made (untreated was considered 100% viability). *** < 0.001; n = 3.
These findings indicate that Gal-3 inhibition not only reduces wound regeneration capacity but also enlarges the wound area, suggesting induction of cell death. The effect was gradual in parental cells compared to controls, while radioresistant cells showed a more pronounced impairment in migration and wound closure.
To confirm these results, a PrestoBlue viability assay was performed at the end of the wound healing experiment. After 48 h of exposure to 10 μM GB1107, both 22RV1-P and 22RV1-RR cells displayed sensitivity (Figure 4C,D).
3.4. Survival Curves Differ upon Gal-3 Inhibition in Parental but Not in Radioresistant 22RV1 Cells
In the clonogenic assay—which measures the ability of individual cells to survive and form colonies after treatment—parental cells exposed to the Gal-3 inhibitor displayed survival curves that differed significantly from those of the control group, confirming that Gal-3 inhibition influences their response to radiation (Figure 5). In contrast, treatment of radioresistant (RR) cells with 5 μM GB1107 did not significantly affect colony formation compared with controls, suggesting reduced sensitivity to Gal-3 inhibition at this concentration.
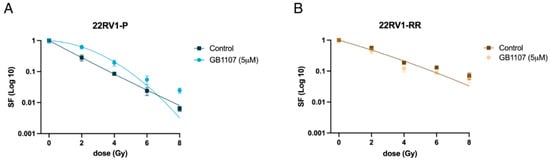
Figure 5.
(A) The survival fraction of parental 22RV1 (P) cells, untreated and treated with GB1107 (5 μM), is represented using the linear-quadratic (LQ) model (S = e−(αD+βD2)). p-value < 0.0001. (B) The survival fraction of 22RV1-RR cells, untreated and treated with GB1107 (5 μM), is represented by a single curve using the linear-quadratic (LQ) model (S = e−(αD+βD2)), as no significant difference was observed between the two conditions (p-value = 0.1258); n = 3.
3.5. PP1α Expression Appears Elevated in Radioresistant Cells
As previously mentioned, PP1α may play a role in the signalling pathways associated with the acquisition of radioresistance. Moreover, its potential link to Gal-3, possibly mediated through p53-dependent mechanisms, makes it a relevant target to explore in this context. Accordingly, the expression of PP1α was evaluated to determine whether alterations in its levels could be associated with the differential radiation responses observed between parental and radioresistant cells. A tendency for higher PP1α expression was observed in radioresistant (RR) cells compared with the parental cell line (Figure 6), although the difference was not statistically significant. This upward trend suggests that PP1α may be upregulated as part of the cellular response associated with resistance to radiation, highlighting its potential involvement in the mechanisms underlying this phenotype.
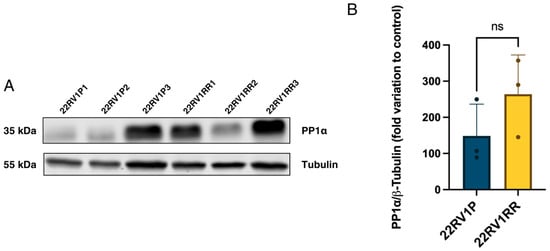
Figure 6.
(A) Western blot showing the variation in PP1α expression between parental cells and radioresistant cells, including a tubulin blot for subsequent normalisation. (B) Variation in PP1α expression in parental 22RV1 cells and radioresistant 22RV1. Values were normalised to tubulin. An unpaired t-test was also performed. p-value = 0.227; n = 3; ns (non-significant).
4. Discussion
Understanding the molecular mechanisms underlying radioresistance is crucial for improving the efficacy of radiotherapy in prostate cancer, as the development of resistant tumour cell populations remains a major barrier to successful treatment. Given its multifaceted roles in cancer biology, Galectin-3 (Gal-3) has emerged as a potential mediator of cellular adaptation to radiation.
Our findings support this hypothesis, showing that Gal-3 expression is upregulated in the radioresistant 22RV1-RR cells compared with the parental line. This observation is consistent with previous reports describing Gal-3’s pleiotropic functions in other tumour types, including its involvement in DNA damage response, apoptosis evasion, and modulation of the tumour microenvironment [14,48]. Pharmacological inhibition with GB1107 reduced cell viability and migration in both lines, confirming Gal-3 functional activity (Figure S1).
Our results indicated that GB1107 effectively radiosensitized parental cells but failed to significantly sensitise the radioresistant (22RV1-RR) subline (Figure 5). Rather than a methodological failure, we interpret this as a biologically meaningful result. It suggests that the adaptations driving radioresistance in 22RV1-RR cells—potentially involving deep metabolic reprogramming or alternative pathway activation—confer a degree of cross-resistance to Gal-3 inhibition. This implies that in established radioresistant tumours, Gal-3 inhibition alone may be insufficient, and combinatorial strategies targeting multiple survival nodes may be required.
Regarding the molecular mechanism, we observed a trend towards increased PP1α expression in RR cells (Figure 6). Based on the literature, we propose a hypothetical model where a PP1α–p53–Gal-3 axis supports resistance, as illustrated in Figure 7 [32,49]. However, we emphasise that our current data is observational and hypothesis-generating. We did not assess p53 phosphorylation status or conduct knockdown experiments in this study; thus, this mechanism remains to be experimentally validated.
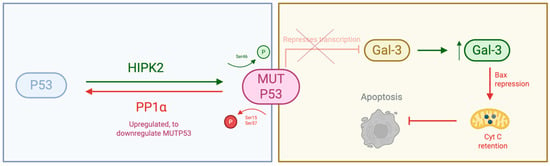
Figure 7.
Hypothetical interaction between PP1α, p53, and Galectin-3 (Gal-3). This schematic illustrates the proposed molecular interplay between PP1α, the tumour suppressor p53, and Galectin-3 (Gal-3) in the context of prostate cancer radioresistance. Created with BioRender.com.
To place our findings in a clinical context, Gal-3 should be viewed alongside other emerging biomarkers of aggressiveness. Recent research highlights the utility of histopathological ratios and specific molecular signatures (e.g., PD-L1, H19) in predicting prognosis [50,51,52,53]. Integrating Gal-3 assessment via immunohistochemistry could further refine risk stratification, identifying patients with a high-risk profile who might require intensified initial therapy.
Clinically, the management of radioresistant prostate cancer remains challenging, with limited salvage options often restricted to systemic therapies with significant side effects. The ability to predict resistance before treatment would be invaluable. In this context, immunohistochemistry (IHC) for Gal-3 could serve as a practical tool for pathologists. Gal-3 is readily detectable by IHC in biopsy specimens; however, its specific utility in predicting radioresistance requires standardisation of scoring systems and validation in prospective cohorts.
As a proof-of-concept study, findings should be validated across multiple cell lines and in vivo models. In this study, we focused on the 22RV1 model due to its relevance as a proxy for castration-resistant disease (CRPC). While the use of a single isogenic pair (parental vs. radioresistant) limits the generalizability of the results, it allows for a controlled evaluation of acquired resistance mechanisms within a specific genetic background. Future studies should extend this work to other models (e.g., PC-3, DU145) and patient-derived xenografts to confirm the universality of Gal-3 as a target.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/therapeutics3010007/s1. Figure S1: Schematic illustration of the proposed effects of Gal-3 inhibition in 22RV1-RR cells.
Author Contributions
Conceptualisation: R.M.R. and M.F. Methodology: R.M.R., B.M. and V.M.-G. Formal Analysis: R.M.R., B.M. and V.M.-G. Investigation: R.M.R., B.M., V.M.-G. and C.J. Resources: M.F. and C.J. Data Curation: R.M.R. and B.M. Writing—Original Draft Preparation: R.M.R. Writing—Review and Editing: R.M.R., B.M., V.M.-G., C.J. and M.F. Visualisation: R.M.R. Supervision: M.F. and C.J. Project Administration: R.M.R. and M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Institute for Biomedicine—iBiMED (UIDB/04501/2020).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank the Fardilha’s Lab at the Institute of Biomedicine (iBiMED) and the Cancer Biology and Epigenetics Group for their support and valuable discussions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Cereser, L.; Evangelista, L.; Giannarini, G.; Girometti, R. Prostate MRI and PSMA-PET in the Primary Diagnosis of Prostate Cancer. Diagnostics 2023, 13, 2697. [Google Scholar] [CrossRef]
- del Pino-Sedeño, T.; Infante-Ventura, D.; de Armas Castellano, A.; de Pablos-Rodríguez, P.; Rueda-Domínguez, A.; Serrano-Aguilar, P.; Trujillo-Martín, M.M. Molecular Biomarkers for the Detection of Clinically Significant Prostate Cancer: A Systematic Review and Meta-Analysis. Eur. Urol. Open Sci. 2022, 46, 105–127. [Google Scholar] [CrossRef]
- Cornford, P.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Brunckhorst, O.; Darraugh, J.; Eberli, D.; De Meerleer, G.; De Santis, M.; Farolfi, A.; et al. EAU-EANM-ESTRO-ESUR-ISUP-SIOG Guidelines on Prostate Cancer—2024 Update. Part I: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2024, 86, 148–163. [Google Scholar] [CrossRef]
- Sekhoacha, M.; Riet, K.; Motloung, P.; Gumenku, L.; Adegoke, A.; Mashele, S. Prostate Cancer Review: Genetics, Diagnosis, Treatment Options, and Alternative Approaches. Molecules 2022, 27, 5730. [Google Scholar] [CrossRef]
- Riaz, I.B.; Naqvi, S.A.A.; He, H.; Asghar, N.; Siddiqi, R.; Liu, H.; Singh, P.; Childs, D.S.; Ravi, P.; Hussain, S.A.; et al. First-Line Systemic Treatment Options for Metastatic Castration-Sensitive Prostate Cancer: A Living Systematic Review and Network Meta-Analysis. JAMA Oncol. 2023, 9, 635. [Google Scholar] [CrossRef]
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation Resistance of Cancer Stem Cells: The 4 R’s of Radiobiology Revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K Pathway Alterations in Cancer: Variations on a Theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer Stem Cells and Signaling Pathways in Radioresistance. Oncotarget 2016, 7, 11002–11017. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.J.; Sibson, N.R.; Kiltie, A.E. Treatment of Breast and Prostate Cancer by Hypofractionated Radiotherapy: Potential Risks and Benefits. Clin. Oncol. 2015, 27, 420–426. [Google Scholar] [CrossRef]
- Macedo-Silva, C.; Benedetti, R.; Ciardiello, F.; Cappabianca, S.; Jerónimo, C.; Altucci, L. Epigenetic Mechanisms Underlying Prostate Cancer Radioresistance. Clin. Epigenetics 2021, 13, 125. [Google Scholar] [CrossRef]
- Peitzsch, C.; Cojoc, M.; Hein, L.; Kurth, I.; Mäbert, K.; Trautmann, F.; Klink, B.; Schröck, E.; Wirth, M.P.; Krause, M.; et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer Res. 2016, 76, 2637–2651. [Google Scholar] [CrossRef]
- De Bari, B.; Fiorentino, A.; Arcangeli, S.; Franco, P.; D’Angelillo, R.M.; Alongi, F. From Radiobiology to Technology: What Is Changing in Radiotherapy for Prostate Cancer. Expert. Rev. Anticancer Ther. 2014, 14, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Macedo-Silva, C.; Miranda-Gonçalves, V.; Tavares, N.T.; Barros-Silva, D.; Lencart, J.; Lobo, J.; Oliveira, Â.; Correia, M.P.; Altucci, L.; Jerónimo, C. Epigenetic Regulation of TP53 Is Involved in Prostate Cancer Radioresistance and DNA Damage Response Signaling. Signal Transduct. Target. Ther. 2023, 8, 395. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Moya, A.; Supiot, S.; Seegers, V.; Lizée, T.; Legouté, F.; Perennec, T.; Calais, G. Mapping of Recurrence Sites Following Adjuvant or Salvage Radiotherapy for Prostate Cancer Patients. Front. Oncol. 2022, 11, 787347. [Google Scholar] [CrossRef] [PubMed]
- Khuntia, D.; Reddy, C.A.; Mahadevan, A.; Klein, E.A.; Kupelian, P.A. Recurrence-Free Survival Rates after External-Beam Radiotherapy for Patients with Clinical T1–T3 Prostate Carcinoma in the Prostate-Specific Antigen Era. Cancer 2004, 100, 1283–1292. [Google Scholar] [CrossRef]
- Dearnaley, D.; Syndikus, I.; Mossop, H.; Khoo, V.; Birtle, A.; Bloomfield, D.; Graham, J.; Kirkbride, P.; Logue, J.; Malik, Z.; et al. Conventional versus Hypofractionated High-Dose Intensity-Modulated Radiotherapy for Prostate Cancer: 5-Year Outcomes of the Randomised, Non-Inferiority, Phase 3 CHHiP Trial. Lancet Oncol. 2016, 17, 1047–1060. [Google Scholar] [CrossRef]
- Cho, L.C.; Timmerman, R.; Kavanagh, B. Hypofractionated External-Beam Radiotherapy for Prostate Cancer. Prostate Cancer 2013, 2013, 103547. [Google Scholar] [CrossRef]
- Freedland, S.J.; Humphreys, E.B.; Mangold, L.A.; Eisenberger, M.; Dorey, F.J.; Walsh, P.C.; Partin, A.W. Risk of Prostate Cancer-Specific Mortality Following Biochemical Recurrence after Radical Prostatectomy. JAMA 2005, 294, 433–439. [Google Scholar] [CrossRef]
- Lima, T.; Macedo-Silva, C.; Felizardo, D.; Fraga, J.; Carneiro, I.; Jerónimo, C.; Henrique, R.; Fardilha, M.; Vitorino, R. Gal-3 Protein Expression and Localization in Prostate Tumours. Curr. Oncol. 2023, 30, 2729–2742. [Google Scholar] [CrossRef]
- Carvalho, R.S.; Fernandes, V.C.; Nepomuceno, T.C.; Rodrigues, D.C.; Woods, N.T.; Suarez-Kurtz, G.; Chammas, R.; Monteiro, A.N.; Carvalho, M.A. Characterization of LGALS3 (Galectin-3) as a Player in DNA Damage Response. Cancer Biol. Ther. 2014, 15, 840. [Google Scholar] [CrossRef]
- Abramovic, I.; Pezelj, I.; Dumbovic, L.; Skara Abramovic, L.; Vodopic, T.; Bulimbasic, S.; Stimac, G.; Bulic-Jakus, F.; Kulis, T.; Katusic Bojanac, A.; et al. LGALS3 CfDNA Methylation in Seminal Fluid as a Novel Prostate Cancer Biomarker Outperforming PSA. Prostate 2024, 84, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Balan, V.; Wang, Y.; Nangia-Makker, P.; Kho, D.; Bajaj, M.; Smith, D.; Heilbrun, L.; Raz, A.; Heath, E. Galectin-3: A Possible Complementary Marker to the PSA Blood Test. Oncotarget 2013, 4, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Filipová, M.; Bojarová, P.; Rodrigues Tavares, M.; Bumba, L.; Elling, L.; Chytil, P.; Gunár, K.; Křen, V.; Etrych, T.; Janoušková, O. Glycopolymers for Efficient Inhibition of Galectin-3: In Vitro Proof of Efficacy Using Suppression of T Lymphocyte Apoptosis and Tumor Cell Migration. Biomacromolecules 2020, 21, 3122–3133. [Google Scholar] [CrossRef] [PubMed]
- Su, W.H.; Chuang, P.C.; Huang, E.Y.; Yang, K.D. Radiation-Induced Increase in Cell Migration and Metastatic Potential of Cervical Cancer Cells Operates via the K-Ras Pathway. Am. J. Pathol. 2012, 180, 862–871. [Google Scholar] [CrossRef]
- Xin, M.; Dong, X.W.; Guo, X.L. Role of the Interaction between Galectin-3 and Cell Adhesion Molecules in Cancer Metastasis. Biomed. Pharmacother. 2015, 69, 179–185. [Google Scholar] [CrossRef]
- Kang, H.G.; Kim, D.H.; Kim, S.J.; Cho, Y.; Jung, J.; Jang, W.; Chun, K.H. Galectin-3 Supports Stemness in Ovarian Cancer Stem Cells by Activation of the Notch1 Intracellular Domain. Oncotarget 2016, 7, 68229. [Google Scholar] [CrossRef]
- Wang, D.; You, D.; Li, L. Galectin-3 Regulates Chemotherapy Sensitivity in Epithelial Ovarian Carcinoma via Regulating Mitochondrial Function. J. Toxicol. Sci. 2019, 44, 47–56. [Google Scholar] [CrossRef]
- Califice, S.; Castronovo, V.; Bracke, M.; Van Den Brǔle, F. Dual Activities of Galectin-3 in Human Prostate Cancer: Tumor Suppression of Nuclear Galectin-3 vs. Tumor Promotion of Cytoplasmic Galectin-3. Oncogene 2004, 23, 7527–7536. [Google Scholar] [CrossRef]
- Nangia-Makker, P.; Balan, V.; Raz, A. Galectin-3 Binding and Metastasis. Methods Mol. Biol. 2012, 878, 251–266. [Google Scholar] [CrossRef]
- Wang, N.; Ma, T.; Yu, B. Targeting Epigenetic Regulators to Overcome Drug Resistance in Cancers. Signal Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef]
- Cecchinelli, B.; Lavra, L.; Rinaldo, C.; Iacovelli, S.; Gurtner, A.; Gasbarri, A.; Ulivieri, A.; Del Prete, F.; Trovato, M.; Piaggio, G.; et al. Repression of the Antiapoptotic Molecule Galectin-3 by Homeodomain-Interacting Protein Kinase 2-Activated P53 Is Required for P53-Induced Apoptosis. Mol. Cell Biol. 2006, 26, 4746–4757. [Google Scholar] [CrossRef]
- Sionov, R.V.; Hayon, I.L.; Haupt, Y. The Regulation of P53 Growth Suppression. In Madame Curie Bioscience Database; Landes Bioscience: Philadelphia, PA, USA, 2013. [Google Scholar]
- Xu, Y. Regulation of P53 Responses by Post-Translational Modifications. Cell Death Differ. 2003, 10, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Wan, G.; Guo, H.; Zhang, X.; Lu, X. Protein Phosphatase 1 Inhibits P53 Signaling by Dephosphorylating and Stabilizing Mdmx. Cell Signal 2012, 25, 796. [Google Scholar] [CrossRef]
- Li, D.W.; Liu, J.P.; Schmid, P.C.; Schlosser, R.; Feng, H.; Liu, W.B.; Yan, Q.; Gong, L.; Sun, S.; Deng, M.; et al. Protein Phosphatase–1 Dephosphorylates P53 At Ser–15 And Ser–37 To Modulate Its Transcriptional And Apoptotic Activities. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2550. [Google Scholar] [CrossRef]
- Lee, S.J.; Lim, C.J.; Min, J.K.; Lee, J.K.; Kim, Y.M.; Lee, J.Y.; Won, M.H.; Kwon, Y.G. Protein Phosphatase 1 Nuclear Targeting Subunit Is a Hypoxia Inducible Gene: Its Role in Post-Translational Modification of P53 and MDM2. Cell Death Differ. 2007, 14, 1106–1116. [Google Scholar] [CrossRef]
- Yoshida, K.; Liu, H.; Miki, Y. Protein Kinase C δ Regulates Ser46 Phosphorylation of P53 Tumor Suppressor in the Apoptotic Response to DNA Damage. J. Biol. Chem. 2006, 281, 5734–5740. [Google Scholar] [CrossRef]
- Kachnic, L.A.; Wu, B.; Wunsch, H.; Mekeel, K.L.; DeFrank, J.S.; Tang, W.; Powell, S.N. The Ability of P53 to Activate Downstream Genes P21(WAF1/Cip1) and MDM2, and Cell Cycle Arrest Following DNA Damage Is Delayed and Attenuated in Scid Cells Deficient in the DNA-Dependent Protein Kinase. J. Biol. Chem. 1999, 274, 13111–13117. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e5. [Google Scholar] [CrossRef]
- Curti, B.D.; Koguchi, Y.; Leidner, R.S.; Rolig, A.S.; Sturgill, E.R.; Sun, Z.; Wu, Y.; Rajamanickam, V.; Bernard, B.; Hilgart-Martiszus, I.; et al. Enhancing Clinical and Immunological Effects of Anti-PD-1 with Belapectin, a Galectin-3 Inhibitor. J. Immunother. Cancer 2021, 9, e002371. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, A.C.; Humphries, D.C.; Herman, K.; Roper, J.A.; Holyer, I.; Mabbitt, J.; Mills, R.; Nilsson, U.J.; Leffler, H.; Pedersen, A.; et al. Effect of GB1107, a Novel Galectin-3 Inhibitor on pro-Fibrotic Signalling in the Liver. Eur. J. Pharmacol. 2024, 985, 177077. [Google Scholar] [CrossRef] [PubMed]
- GB1107|Galectin-3 Inhibitor|MedChemExpress. Available online: https://www.medchemexpress.com/GB1107.html?utm_source=google&utm_medium=CPC&utm_campaign=Europe&utm_term=HY-114409&utm_content=GB1107&gad_source=1 (accessed on 21 May 2025).
- Sturgill, E.R.; Rolig, A.S.; Linch, S.N.; Mick, C.; Kasiewicz, M.J.; Sun, Z.; Traber, P.G.; Shlevin, H.; Redmond, W.L. Galectin-3 Inhibition with Belapectin Combined with Anti-OX40 Therapy Reprograms the Tumor Microenvironment to Favor Anti-Tumor Immunity. Oncoimmunology 2021, 10, 1892265. [Google Scholar] [CrossRef] [PubMed]
- Macedo-Silva, C.; Miranda-Gonçalves, V.; Lameirinhas, A.; Lencart, J.; Pereira, A.; Lobo, J.; Guimarães, R.; Martins, A.T.; Henrique, R.; Bravo, I.; et al. JmjC-KDMs KDM3A and KDM6B Modulate Radioresistance under Hypoxic Conditions in Esophageal Squamous Cell Carcinoma. Cell Death Dis. 2020, 11, 1068. [Google Scholar] [CrossRef]
- Sramkoski, R.M.; Pretlow, T.G.; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A New Human Prostate Carcinoma Cell Line, 22Rv1. In Vitro Cell Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Ghazanfarpour, S.; Sheikhsofla, A.; Pourrahimi, M.; Sharma, S.; Skomra, A.; Sharikova, A.; Schwartz, S.A.; Mahajan, S.D.; Khmaladze, A.; Aalinkeel, R. Raman Spectroscopic Modality to Examine Therapeutic Efficacy of Galectin-3 Inhibitor in Prostate Cancer. Biochem. Biophys. Res. Commun. 2025, 757, 151646. [Google Scholar] [CrossRef]
- Zar, J.H. Biostatistical Analysis. 5th Edition, Pearson Prentice Hall, New Jersey—References—Scientific Research Publishing. 2010. Available online: https://www.scirp.org/reference/referencespapers?referenceid=1246378 (accessed on 21 May 2025).
- Ruvolo, P.P. Galectin 3 as a Guardian of the Tumor Microenvironment. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 427–437. [Google Scholar] [CrossRef]
- Vlajnic, T.; Bubendorf, L. Molecular Pathology of Prostate Cancer: A Practical Approach. Pathology 2021, 53, 36–43. [Google Scholar] [CrossRef]
- Kiełb, P.; Kowalczyk, K.; Gurwin, A.; Nowak, Ł.; Krajewski, W.; Sosnowski, R.; Szydełko, T.; Małkiewicz, B. Novel Histopathological Biomarkers in Prostate Cancer: Implications and Perspectives. Biomedicines 2023, 11, 1552. [Google Scholar] [CrossRef]
- Pecci, V.; Troisi, F.; Aiello, A.; De Martino, S.; Carlino, A.; Fiorentino, V.; Ripoli, C.; Rotili, D.; Pierconti, F.; Martini, M.; et al. Targeting of H19/Cell Adhesion Molecules Circuitry by GSK-J4 Epidrug Inhibits Metastatic Progression in Prostate Cancer. Cancer Cell Int. 2024, 24, 56. [Google Scholar] [CrossRef]
- Fiorentino, V.; Pepe, L.; Pizzimenti, C.; Zuccalà, V.; Pepe, P.; Cianci, V.; Mondello, C.; Tuccari, G.; Fadda, G.; Giuffrè, G.; et al. PD-L1 Expression in Prostate Cancer and Gleason Grade Group: Is There Any Relationship? Findings from a Multi-Institutional Cohort. Pathol. Res. Pract. 2025, 269, 155916. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.