
Optimization of Lithium Metal Anode Performance: Investigating the Interfacial Dynamics and Reductive Mechanism of Asymmetric Sulfonylimide Salts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Details
2.1. Classical Molecular Dynamics (cMD) Simulations
2.2. AIMD and RMD Simulations
3. Results and Discussion
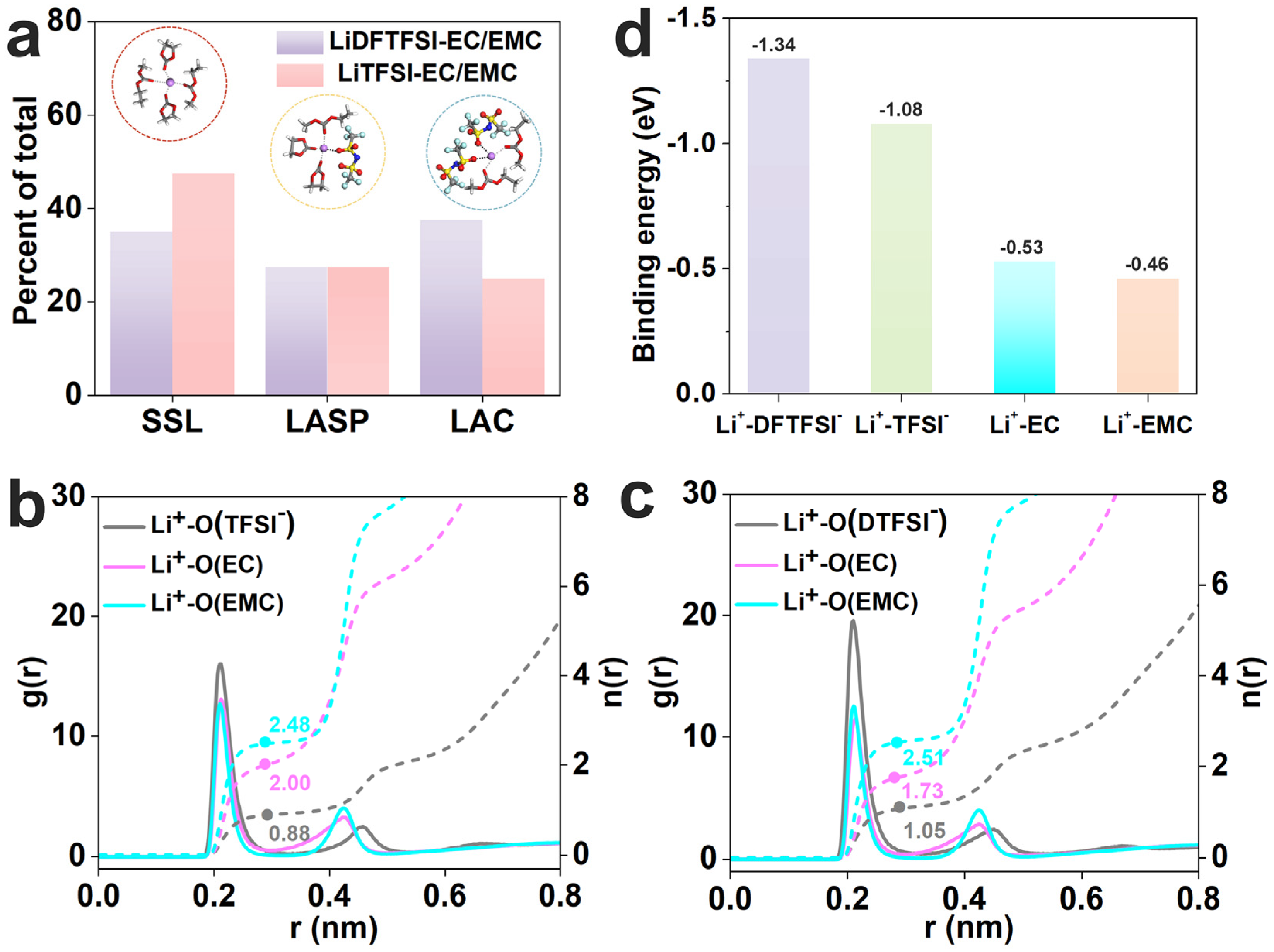
3.1. Electrolyte Solvation Structure Analysis
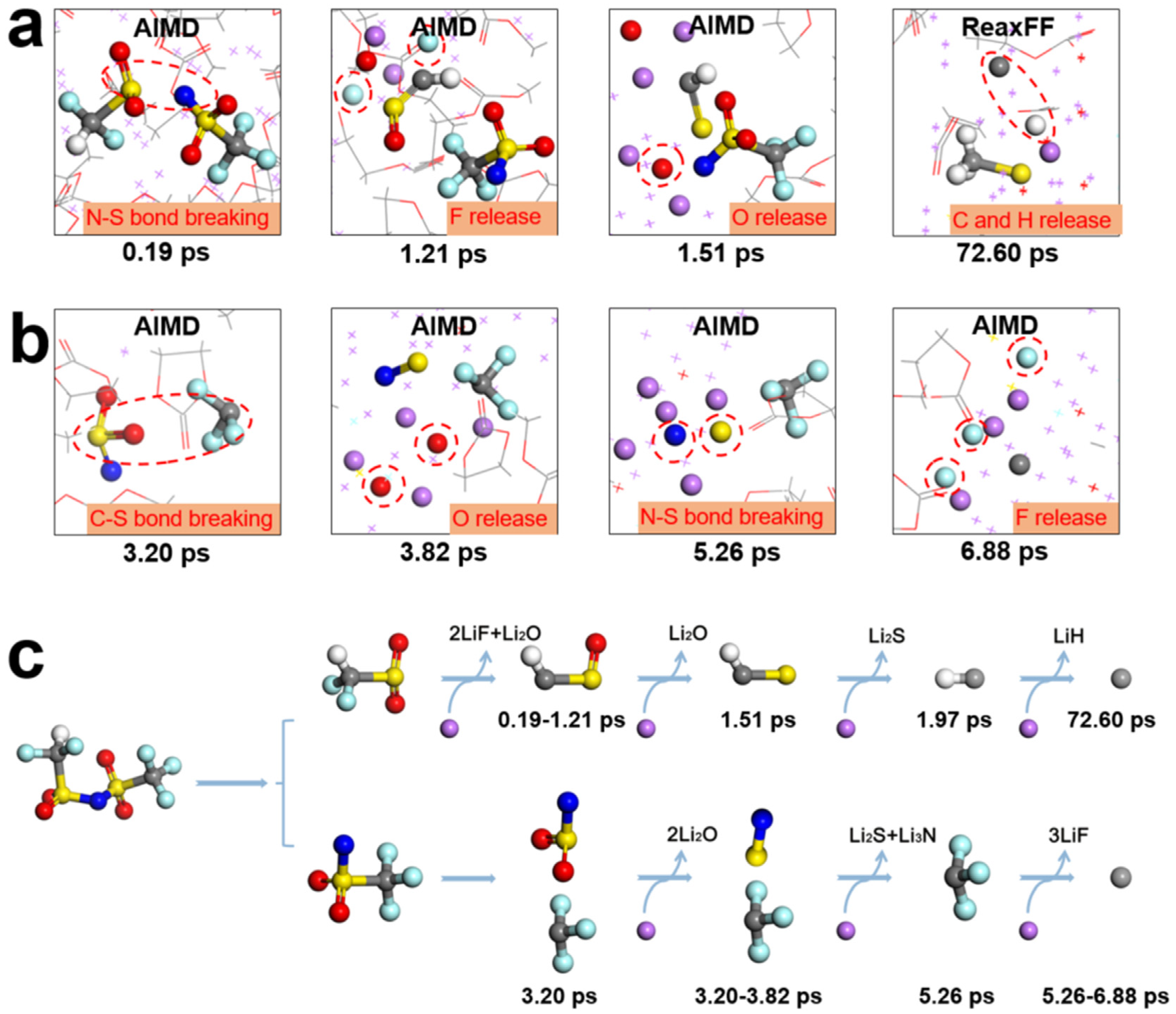
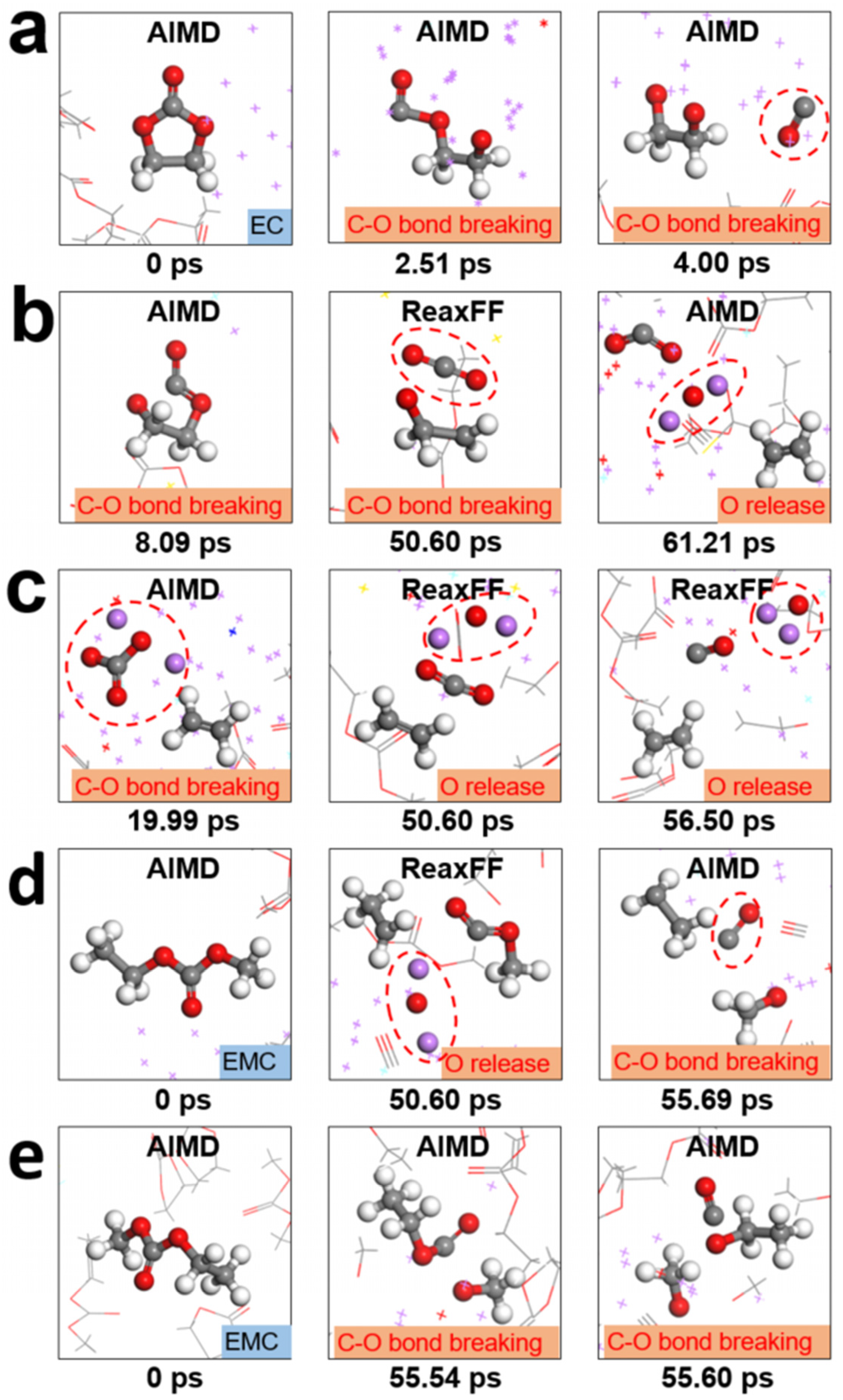
3.2. The Underlying Mechanism of Electrolyte Reduction and SEI Formation
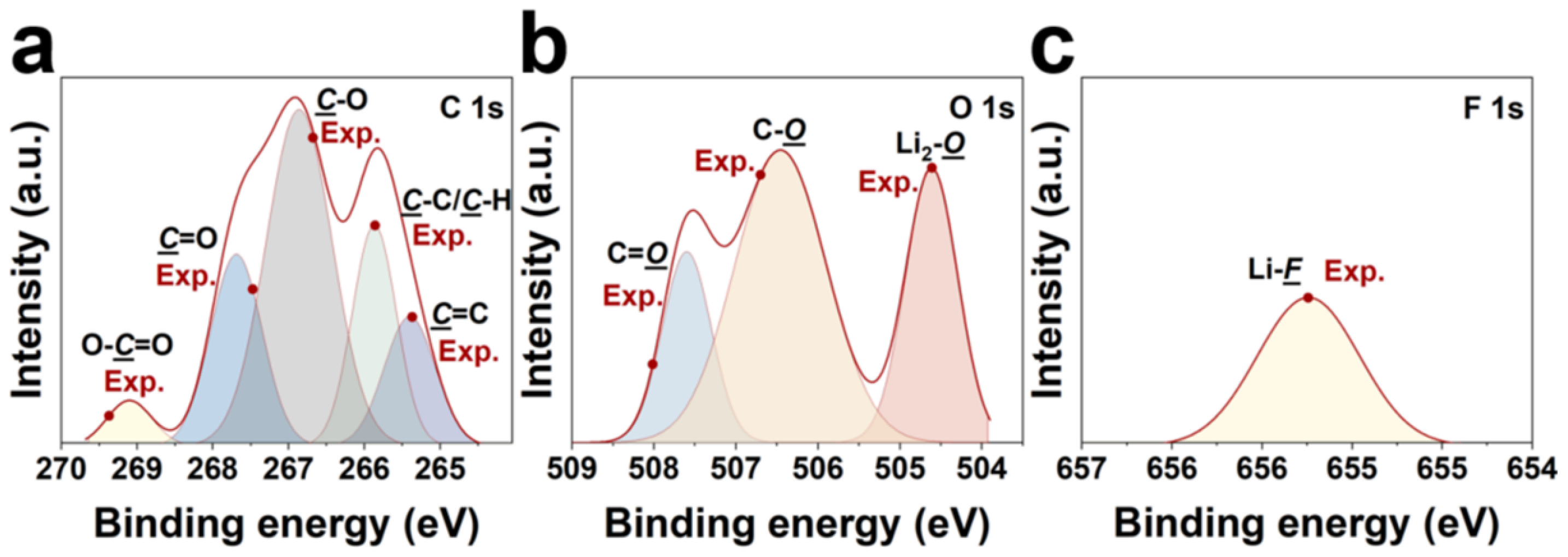
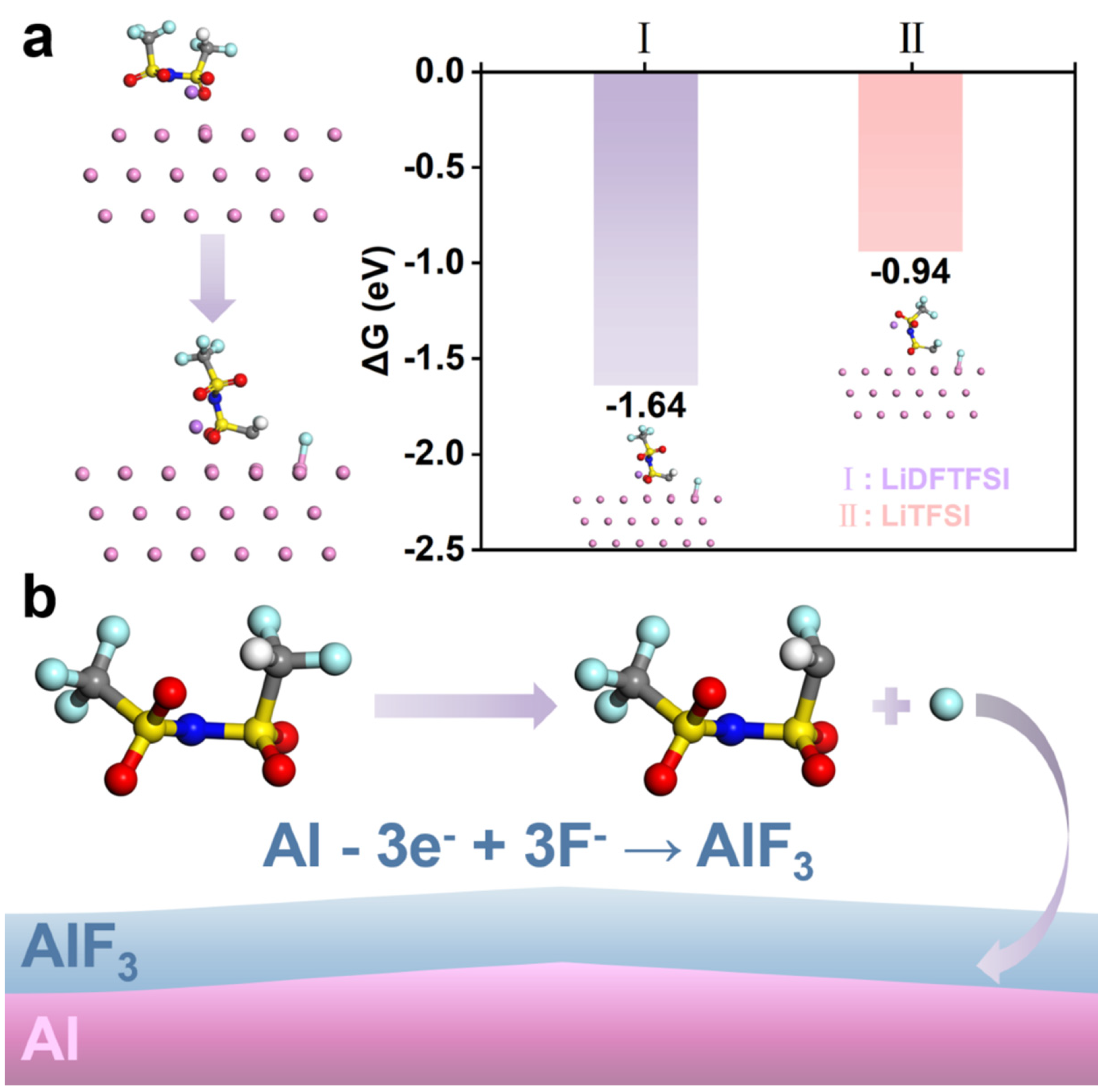
3.3. DFTFSI− Passivated Al
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Winter, M.; Barnett, B.; Xu, K. Before Li ion batteries. Chem. Rev. 2018, 118, 11433–11456. [Google Scholar] [CrossRef] [PubMed]
- Larcher, D.; Tarascon, J.M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Liu, Y.; Cui, Y. Reviving the lithium metal anode for high-energy batteries. Nat. Nanotechnol. 2017, 12, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.; Kamath, H.; Tarascon, J.-M. Electrical energy storage for the grid: A battery of choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Kothalawala, V.N.; Suzuki, K.; Nokelainen, J.; Hyvönen, A.; Makkonen, I.; Barbiellini, B.; Hafiz, H.; Tynjälä, P.; Laine, P.; Välikangas, J.; et al. Compton scattering study of strong orbital delocalization in a LiNiO2 cathode. Phys. Rev. B 2024, 109, 035139–035143. [Google Scholar] [CrossRef]
- Kothalawala, V.N.; Sasikala Devi, A.A.; Nokelainen, J.; Alatalo, M.; Barbiellini, B.; Hu, T.; Lassi, U.; Suzuki, K.; Sakurai, H.; Bansil, A. First principles calculations of the optical response of LiNiO2. Condens. Matter 2022, 7, 54. [Google Scholar] [CrossRef]
- Takenaka, N.; Bouibes, A.; Yamada, Y.; Nagaoka, M.; Yamada, A. Frontiers in theoretical analysis of solid electrolyte interphase formation mechanism. Adv. Mater. 2021, 33, 2100574–2100588. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Yun, X.; Chen, Y.; Guo, X.; Gao, P.; Zhou, G.; Zheng, C. Insights into the solvation chemistry in liquid electrolytes for lithium-based rechargeable batteries. Chem. Soc. Rev. 2023, 52, 5255–5316. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Singh, R.; Feng, S.; Li, J.; Xiao, J.; Bao, J.; Xu, Z.; Lu, D. Understanding of low-porosity sulfur electrode for high-energy lithium-sulfur batteries. Adv. Energy Mater. 2023, 13, 2203386–2203393. [Google Scholar] [CrossRef]
- Wang, H.; Yu, Z.; Kong, X.; Kim, S.C.; Boyle, D.T.; Qin, J.; Bao, Z.; Cui, Y. Liquid electrolyte: The nexus of practical lithium metal batteries. Joule 2022, 6, 588–616. [Google Scholar] [CrossRef]
- Li, M.; Wang, C.; Davey, K.; Li, J.; Li, G.; Zhang, S.; Mao, J.; Guo, Z. Recent progress in electrolyte design for advanced lithium metal batteries. SmartMat 2023, 4, e1185. [Google Scholar] [CrossRef]
- Hobold, G.M.; Lopez, J.; Guo, R.; Minafra, N.; Banerjee, A.; Shirley Meng, Y.; Shao-Horn, Y.; Gallant, B.M. Moving beyond 99.9% coulombic efficiency for lithium anodes in liquid electrolytes. Nat. Energy 2021, 6, 951–960. [Google Scholar] [CrossRef]
- Liu, J.; Bao, Z.; Cui, Y.; Dufek, E.J.; Goodenough, J.B.; Khalifah, P.; Li, Q.; Liaw, B.Y.; Liu, P.; Manthiram, A.; et al. Pathways for practical high-energy long-cycling lithium metal batteries. Nat. Energy 2019, 4, 180–186. [Google Scholar] [CrossRef]
- Nanda, S.; Gupta, A.; Manthiram, A. Anode-Free Full Cells: A Pathway to High-energy density lithium-metal batteries. Adv. Energy Mater. 2021, 11, 2000804. [Google Scholar] [CrossRef]
- Jie, Y.; Ren, X.; Cao, R.; Cai, W.; Jiao, S. Advanced liquid electrolytes for rechargeable Li metal batteries. Adv. Funct. Mater. 2020, 30, 1910777. [Google Scholar] [CrossRef]
- Jagger, B.; Pasta, M. Solid electrolyte interphases in lithium metal batteries. Joule 2023, 7, 2228–2244. [Google Scholar] [CrossRef]
- Qi, M.; Xie, L.; Han, Q.; Zhu, L.; Chen, L.; Cao, X. An overview of the key challenges and strategies for lithium metal anodes. J. Energy Storage 2022, 47, 103641. [Google Scholar] [CrossRef]
- Cheng, X.-B.; Zhang, R.; Zhao, C.-Z.; Zhang, Q. Toward safe lithium metal anode in rechargeable batteries: A review. Chem. Rev. 2017, 117, 10403–10473. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, Z.; Zhao, C.; Verhallen, T.; Tabor, D.P.; Liu, M.; Ooms, F.; Kang, F.; Aspuru-Guzik, A.; Hu, Y.-S.; et al. Interface chemistry of an amide electrolyte for highly reversible lithium metal batteries. Nat. Commun. 2020, 11, 4188. [Google Scholar] [CrossRef]
- Zhai, P.; Liu, L.; Gu, X.; Wang, T.; Gong, Y. Interface engineering for lithium metal anodes in liquid electrolyte. Adv. Energy Mater. 2020, 10, 2001257. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, J.-G.; Xu, W. Advancing lithium metal batteries. Joule 2018, 2, 833–845. [Google Scholar] [CrossRef]
- Liu, S.; Ma, Y.; Zhou, Z.; Lou, S.; Huo, H.; Zuo, P.; Wang, J.; Du, C.; Yin, G.; Gao, Y. Inducing uniform lithium nucleation by integrated lithium-rich li-in anode with lithiophilic 3D framework. Energy Storage Mater. 2020, 33, 423–431. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, W.; Hong, L.; Xu, W.; Yang, H.; Wang, F.; Duan, H.; Tang, M.; Jiang, H. Stress-driven lithium dendrite growth mechanism and dendrite mitigation by electroplating on soft substrates. Nat. Energy 2018, 3, 227–235. [Google Scholar] [CrossRef]
- Wang, T.; Zhai, P.; Legut, D.; Wang, L.; Liu, X.; Li, B.; Dong, C.; Fan, Y.; Gong, Y.; Zhang, Q. S-doped graphene-regional nucleation mechanism for dendrite-free lithium metal anodes. Adv. Energy Mater. 2019, 9, 1804000. [Google Scholar] [CrossRef]
- Zhao, J.; Liao, L.; Shi, F.; Lei, T.; Chen, G.; Pei, A.; Sun, J.; Yan, K.; Zhou, G.; Xie, J.; et al. Surface fluorination of reactive battery anode materials for enhanced stability. J. Am. Chem. Soc. 2017, 139, 11550–11558. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, D.; Yuen, P.Y.; Liu, K.; Xie, J.; Dauskardt, R.H.; Cui, Y. An artificial solid electrolyte interphase with high Li-ion conductivity, mechanical strength, and flexibility for stable lithium metal anodes. Adv. Mater. 2017, 29, 1605531. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Engelhard, M.H.; Mei, D.; Jiao, S.; Polzin, B.J.; Zhang, J.-G.; Xu, W. Electrolyte additive enabled fast charging and stable cycling lithium metal batteries. Nat. Energy 2017, 2, 17012. [Google Scholar] [CrossRef]
- Weber, R.; Genovese, M.; Louli, A.J.; Hames, S.; Martin, C.; Hill, I.G.; Dahn, J.R. Long cycle life and dendrite-free lithium morphology in anode-free lithium pouch cells enabled by a dual-salt liquid electrolyte. Nat. Energy 2019, 4, 683–689. [Google Scholar] [CrossRef]
- Pham, T.D.; Bin Faheem, A.; Lee, K.-K. Design of a LiF-rich solid electrolyte interphase layer through highly concentrated LiFSI–THF electrolyte for stable lithium metal batteries. Small 2021, 17, 2103375. [Google Scholar] [CrossRef]
- Jiao, S.; Ren, X.; Cao, R.; Engelhard, M.H.; Liu, Y.; Hu, D.; Mei, D.; Zheng, J.; Zhao, W.; Li, Q.; et al. Stable cycling of high-voltage lithium metal batteries in ether electrolytes. Nat. Energy 2018, 3, 739–746. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, H.; Kong, X.; Huang, W.; Tsao, Y.; Mackanic, D.G.; Wang, K.; Wang, X.; Huang, W.; Choudhury, S.; et al. Molecular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries. Nat. Energy 2020, 5, 526–533. [Google Scholar] [CrossRef]
- Yu, Z.; Rudnicki, P.E.; Zhang, Z.; Huang, Z.; Celik, H.; Oyakhire, S.T.; Chen, Y.; Kong, X.; Kim, S.C.; Xiao, X.; et al. Rational solvent molecule tuning for high-performance lithium metal battery electrolytes. Nat. Energy 2022, 7, 94–106. [Google Scholar] [CrossRef]
- Zhang, X.-Q.; Cheng, X.-B.; Chen, X.; Yan, C.; Zhang, Q. Fluoroethylene carbonate additives to render uniform Li deposits in lithium metal batteries. Adv. Funct. Mater. 2017, 27, 1605989. [Google Scholar] [CrossRef]
- Hou, T.; Yang, G.; Rajput, N.N.; Self, J.; Park, S.-W.; Nanda, J.; Persson, K.A. The influence of FEC on the solvation structure and reduction reaction of LiPF6/EC electrolytes and its implication for solid electrolyte interphase formation. Nano Energy 2019, 64, 103881. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Wang, H.; Tu, W.; Zhao, Y.; Li, S.; Liu, Q.; Wu, J.; Fu, Y.; Han, C.; et al. Hybrid electrolyte with dual-anion-aggregated solvation sheath for stabilizing high-voltage lithium-metal batteries. Adv. Mater. 2021, 33, 2007945. [Google Scholar] [CrossRef]
- Fu, J.; Ji, X.; Chen, J.; Chen, L.; Fan, X.; Mu, D.; Wang, C. Lithium nitrate regulated sulfone electrolytes for lithium metal batteries. Angew. Chem. Int. Ed. 2020, 59, 22194–22201. [Google Scholar] [CrossRef]
- Yang, F.; Wang, P.; Huang, Q.; Luo, J.; Hu, R.; Huang, Q.; Mao, C.; Yang, L.; Liang, G.; Li, Y.; et al. Saccharin sodium coupling fluorinated solvent enabled stable interface for high--voltage Li-metal batteries. Small 2024, e2311961. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Lee, J.; Roh, Y.; Baek, J.; Shin, D.; Yoon, J.; Ha, H.; Kim, J.; Kim, H. An electron-deficient carbon current collector for anode-free li-metal batteries. Nat. Commum. 2021, 12, 5537–5549. [Google Scholar] [CrossRef]
- Mäntymäki, M.; Ritala, M.; Leskelä, M. Metal fluorides as lithium-ion battery materials: An atomic layer deposition perspective. Coatings 2018, 8, 277–316. [Google Scholar] [CrossRef]
- Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 2004, 104, 4303–4418. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, W.; Guan, Y.; Yu, B.; Zhao, H.; Liu, W. Electrochemical characteristics of LiMxFe1−xPO4 cathode with LiBOB based electrolytes. Electrochim. Acta 2007, 52, 4907–4910. [Google Scholar] [CrossRef]
- Zhang, H.; Oteo, U.; Judez, X.; Eshetu, G.G.; Martinez-Ibañez, M.; Carrasco, J.; Li, C.; Armand, M. Designer anion enabling solid-state lithium-sulfur batteries. Joule 2019, 3, 1689–1702. [Google Scholar] [CrossRef]
- Qiao, L.; Oteo, U.; Martinez-Ibañez, M.; Santiago, A.; Cid, R.; Sanchez-Diez, E.; Lobato, E.; Meabe, L.; Armand, M.; Zhang, H. Stable non-corrosive sulfonimide salt for 4-V-class lithium metal batteries. Nat. Mater. 2022, 21, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, P.; Wu, Y.; Yang, H.; Xie, M.; Huai, L.; Goddard, W.A., III; Cheng, T. The DFT-ReaxFF hybrid reactive dynamics method with application to the reductive decomposition reaction of the TFSI and DOL electrolyte at a lithium-metal anode surface. J. Phys. Chem. Lett. 2021, 12, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. B 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Qi, F.; Yu, P.; Zhou, Q.; Liu, Y.; Sun, Q.; Ma, B.; Ren, X.; Cheng, T. Preferential decomposition of the major anion in a dual-salt electrolyte facilitates the formation of organic-inorganic composite solid electrolyte interphase. J. Chem. Phys. 2023, 158, 104704. [Google Scholar] [CrossRef]
- Chen, Y.; He, Q.; Zhao, Y.; Zhou, W.; Xiao, P.; Gao, P.; Tavajohi, N.; Tu, J.; Li, B.; He, X.; et al. Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery. Nat. Commun. 2023, 14, 8326. [Google Scholar] [CrossRef]
- Cheng, H.; Sun, Q.; Li, L.; Zou, Y.; Wang, Y.; Cai, T.; Zhao, F.; Liu, G.; Ma, Z.; Wahyudi, W.; et al. Emerging era of electrolyte solvation structure and interfacial model in batteries. ACS Energy Lett. 2022, 7, 490–513. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Y.; Sun, Q.; Ma, B.; Yu, P.; Xu, L.; Xie, M.; Yang, H.; Cheng, T. Formation of linear oligomers in solid electrolyte interphase via two-electron reduction of ethylene carbonate. Adv. Theory Simul. 2022, 5, 2100612. [Google Scholar] [CrossRef]
- Zhao, Q.; Stalin, S.; Archer, L.A. Stabilizing metal battery anodes through the design of solid electrolyte interphases. Joule 2021, 5, 1119–1142. [Google Scholar] [CrossRef]
- Gong, C.; Pu, S.D.; Gao, X.; Yang, S.; Liu, J.; Ning, Z.; Rees, G.J.; Capone, I.; Pi, L.; Liu, B.; et al. Revealing the role of fluoride-rich battery electrode interphases by operando transmission electron microscopy. Adv. Energy Mater. 2021, 11, 2003118. [Google Scholar] [CrossRef]
- Stephan, A.M.; Prem Kumar, T.; Thomas, S.; Bongiovanni, R.; Nair, J.R.; Angulakshmi, N.; Pollicino, A. Ca3(PO4)2-incorporated poly(ethylene oxide)-based nanocomposite electrolytes for lithium batteries. Part II. Interfacial properties investigated by XPS and a.c. impedance studies. J. Appl. Polym. Sci. 2012, 124, 3255–3263. [Google Scholar] [CrossRef]
- Schulz, N.; Hausbrand, R.; Dimesso, L.; Jaegermann, W. XPS-Surface Analysis of SEI layers on Li-ion cathodes: Part I. Investigation of initial surface chemistry. J. Electrochem. Soc. 2018, 165, A819. [Google Scholar] [CrossRef]
- Rustomji, C.S.; Yang, Y.; Kim, T.K.; Mac, J.; Kim, Y.J.; Caldwell, E.; Chung, H.; Meng, Y.S. Liquefied gas electrolytes for electrochemical energy storage devices. Science 2017, 356, eaal4263. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; He, F.; Xie, J.; Xue, L. Calibration of binding energy positions with C1s for XPS results. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2020, 35, 711–718. [Google Scholar] [CrossRef]
- Yang, S.; Zhong, J.; Li, S.; Li, B. Revisiting aluminum current collector in lithium-ion batteries: Corrosion and countermeasures. J. Energy Chem. 2024, 89, 610–634. [Google Scholar] [CrossRef]
- Ma, T.; Xu, G.; Li, Y.; Wang, L.; He, X.; Zheng, J.; Liu, J.; Engelhard, M.; Zapol, P.; Curtiss, L.; et al. Revisiting the corrosion of the aluminum current collector in lithium-ion batteries. J. Phys. Chem. Lett. 2017, 8, 1072–1077. [Google Scholar] [CrossRef]
- Rodríguez, S.; Candia, A.; Stankovic, I.; Passeggi, M.; Ruano, G. Study of in-plane and interlayer interactions during aluminum fluoride intercalation in graphite: Implications for the development of rechargeable batteries. ACS Appl. Nano Mater. 2023, 6, 16977–16985. [Google Scholar] [CrossRef]
- Köhler, L.; Kresse, G. Density functional study of CO on Rh(111). Phys. Rev. B 2004, 70, 165405. [Google Scholar] [CrossRef]
- Yang, H.; Negreiros, F.R.; Sun, Q.; Xie, M.; Sementa, L.; Stener, M.; Ye, Y.; Fortunelli, A.; Goddard, W.A., III; Cheng, T. Predictions of chemical shifts for reactive intermediates in CO2 reduction under operando conditions. ACS Appl. Mater. Interfaces 2021, 13, 31554–31560. [Google Scholar] [CrossRef]
- Qian, J.; Baskin, A.; Liu, Z.; Prendergast, D.; Crumlin, E.J. Addressing the sensitivity of signals from solid/liquid ambient pressure XPS (APXPS) measurement. J. Chem. Phys. 2020, 153, 044709. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, S.; Yin, T.; Bian, L.; Liu, Y.; Cheng, T. Optimization of Lithium Metal Anode Performance: Investigating the Interfacial Dynamics and Reductive Mechanism of Asymmetric Sulfonylimide Salts. Batteries 2024, 10, 180. https://doi.org/10.3390/batteries10060180
Feng S, Yin T, Bian L, Liu Y, Cheng T. Optimization of Lithium Metal Anode Performance: Investigating the Interfacial Dynamics and Reductive Mechanism of Asymmetric Sulfonylimide Salts. Batteries. 2024; 10(6):180. https://doi.org/10.3390/batteries10060180
Chicago/Turabian StyleFeng, Shuang, Tianxiu Yin, Letao Bian, Yue Liu, and Tao Cheng. 2024. "Optimization of Lithium Metal Anode Performance: Investigating the Interfacial Dynamics and Reductive Mechanism of Asymmetric Sulfonylimide Salts" Batteries 10, no. 6: 180. https://doi.org/10.3390/batteries10060180
APA StyleFeng, S., Yin, T., Bian, L., Liu, Y., & Cheng, T. (2024). Optimization of Lithium Metal Anode Performance: Investigating the Interfacial Dynamics and Reductive Mechanism of Asymmetric Sulfonylimide Salts. Batteries, 10(6), 180. https://doi.org/10.3390/batteries10060180