Abstract
Dragon fruit comprises a wide variety of species that are rich in nutritional value and have great economic potential; however, numerous studies have focused on their nutritional and commercial quality. In contrast, few studies have addressed their flavor quality, particularly with respect to the regulatory networks responsible for their flavor-related substance contents. To this end, we sequenced the transcriptomes and metabolomes of red-skin/white-fleshed and red-skin/red-fleshed dragon fruit at different timepoints during fruit development. RNA-seq and metabolome data were used to divide the seven developmental stages of the dragon fruit into four categories (young fruit, expansion, maturity, and senescence). In all, 16,827 differentially expressed genes (DEGs), including 958 transcription factors, were identified and grouped into 10 clusters, and the pathways in each cluster were annotated. Additionally, 318 differentially accumulated metabolites (DAMs) were identified, including 88 common metabolites. The main flavor-related substances and the key genes regulating them were determined via joint analysis via RNA-seq and metabolomics. Furthermore, 10 volatile active components related to green flavors and aromas were screened according to the relative odor activity value (ROAV), and 15 candidate genes related to key flavor compounds were screened via WGCNA, 3 of which encoded transcription factors. In conclusion, our results provide a theoretical basis for an in-depth understanding of the volatile flavor compounds in dragon fruit and provide new genetic resources for the subsequent study of fruit flavor compounds.
1. Introduction
Dragon fruit (Selenicereus spp.) is a perennial climbing species of the genus Selenicereus within Cactaceae [1]. Native to tropical and subtropical regions of Mexico and Central and South America, dragon fruit has become widely popular only in recent years, being favored by growers and consumers because of its ease of planting, unique appearance, rich flavor, and high nutrient contents, particularly in terms of minerals (calcium, potassium, phosphorus, sodium, iron, and zinc), carbohydrates, fibers, proteins, vitamins (B1, B2, B3, C, niacin, pyridine, and cobalamin), and antioxidants (plant albumin, flavonoids, phenolic compounds, and betaine) [2]. The high fiber content in the fruit includes insoluble and soluble fibers that are mainly composed of pectin, which favors digestion. In turn, studies on the metabolic components of dragon fruit peel and flesh have shown that the main substances associated with fruit color include betalains, flavonoids, and very small amounts of anthocyanin [3]. Among these, red-skin/red flesh dragon fruit gradually accumulate large amounts of betalain during the ripening period. Betalain xanthine also accumulates in the fruit and peel during the ripening period, but not as significantly as betalain does [4]. As living standards continue to improve, consumer demand for dragon fruit is on the rise, not only for fresh fruits with high nutritional value but also for those with a strong aroma. Indeed, aroma is a key factor determining the characteristics of fruit flavor, which in turn affects consumer acceptance; in particular, volatile flavor-related substances are important components of the aroma [5]. Furthermore, the content and types of volatile flavor-related substances vary greatly among different fruits, such that the resulting types of aromas produced by fruits also differ [6]. In general, the higher the content of volatile flavor-related substances, and the greater the number of types of these substances is, the stronger the overall aroma of the fruit [7].
At present, volatile flavor-related substances in fruits are analyzed via gas chromatography–mass spectrometry (GC–MS), which combines the high separation ability of gas chromatography with the high identification ability of mass spectrometry to achieve a highly reliable and reproducible quantitative analysis of flavor substances in complex samples [8]. Furthermore, transcriptomics is often combined with metabolomics for a more comprehensive analysis of the regulatory networks and molecular mechanisms underlying the role of important metabolites [9,10]. For example, a combination of transcriptomics and metabolomics analysis revealed that red light had a greater effect on some catechins and improved the quality of tea [11]. Similarly, combined transcriptomic and metabolomic analyses led to the identification of the catalytically active linalool synthase CsLIN, thus providing new insights into the potential causes of aroma formation in oolong tea varieties [12]. In turn, metabolomic and transcriptomic techniques used to analyze the differences between diploid and tetraploid ‘Ziyang Xiangcheng’ plants revealed that stress-related metabolites such as sucrose, proline, and GABA were significantly upregulated in tetraploid materials [13]. Additionally, metabolomic and transcriptomic analyses of volatile flavor-related substances in tomato fruits revealed that the volatile contents affecting fruit flavor significantly increased from fruit breaking to ripening [14]. Furthermore, a total of 215 aromatic compounds were identified in grape pulp and peel, with significant differences in the number of terpene compounds between the pulp and peel [15]. Volatile and transcriptome analyses have revealed that limonene terpene synthase (CaTPS10-like) is a major contributor to the characteristic aroma (limonene) of Arabica coffee [16].
Dragon fruit trees cultivated on a large scale include three main species: red skin/red flesh (Selenicereus monacanthus), red skin/white flesh (Selenicereus undatus), and yellow skin/white flesh (Selenicereus megalanthus) [17]. Red skin/red flesh dragon fruit (RF) and red skin/white flesh dragon fruit (WF) are the two main cultivated species. In actual production, WF has a beautiful shape and high yield, whereas RF has bright colors and is more popular among consumers [18]. The cultivation of dragon fruit, a high-quality economic crop, has increased in recent years. Furthermore, owing to the wide variety of dragon fruit, its rich nutritional value, and enormous economic potential, the need for germplasm optimization is paramount. At present, there are many reports on the quality of dragon fruit, most of which focus on nutritional and commodity qualities [19,20,21]. In contrast, relatively few studies on flavor quality have focused on the composition, content, and regulatory networks of volatile flavor-related substances. Additionally, there are few reports on the mechanisms of the biosynthesis of volatile flavor-related substances, an issue that warrants further research. Screening key metabolites and genes is considered to be the key to solving this problem. Therefore, in this study, we conducted headspace solid-phase microextraction gas chromatography–mass spectrometry (HS-SPME-GC-MS) and RNA-seq on RF and WF at seven developmental stages. Using bioinformatic methods, the regular characteristics of the fruit development process were analyzed from the perspective of transcription and metabolomics, revealing the metabolite regulatory network genes responsible for the biosynthesis of volatile flavor-related substances during the development of dragon fruit and providing a sound theoretical basis for the creation of novel dragon fruit germplasms. The study of volatile flavor-related substances during the development of dragon fruit will not only guide the breeding of new dragon fruit varieties from a molecular perspective but also enable a deeper exploration of the functions of volatile flavor-related substances, thereby providing a reference for processing and utilization.
2. Materials and Methods
2.1. Plant Materials
Two fruit varieties were used in this study, namely, Yulong No. 1 (red skin/white flesh, WF) and Jindu No. 1 (red skin/red flesh, RF). The fruit samples were taken from 4-year-old fruit trees in the same field as the pitaya base in Changtang, Nanning, Guangxi, China. The field management (including irrigation, fertilization, and pesticide application) process was consistent. There were many rainy days from August to September 2023, with an average daily maximum temperature of 31.7 °C and a minimum temperature of 24.7 °C. On 20 August 2023, 80 flowers were marked at the flowering period of the two varieties (blooming at the same time), and fruit samples of the two varieties were collected at 7 different developmental stages (7, 14, 21, 28, 33, 36, and 39 days after flowering (DAF)) from 27 August to 28 September 2023 (Figure 1). Each sampling was performed with 3 independent biological replicates, and each replicate was taken from 3 fruits of 3 different plants with basically the same weight and size and no obvious diseases or insect pests. After sampling, the samples were placed in an ice box and taken back to the laboratory for processing. The fruit was peeled first, and then the pulp was divided into equal parts according to the size of the fruit. An appropriate amount of pulp was taken for grinding. The fruits were ground into powder via liquid nitrogen and a tissue grinder (Shanghai Jingxin Industrial Development Co., Ltd., Shanghai, China). The samples were then stored at −80 °C for flavoromic and transcriptomic analysis.

Figure 1.
Fruit phenotypes of Yulong 1 (red skin/white flesh, WF) and Jindu 1 (red skin/red flesh, RF).
2.2. RNA-Seq Sequencing and Analysis
RNA was extracted via a polysaccharide and polyphenol plant total RNA extraction kit (each sample was guaranteed to be approximately 100 mg), and the extraction process was carried out according to the instructions. After RNA extraction, a Nanodrop device was used to determine the RNA purity (OD260/280) and concentration, and to determine whether the nucleic acid absorption peak was normal. The Agilent 2100 accurately detected the integrity of the RNA, and the detection indicators included a RIN value greater than 7, a 28S/18S ratio, whether the baseline of the spectrum was increased, and the 5S peak. After the sample was tested, it was transported to Xinjiang Aidesen Biological Co., Ltd. (Urumqi, China) for RNA-seq via dry ice. After the raw sequences were obtained, fastp software (version 0.23.4) was used to remove the adapter sequence, filter out low-quality (Q ≤ 20) and N sequences with a ratio greater than 10%, and obtain clean reads for subsequent analysis [22]. HISAT2 was used to map clean reads to the reference genome of ‘GHB’ pitaya (http://pitayagenomic.com/download.php, accessed on 3 March 2024), and fragments per kilobase of transcription per million mapped reads (FPKM) were used to characterize expression levels [23]. The read counts of the genes were used as input data for differential expression analysis, and the p value and fold-change values were calculated via edgeR software (version 4.6.2) [24]. Setting the FDR standard to less than 0.01 ensured that fewer than 1% of the DEGs screened were caused by random differences. FDR < 0.01 and |log2fold change| > 1 were used as the criteria for screening DEGs. Functional annotation was performed via the KEGG database (http://www.genome.jp/kegg/, accessed on 6 March 2024). The protein sequences of the DEGs were submitted to the PlantTFDB (https://planttfdb.gao-lab.org/prediction.php, accessed on 6 March 2024) for analysis and the prediction of differentially expressed transcription factors (TFs).
2.3. Flavourome Sequencing and Analysis
Five grams of each pitaya sample was weighed, frozen, ground with liquid nitrogen, added to a 10 mL headspace bottle with 1 µL of 0.025 mL/L o-dichlorobenzene solution as the internal standard, and mixed thoroughly. The headspace bottle was placed in a 55 °C water bath, where the extraction was balanced with magnetic stirring for 15 min; the aged extraction head was inserted into the headspace bottle, the mixture was heated at 55 °C for 30 min, the extraction fiber was quickly retracted, the extraction head was inserted into the gas chromatography injection port, and the mixture was analyzed at 250 °C for 3 min.
The detection platform was an Agilent 7890 gas chromatograph (Agilent Technologies Inc., Santa Clara, CA, USA) in tandem with a LECO PEGASUS HT high-resolution mass spectrometer. Sample detection was performed according to the following parameters. The derivatized substances were separated under a constant flow (1 mL/min) of helium, and each sample was separately added to a 20 mL headspace bottle and sealed with a cap. The injection port temperature was set at 260 °C, and the heating program started at 40 °C for 5 min, increased to 220 °C at 5 °C/min, then to 250 °C at 20 °C/min, and was maintained at that point for 2.5 min. ChromaTOF software (version 4.3x) from LECO Corporation and the LECO-Fiehn Rtx5 database were used for raw peak extraction, data baseline filtering, the calibration of the baseline, peak alignment, deconvolution analysis, peak identification, and the integration of peak area1. Both mass spectrum matching and retention index matching were considered in metabolite identification. Peaks detected in <50% of the QC samples or with an RSD > 30% were removed from the QC samples [25]. On the basis of the metabolite content data matrix, correlations and principal component analysis (PCA) were performed for each sample via the R language. The R language package ropls was used to perform OPLS-DA modeling, and 200 permutation tests were performed to verify the reliability of the model. The VIP value of the model was calculated via multiple cross-validations. The method of combining the difference multiple, the p value, and the VIP value of the OPLS-DA model was adopted in order to screen the differentially accumulated metabolites (DAMs). The screening criteria were a fold change ≥2 or ≤0.5, p < 0.05, and VIP > 1.
2.4. RNA-Seq–Metabolome Joint Analysis
The correlation between the DEGs and metabolites was calculated by fitting the k-means clustering results for the DEGs. The clustering effect was evaluated on the basis of the sum of squared errors (SSE), which calculates the sum of the squared distances from each data point to the center of the cluster to which it belongs. As the K value (the number of clusters) increases, the SSE gradually decreases because more clusters can better fit the data. The correlation between the DEGs and differentially abundant metabolites in each cluster was represented by a network diagram. The correlation results between the DEGs and DAMs with an absolute value of the Pearson correlation coefficient greater than 0.80 and a p value less than 0.05 in each cluster were plotted. The permutation test was used to correct the p value. The scores were normalized at the row level and visualized via heatmaps.
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
The R language WGCNA package was used to analyze the gene expression profiles of the identified DEGs via the dynamic branch-cutting method [26]. In this study, β = 7 was selected as the weighting coefficient (Figure S1). The network was constructed via the blockwise modules of the automatic network construction function. Multiple valid modules were obtained, and the number of genes in each module was different. The modules with a similarity coefficient of 0.75 were merged with minModuleSize = 30 and Merge Cut Height = 0.25 as the standard. The correlation coefficients among the module characteristic vector ME (Module Eigengene), hormone content, and different sampling timepoints were calculated. Finally, Cytoscape (version 3.10.1) was used to visualize the co-expression network [27].
2.6. qRT–PCR
On the basis of the cDNA information of the genes, Primer 5.0 software was used to design primers (Table S1) at specific regions of the 5′ or 3′ ends of the gene sequence. Total RNA was extracted via the RNAprep Pure Polysaccharide and Polyphenol Plant Total RNA Extraction Kit (TIANGEN, Beijing, China) in February 2024, and each sample was analyzed three times. The concentration of each RNA sample was determined via a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The RNA was reverse-transcribed via the M-MLV RTase cDNA Synthesis Kit (TaKaRa, Kyoto, Japan) to obtain cDNA. Real-time PCR amplification was performed on the Bio-Rad CFX96 Real-time System. The iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) kit was used, and the method provided was followed, with a total system volume of 20 μL. The reaction procedure was 95 °C predenaturation for 30 s, 95 °C denaturation for 5 s, 57 °C annealing for 5 s, and 72 °C extension for 34 s, for 40 cycles. The results were analyzed via relative quantitative analysis via the 2−ΔΔCt method, and the internal reference gene used was Actin [28].
3. Results
3.1. Global RNA-Seq Analysis During Dragon Fruit Development
After filtering the RNA-seq raw data from the 42 samples from the two dragon varieties and the seven fruit development stages, 273.11 Gb of clean data were obtained. The clean data for each sample exceeded 4.84 Gb, the proportion of Q30 bases exceeded 85.31%, and the alignment rate with the reference genome exceeded 77.84% (Table S1). Subsequently, correlation analysis was performed on the RNA-Seq data from the 42 samples. Firstly, the correlation coefficient among the three biological replicates of the same sample exceeded 0.98, indicating that the transcriptome data were reliable and reproducible (Figure S1). Secondly, to analyze the transcriptome dynamics of dragon fruit growth, we performed hierarchical clustering and PCA for samples from the seven phenological developmental stages of the two fruit types under study. Our RNA-seq data divided the seven fruit developmental stages into only four (young, expanding, maturing, and senescent fruit) (Figure 2a,b). Samples collected at the earliest sampling timepoint (7 DAF) constituted the first group, representing the young fruit stage. In turn, samples collected between 14 and 21 DAF constituted the second group and represented the fruit expansion stage. Meanwhile, samples collected between 28 and 36 DAF constituted the third group, corresponding to the fruit ripening stage, and lastly, samples collected at 39 DAF constituted the fourth group, corresponding to the fruit senescence stage. Fruit growth at each of the four stages is highly similar for the two varieties, indicating that their transcriptional regulation patterns must also be highly similar. Samples clustered more closely between WF at 28 DAF and RF at 33 DAF, and between RF at 28 DAF and WF at 36 DAF. These findings suggest that the difference in fruit color between WF and RF may have been caused by developmental differences after 28 DAF.
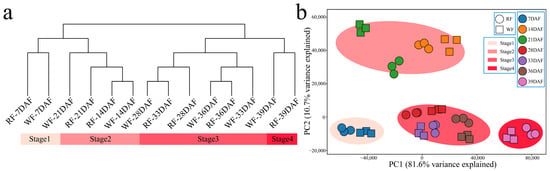
Figure 2.
Clustering and PCA of RNA-seq data. (a) Cluster dendrogram of dragon fruit RNA-seq data; (b) PCA of dragon fruit RNA-seq data.
3.2. Differential Gene Expression Analysis
To investigate the transcriptional differences between the two varieties at different stages of fruit development, 16,827 DEGs were identified in the same variety at different stages of fruit development and at the same developmental stage in the two different varieties, including 4929 common DEGs (Figure 3a). In all, 10,927 DEGs were identified in the RF material, including 864 unique DEGs. In turn, a total of 13,065 DEGs were identified in WF, including 2742 unique DEGs. Further, a total of 9201 DEGs were identified between the RF and WF, including 1784 unique DEGs. Additionally, GO enrichment analysis of all 16,827 DEGs revealed that they were significantly annotated in biological processes including carbohydrate biosynthesis, auxin-activated signaling, fatty acid biosynthesis, starch biosynthesis, polysaccharide metabolism, fruit development, galactose catabolism, light responses, auxin transport, and fruit morphogenesis (Figure 3b). Meanwhile, KEGG annotation included lipid metabolism, glycolysis/gluconeogenesis, carbohydrate metabolism, starch and sucrose metabolism, galactose metabolism, carbon fixation in photosynthetic organisms, the pentose phosphate pathway, fatty acid biosynthesis, ABC transporters, fructose and mannose metabolism, plant hormone-signal transduction, and the Cytochrome P450 pathway (Figure 3c).
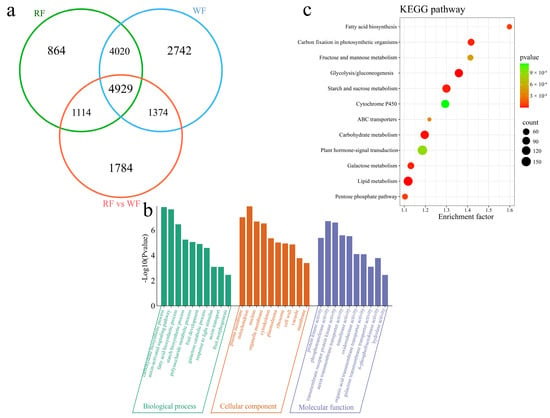
Figure 3.
Differential expression analysis. (a) Venn diagram of DEGs at different developmental stages of same variety and at same developmental stage of different varieties; (b) DEG GO enrichment analysis; (c) DEG KEGG enrichment analysis.
The k-means clustering algorithm was used to cluster the detected 16,827 DEGs into 10 clusters, and the pathways in each cluster were annotated (Figure 4a,b). The DEGs in cluster 1 had the highest expression level at 7 DAF, which gradually decreased with fruit development, and were significantly annotated in carbohydrate metabolism, lipid metabolism, and the biosynthesis of unsaturated fatty acids. In turn, the DEGs in cluster 2 showed an unchanged expression level during fruit development, but their expression level in RF was higher than that in WF, and they were significantly annotated for the biosynthesis of unsaturated fatty acids and galactose metabolism. Meanwhile, the DEGs in cluster 3 showed the highest expression level at 14 DAF, which gradually decreased with fruit development, and were significantly annotated in plant hormone-signal transduction and betalain biosynthesis. As for the DEGs in cluster 4, these showed the highest expression level at 7 DAF, the lowest expression level at 21 DAF, and were significantly annotated in ribosome biogenesis and mitochondrial biogenesis. Additionally, the expression of the DEGs in cluster 5 gradually increased with fruit development, reaching a maximum value at 30 DAF, and they were significantly annotated in carbon fixation in photosynthetic organisms and glycolysis/gluconeogenesis. Further, the DEGs in cluster 6 showed the highest expression level only at 21 DAF, their expression level in WF was higher than that in RF, and they were significantly annotated in galactose metabolism and phenylpropanoid biosynthesis. As for the DEGs in cluster 7, they were highly expressed from 21 to 39 DAF and significantly annotated in the anthocyanin biosynthesis and Cytochrome P450 pathways. With respect to cluster 8, these DEGs showed the highest expression level at 21 DAF, their expression level in RF was higher than that in WF, and they were significantly annotated in plant hormone-signal transduction, phenylpropanoid biosynthesis, and the Cytochrome P450 pathway. In turn, the DEGs in cluster 9 showed their highest expression levels at 7 and 14 DAF, their expression level remained basically unchanged through fruit development, and they were significantly annotated for plant hormone-signal transduction, starch and sucrose metabolism, and vitamin B6 metabolism. Finally, cluster 10 comprised DEGs specifically expressed at high levels in the interval between 33 and 39 DAF and significantly annotated in the folate biosynthesis and glycerophospholipid metabolism pathways.
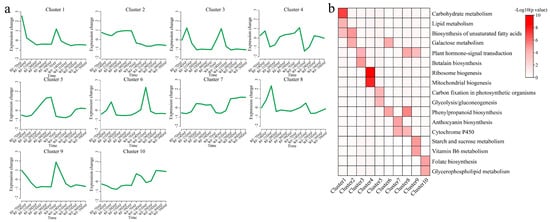
Figure 4.
k-means clustering and KEGG pathway annotation of DEGs. (a) k-means clustering line graph of DEGs; (b) KEGG pathway annotation of each cluster.
3.3. Transcription Factor Analysis
There were 958 differentially expressed TFs among the 16,827 DEGs identified at the same developmental stage between the two plant materials and at different developmental stages in the same material. Further, we identified 958 differentially expressed TFs, including, MYB, ERF, C2H2, bZIP, WRKY, NAC, HD-ZIP, GRAS, B3, C3H, and G2-like (Figure 5a). Using the k-means clustering method, four statistically significant clusters were identified for the 958 TFs (Figure 5b). The TFs in cluster 1 showed the highest expression level at 28–39 DAF, and their expression level in WF was higher than that in RF. The top four TFs with the largest fold differences were HU06G01182 (MYB), HU03G01158 (bHLH), HU09G01092 (bHLH), and HU08G02214 (ERF). Meanwhile, the TFs in cluster 2 showed an increased expression level in RF as fruit development proceeded, but remained unchanged in WF. The four TFs with the largest fold differences in this case were HU10G01811 (MYB), HU02G03012 (WRKY), HU01G02271 (TCP), and HU09G01086 (bHLH). In turn, the TFs in cluster 3 had the highest expression level at 7 DAF, and their expression level gradually decreased in RF and WF with fruit development. The top four TFs with the largest fold differences were HU01G01125 (C3H), HU07G00998 (ERF), HU03G01421 (MYB), and HU08G00532 (ERF). Lastly, cluster 4 showed the highest expression levels at 14 and 21 DAF, and the top four TFs with the largest fold differences were HU03G00583 (ERF), HU11G01126 (MYB), HU06G01450 (B3), and HU06G02562 (WRKY).
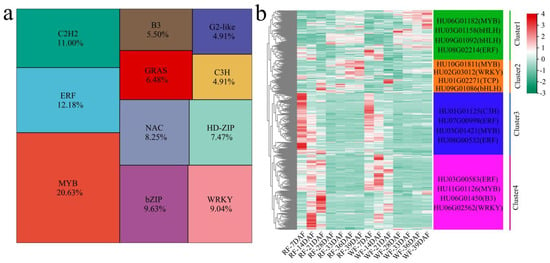
Figure 5.
Identification and clustering of differentially expressed TFs. (a) Percentage of differentially expressed TFs; (b) clustering heat map of differentially expressed TFs and TOP TFs.
3.4. Metabolome Overall Analysis
To explore the changing characteristics of volatile flavor-related substances during the development of dragon fruit, metabolome sequencing was performed on 42 samples from the seven fruit developmental stages of the two varieties, and a total of 837 volatile flavor-related metabolites were detected. Through the correlation analysis of the samples, it was found that the correlation among the replicates was very high (r > 0.96), indicating that the error among the replicates was negligible, the sample quality was good, and that the following step of the analysis could be performed (Figure 6a). Through PCA, we found that the samples of the seven fruit developmental stages of the two varieties were clustered into four categories (7, 14–21, 28–36, and 39 DAF), which was consistent with the results of RNA-seq (Figure 6b).
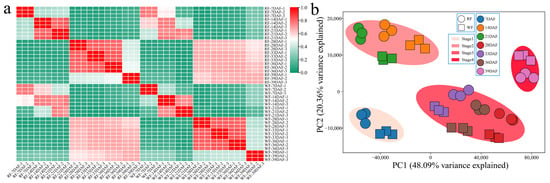
Figure 6.
Metabolome correlation and PCA. (a) Metabolome correlation heat map; (b) PCA of dragon fruit metabolome.
3.5. Differential Metabolite Analysis
To investigate the transcriptional differences between the two varieties at different stages of fruit development, 318 differentially accumulated metabolites (DAMs) were identified at different stages of fruit development in the same variety and at the same developmental stage in different varieties, including 88 common ones (Figure 7a). In all, 212 DAMs were identified in the RF material, including 32 unique ones. Similarly, a total of 202 DAMs were identified in the WF material, including 21 unique DAMs. Furthermore, 240 DAMs were identified as common to RF and WF, including 17 unique ones. The 318 DAMs were grouped into nine clusters using the k-means clustering algorithm, and the number of metabolites in each cluster was counted (Figure 7b). Cluster 1 showed the highest DAM content at 36 DAF in RF and at 39 DAF in WF, and contained 48 volatile flavor-related metabolites; meanwhile, cluster 2 showed the highest DAM content at 14 DAF, including 25 volatile flavor-related metabolites, and then gradually decreased with fruit development; in turn, cluster 3 showed the highest DAM content at 21 DAF in WF, including 32 volatile flavor-related metabolites, and remained unchanged in RF. Meanwhile, cluster 4 showed the highest DAM content at 7 DAF in WF, including 22 volatile flavor-related metabolites, and remained unchanged in RF. As for cluster 5, this cluster showed the highest content at 7 DAF, including 30 volatile flavor-related metabolites, which then gradually decreased with fruit development; cluster 6 showed a gradually increasing DAM content over 28–39 DAF in WF, including 62 volatile flavor-related metabolites, and remained basically unchanged in RF. As for cluster 7, this cluster showed the highest DAM content at 39 DAF in RF, including 36 volatile flavor-related metabolites, and remained unchanged in WF. With regard to cluster 8, this cluster showed the highest DAM content at 14 DAF, including 37 volatile flavor-related metabolites. Lastly, cluster 9 showed the highest DAM content at 28 DAF in WF, including 26 volatile flavor-related metabolites, and remained unchanged in RF. Furthermore, we screened the 18 metabolites with the largest fold differences and found that nine metabolites decreased in content, including the following: acetaldehyde, isoamyl alcohol, d-glucuronic acid, 3,3-dimethyl-2-butanol, 2,3-buranedione, 2-phenyl-2-butenal, 2,3-octanedione, 2-undecenal, and 2-ethylfuran. In contrast, nine metabolites increased in content, including the following: 3,5-octadien-2-one, 2-methylfuran, 2-octen-1-ol, (2z)-tridecan-2-ol, 1-tridecanol, didodecyldimethylammonium, 2-pentanone, methyl mercaptan, and l-2,4-diaminobutyric acid (Figure 7c).
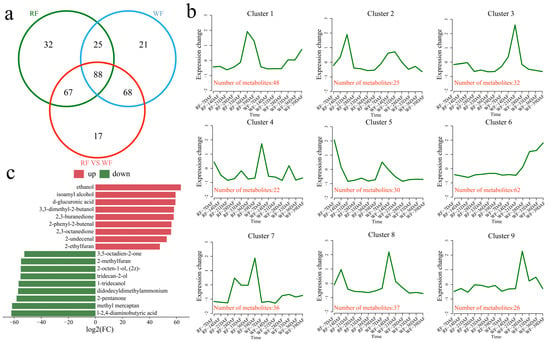
Figure 7.
Identification and clustering line graph of DAMs. (a) Venn diagram of DAMs at different developmental stages of same variety and at same developmental stage of two different varieties; (b) DAM clustering line graph; (c) TOP metabolite difference fold bar graph.
3.6. Transcriptome–Metabolome Joint Analysis
To explore the relationship between the DEGs and DAMs, the correlation between the 18 metabolites with the largest fold differences and 10 clusters was calculated (Figure 8a). Isoamyl alcohol was highly and positively correlated with cluster 5 (r = 0.83) and 2-pentanone was highly and negatively correlated with cluster 5 (r = −0.87); acetaldehyde was highly and positively correlated with cluster 6 (r = 0.78) and 2-methylfuran was highly and negatively correlated with cluster 6 (r = −0.75); 3,5-octadien-2-one was highly and positively correlated with cluster 3 (r = 0.83) and 3,3-dimethyl-2-butanol was highly and negatively correlated with cluster 3 (r = −0.78); 2,3-octanedione was highly and positively correlated with cluster 9 (r = 0.77) and didodecyldimethylammonium was highly and negatively correlated with cluster 3 (r = −0.82); and methyl mercaptan was highly and negatively correlated with cluster7 (r = −0.88) and d-glucuronic acid was highly and positively correlated with cluster3 (r = 0.72). The network between the DEGs and DAMs in each cluster was constructed by the screening criteria of PCC ≥ 0.80 and p < 0.05 (Figure 8b). Isoamyl alcohol was significantly and positively correlated with eight genes and 2-pentanone was significantly and negatively correlated with ten genes; 3,3-dimethyl-2-butanol was significantly and positively correlated with nine genes and 3,5-octadien-2-one was significantly and negatively correlated with six genes; d-Glucuronic acid was significantly and positively correlated with seven genes, whereas methyl mercaptan was significantly and negatively correlated with seven genes; 2,3-octanedione was significantly and positively correlated with twelve genes, whereas didodecyldimethylammonium was significantly and negatively correlated with eight genes; and acetaldehyde was significantly and positively correlated with eight genes, and 2-methylfuran was significantly and negatively correlated with six genes.
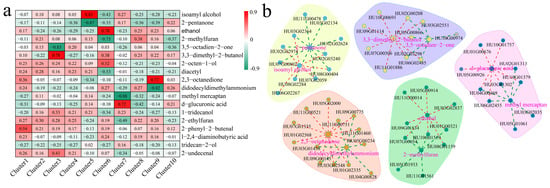
Figure 8.
Correlation analysis between DAMs and DEGs. (a) Heat map of correlation analysis between metabolites with largest fold difference and DEG clusters; (b) network diagram of correlation between metabolites and genes.
3.7. WGCNA
Relative odor activity value (ROAV) is often used to assess the contribution of a compound to an overall aroma. The greater the ROAV of a component, the greater the contribution of that component to the aroma system. ROAV screening showed that the main volatile active components of green flavor mainly included hexaldehyde, trans-2-hexenal, trans-2-nonenal, trans-2,cis-6-nonadienal, and 2-isobutyl-3-methoxypyrazine, and the main active components of flavor mainly included 3-methylbutanal. phenylacetaldehyde, 2-methylbutyraldehyde, 2,4-nonadienal, and ethyl oenanthate. Based on the 16,827 detected DEGs, 18 co-expression modules were obtained using WGCNA (Figure 9a). The correlations between the modules and different sampling timepoints during dragon fruit development were calculated (Figure 9b). The magenta module was significantly highly correlated with 3-methylbutanal and phenylacetaldehyde, the yellow module was significantly highly correlated with 3-methylbutanal and 2-methylbutyraldehyde, and the turquoise module was significantly highly correlated with trans-2 and cis-6-nonadienal. Each module identified 5 genes with the highest connectivity as hub genes, resulting in 15 hub genes (Figure 9c). The candidate genes were functionally annotated in the COG, GO, KEGG, KOG, NR, Pfam, and Swiss-Prot databases (Table 1). HU01G01237 encodes the MYB transcription factor, which is mainly involved in pollen development. HU08G02295 encodes GRAS transcription factors, which are mainly involved in aromatic compound biosynthetic processes. HU11G01794 encodes the BES1 transcription factor, which is mainly involved in the polysaccharide catabolic process. HU09G00745 encodes betaine aldehyde dehydrogenase (BADH), which is mainly involved in the gamma-aminobutyric acid catabolic process. HU09G01998 encodes metacaspase-9 (MCA9), which is mainly involved in the nitrogen compound metabolic process. HU08G01269 encodes 3-Hydroxy-3-Methylglutaryl-CoA Lyase (HMGCL), which is mainly involved in the organic substance metabolic process. HU09G00220 encodes NIMA-related kinase 5 (NEK5) and is mainly involved in tissue development. HU11G00938 encodes galacturonosyltransferase-like (GATL) and is mainly involved in pollen tube growth. HU04G01313 encodes pyrophosphatase (PPA), which is mainly involved in the pphosphate-containing compound metabolic process. HU09G00484 encodes transparent testa10 (TT10), which is mainly involved in aromatic compound metabolic process. HU05G01229 encodes xyloglucan endotransglucosylase/hydrolase (XTH), which is mainly involved in the glucan metabolic process. HU02G00110 encodes ketoacyl reductase (KCR), which is mainly involved in the very-long-chain fatty acid metabolic process. HU09G01988 encodes apical spikelet degeneration 1 (ASD1), which is mainly involved in the L-arabinose metabolic process. HU03G01746 encodes GDSL-type esterase/lipase protein (GELP), which is mainly involved in the lipid metabolic process. HU03G01569 encodes beta-hexosaminidase (HOX), which is primarily involved in the carbohydrate metabolic process.
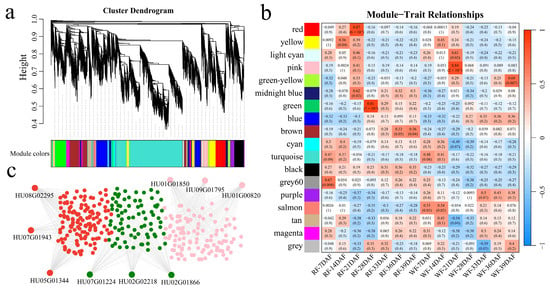
Figure 9.
WGCNA. (a) WGCNA clustering dendrogram. Different colors represent different modules. (b) Heat map of correlation and significance analysis between modules and key flavor compounds of dragon fruit; (c) gene network diagram of magenta, yellow, and turquoise modules.

Table 1.
Functional annotations of candidate genes.
Fifteen genes were selected for three independent repetitions of qRT–PCR to further verify the reliability of the transcriptome data. The transcriptome data of these 15 genes were significantly correlated with the fold change in the qRT–PCR data (R = 0.90, p < 0.01), indicating that the transcriptome sequencing data were reliable (Figure 10).
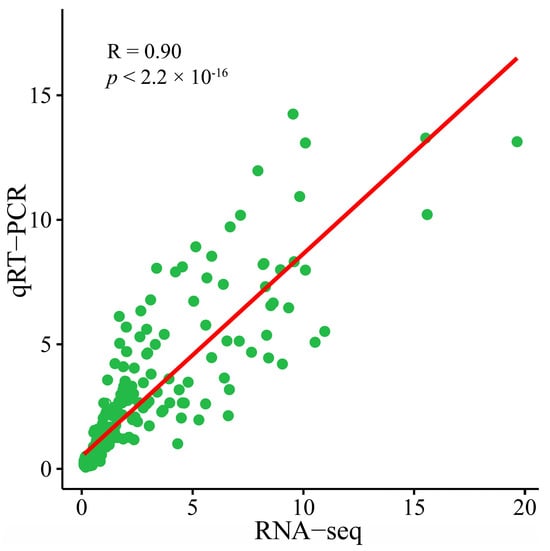
Figure 10.
Scatter plot of correlation and significance of RNA-seq and qRT–PCR results.
4. Discussion
Dragon fruit is delicious and rich in nutrients such as soluble sugar, organic acid, betaine, and mineral elements required by the human body, and is widely loved by consumers. Furthermore, it has a high value as a research model to study the regulatory networks that control volatile flavor-related substance biosynthesis. From fruit setting to fruit maturity, the fruit appearance and contents reflect the external environment and crop management practices experienced by the plant [29]. To date, research on the nutritional components and functional substances present in dragon fruit has focused mainly on organic acid, flavonoid, and betaine contents, whereas studies on flavor quality, including the composition, content, and regulatory network of volatile flavor-related substances, are scarce, and there are even fewer reports on the metabolic mechanisms responsible for the biosynthesis of volatile flavor-related substances that need to be monitored [30,31]. In this study, RNA-seq and HS-SPME-GC-MS were performed at seven timepoints during fruit development from Yulong No. 1 (red skin/white flesh, WF) and Jindu No. 1 (red skin/red flesh, RF) trees. Cluster analysis and PCA of the RNA-seq data revealed that these time-dependent processes could be clearly divided into four different groups, corresponding to young, expanding, mature, and senescent fruits. We found that the difference in fruit color between WF and RF was caused mainly by developmental differences occurring after 28 DAF. The period from 28 to 33 DAF corresponds to the mature stage (color change period) of fruit development, at which point the flesh begins to gradually turn light red and red as the maturation process progresses. The difference in dragon fruit flesh color is caused by the difference in the accumulation of betalains and anthocyanins in cells, which accumulate in the flesh, thereby gradually turning red [32]. We also found that genes specifically expressed at this stage were significantly annotated in the anthocyanin biosynthesis and Cytochrome P450 pathways. The results of the comprehensive analysis reported herein show that the period beginning at 28 DAF and continuing thereafter is the most important period for color change in dragon fruit, as it determines the final fruit color. These findings provide an important theoretical basis for analyzing the mechanism of dragon fruit color evolution during the fruit maturation period.
Fruit aroma is an important indicator of postharvest fruit quality and mainly includes a variety of volatile substances, such as aldehydes, alcohols, esters, terpenes, and some sulfur-containing compounds [33]. Analysis of the characteristic aroma components of fruits can help us understand their fragrance and provide a basis for the synthesis of specific flavors and fragrances [6]. Furthermore, more than 100 kinds of aroma-related substances have been identified in many fruits, such as apples, strawberries, mangoes, tomatoes, and bananas [34,35,36,37]. To date, 34 volatile aromatic substances have been found in dragon fruits, including aldehydes, hydrocarbons, alcohols, ketones, esters, and furan compounds, of which, aldehydes are the most abundant, with fragrant hexanal accounting for 92% of the total amount of this type of compound [38]. Our data also revealed that acetaldehyde is one of the substances with the greatest variation in content in dragon fruit. Although there are few reports on the regulatory mechanism of acetaldehyde biosynthesis in plants, in this study, the correlation analysis of the DEGs and DAMs revealed that HU11G00014, HU01G00321, HU03G02837, HU07G00054, HU08G01159, HU09G01834, HU10G01559, and HU11G00014 were significantly and highly correlated with changes in acetaldehyde content. These genes can be used as the focus of future research on acetaldehyde synthesis in dragon fruit.
The aroma sensation produced by fruits is not the result of one or two compounds acting alone, but rather the result of the combined action of multiple aroma components [39]. Studies have shown that the content of aldehydes in commercially ripe pears is the highest, followed by that of alcohols and esters [40]. For example, the volatile flavor-related substances present in strawberries include mainly esters, ketones, and alcohols, and more than 360 volatile flavor-related substances have been identified [41]. Moreover, among the aroma substances found in peach fruit, acetic acid, linalool, and sucrose play positive roles, whereas benzaldehyde and histidine play negative roles in fruit flavor [42]. However, dynamic changes in volatile aroma-related substances during dragon fruit development are relatively rare. Our findings that hexaldehyde, trans-2-hexenal, trans-2-nonenal, trans-2,cis-6-nonadienal, 2-isobutyl-3-methoxypyrazine, 3-methylbutanal, phenylacetaldehyde, 2-methylbutyraldehyde, 2,4-nonadienal, and ethyl oenanthate are the main volatile flavor compounds in dragon fruit provide 15 candidate genes that may regulate these important flavor compounds, which can provide a reference for later studies of the mechanism of dragon fruit flavor compounds and further explore the metabolic process of volatile aroma compounds in dragon fruit from the perspective of molecular biology.
Pollen development is a crucial part of the plant growth process and directly affects the reproductive success and yield of plants [43]. In addition to influencing the fruit setting rate, fruit size, color, and other indicators of multiple fruits, pollen also affects the aroma components of fruits [44]. The volatile components in ‘Fuji’ bagged apples are affected by the pollen intuitionistic effect, the volatile compounds and characteristic aroma components in the pollinated fruits of ‘Golden Crown’ apples increase, the content of alcohol in ‘Fuji’ apples pollinated by ‘New Red Star’ and ‘Ruby’ apples significantly increases, and the content of aldehydes significantly increases in the pollinated fruits of ‘New Red Star’, ‘Golden Crown’, and ‘Gala’ apples [45]. The main components of the 10 dragon fruit aromas we screened were mainly aldehydes. Among the selected candidate genes, HU01G01237 and HU11G00938 are involved mainly in the development of pollen. Sugar metabolism is the core of primary carbohydrate metabolism in fruits and is the substrate of various physiological metabolisms. All photosynthetic products are carbohydrates, which play a key role in regulating carbohydrate distribution and coordinating intercellular signal transduction and environmental adaptability [46]. Key genes such as glucose-metabolizing enzymes and sugar transport pathways in fruit can regulate the formation of fruit aroma by affecting enzymes and genes related to volatile organic compound synthesis [47]. In addition, the sugar signaling pathway and regulatory network in fruit are involved in the formation of fruit aroma [48]. We also screened candidate genes that control glucose metabolism and carbohydrate metabolism. The major rice flavor gene BADH2 (LOC_Os08g32870) was subsequently cloned by mapping. The deletion of 8 bp in exon 7 and mutation of three bases in BADH2 resulted in the inactivation of the gene and ultimately caused the accumulation of 2-acetyl-1-pyrroline [49]. Through the sequence analysis of rice resource materials, 15 haplotypes of functional mutations have been found in the coding region of the BADH2 gene [50,51,52,53]. We also found that HU09G00745, encoding BADH, is also expressed in dragon fruit and is involved mainly in the gamma-aminobutyric acid catabolic process. HU09G00745 can be used as the focus of later dragon fruit aroma research, although its specific function needs to be further verified. In summary, RNA-seq combined with GC–MS analysis at different stages of dragon fruit development highlighted the roles of flavonoid biosynthesis, photosynthesis, and glucose metabolism in fruit development, and 15 candidate genes related to key flavor compounds were screened via WGCNA, which can be used as the focus of future research.
5. Conclusions
A total of 16,827 DEGs (958 TFs) were identified and grouped into 10 clusters via RNA-seq and HS-SPME-GC-MS analysis. A total of 318 DAMs, including 88 common DAMs, were identified via metabolomics. The main volatile flavor-related substances in dragon fruit and the key genes responsible for their regulation were determined via joint RNA-seq and metabolomics analyses. In addition, 10 volatile main components related to green flavor and aroma were screened using ROAV, and 15 candidate genes related to key flavor compounds were screened via WGCNA. Our study highlights the importance of 10 major volatile flavor compounds in pitaya aroma, which is essential for new cultivar improvement strategies and technologies that consider flavor compounds in the future. In addition, flavonoid biosynthesis, photosynthesis, and sugar metabolism may play important roles in fruit development, but further experimental verification is needed.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/horticulturae11060599/s1, Table S1. All primers used in this study. Table S2. RNA-seq data statistics. Figure S1. WGCNA power value scatter plot. Figure S2. Correlation analysis of RNA-seq samples.
Author Contributions
Conceptualization, H.-L.L. and G.-D.L.; methodology, Z.-J.W. and R.-W.J.; software, Z.-J.W., Z.-J.H. and L.-F.H.; validation, Z.-J.W., X.-Y.Y., L.-F.H., H.-Y.D. and C.-A.L.; formal analysis, Z.-J.W., X.-Y.Y., G.-F.L., S.-T.W. and Z.-Y.L.; investigation, Z.-J.H., G.-F.L., S.-T.W., C.-A.L. and Z.-Y.L.; data curation, Z.-J.H., H.-Y.D., G.-F.L. and Z.-Y.L.; writing—original draft preparation, Z.-J.W. and R.-W.J.; writing—review and editing, Z.-J.W., Z.-J.H., X.-Y.Y., L.-F.H., H.-Y.D., G.-F.L., S.-T.W., C.-A.L., Z.-Y.L., R.-W.J., H.-L.L. and G.-D.L.; visualization, X.-Y.Y., L.-F.H., H.-Y.D., S.-T.W. and C.-A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (32202464), Guangxi Natural Science Foundation (2023GXNSFBA026055), Project of Dragon Fruit Breeding and Cultivation Position of China Agriculture Research System Guangxi Innovation Team—Specialty Fruits (nycytxgxcxtd-2024-17-04), and the Basic Scientific Research Business Special Project of Guangxi Academy of Agricultural Sciences (GuiNongKe 2025YP042, GuiNongKe 2025YP046).
Data Availability Statement
The RNA-seq data presented in the study are deposited in the NCBI repository under accession number PRJNA1262708.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shah, K.; Chen, J.; Chen, J.; Qin, Y. Pitaya nutrition, biology, and biotechnology: A review. Int. J. Mol. Sci. 2023, 24, 13986. [Google Scholar] [CrossRef] [PubMed]
- Arivalagan, M.; Karunakaran, G.; Roy, T.K.; Dinsha, M.; Sindhu, B.C.; Shilpashree, V.M.; Satisha, G.C.; Shivashankara, K.S. Biochemical and nutritional characterization of dragon fruit (Hylocereus species). Food Chem. 2021, 353, 129426. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Gao, H.; You, Z.; Zhang, Z.; Zhu, H.; He, M.; He, J.; Duan, X.; Jiang, Y.; Yun, Z. Multiple metabolomics comparatively investigated the pulp breakdown of four dragon fruit cultivars during postharvest storage. Food Res. Int. 2023, 164, 112410. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.; Luo, D.; Ba, L. Advances in the Understanding of Postharvest Physiological Changes and the Storage and Preservation of Pitaya. Foods 2024, 13, 1307. [Google Scholar] [CrossRef]
- Sukaew, T. The Current and Emerging Research Related Aroma and Flavor. In Aroma and Flavor in Product Development: Characterization, Perception, and Application; Springer: Cham, Switzerland, 2024; pp. 329–369. [Google Scholar] [CrossRef]
- Xu, L.; Zang, E.; Sun, S.; Li, M. Main flavor compounds and molecular regulation mechanisms in fruits and vegetables. Crit. Rev. Food Sci. Nutr. 2023, 63, 11859–11879. [Google Scholar] [CrossRef]
- Zhao, Z.; Hao, Y.; Liu, Y.; Shi, Y.; Lin, X.; Wang, L.; Wen, P.; Hu, X.; Li, J. Comprehensive evaluation of aroma and taste properties of different parts from the wampee fruit. Food Chem. X 2023, 19, 100835. [Google Scholar] [CrossRef]
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. [Google Scholar] [CrossRef]
- Li, C.; Zhao, J.; Liu, Z.; Yang, Y.; Lai, C.; Ma, J.; Aierxi, A. Comparative Transcriptomic Analysis of Gossypium hirsutum Fiber Development in Mutant Materials (xin w 139) Provides New Insights into Cotton Fiber Development. Plants 2024, 13, 1127. [Google Scholar] [CrossRef]
- Sun, J.; Wang, Y.; Zhang, X.; Cheng, Z.; Song, Y.; Li, H.; Wang, N.; Liu, S.; Cao, Z.; Li, H.; et al. Transcriptomic and Metabolomic Analyses Reveal the Role of Phenylalanine Metabolism in the Maize Response to Stalk Rot Caused by Fusarium proliferatum. Int. J. Mol. Sci. 2024, 25, 1492. [Google Scholar] [CrossRef]
- Li, Y.; He, C.; Yu, X.; Zhou, J.; Ran, W.; Chen, Y.; Ni, D. Effects of red-light withering on the taste of black tea as revealed by non-targeted metabolomics and transcriptomics analysis. LWT 2021, 147, 111620. [Google Scholar] [CrossRef]
- Kianersi, F.; Amin Azarm, D.; Fatemi, F.; Pour-Aboughadareh, A.; Poczai, P. Methyl Jasmonate Induces Genes Involved in Linalool Accumulation and Increases the Content of Phenolics in Two Iranian Coriander (Coriandrum sativum L.) Ecotypes. Genes 2022, 13, 1717. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.Q.; Tu, H.; Liang, W.J.; Long, J.M.; Wu, X.M.; Zhang, H.Y.; Guo, W.W. Comparative metabolic and transcriptional analysis of a doubled diploid and its diploid citrus rootstock (C. junos cv. Ziyang xiangcheng) suggests its potential value for stress resistance improvement. BMC Plant Biol. 2015, 15, 89. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, J.; Wang, L.; Lu, X.; Zhang, X.; Cui, X.; Wang, H. Integration of transcriptome and metabolome reveals regulatory mechanisms of volatile flavor formation during tomato fruit ripening. Hortic. Plant J. 2025, 11, 680–692. [Google Scholar] [CrossRef]
- Li, Y.; He, L.; Song, Y.; Zhang, P.; Chen, D.; Guan, L.; Liu, S. Comprehensive study of volatile compounds and transcriptome data providing genes for grape aroma. BMC Plant Biol. 2023, 23, 171. [Google Scholar] [CrossRef]
- Marie, L.; Breitler, J.C.; Bamogo, P.K.A.; Bordeaux, M.; Lacombe, S.; Rios, M.; Lebrun, M.; Boulanger, R.; Lefort, E.; Nakamura, S.; et al. Combined sensory, volatilome and transcriptome analyses identify a limonene terpene synthase as a major contributor to the characteristic aroma of a Coffea arabica L. specialty coffee. BMC Plant Biol. 2024, 24, 238. [Google Scholar] [CrossRef]
- Coelho, V.S.; de Moura, D.G.; Aguiar, L.L.; Ribeiro, L.V.; Silva, V.D.M.; da Veiga Correia, V.T.; Melo, A.C.; Silva, M.R.; de Paula, A.C.C.F.F.; de Araújo, R.L.B.; et al. The Profile of Phenolic Compounds Identified in Pitaya Fruits, Health Effects, and Food Applications: An Integrative Review. Plants 2024, 13, 3020. [Google Scholar] [CrossRef]
- Singh, A.; Swami, S.; Panwar, N.R.; Kumar, M.; Shukla, A.K.; Rouphael, Y.; Sabatino, L.; Kumar, P. Development changes in the physicochemical composition and mineral profile of red-fleshed dragon fruit grown under semi-arid conditions. Agronomy 2022, 12, 355. [Google Scholar] [CrossRef]
- Sarwar, G.; Anwar, T.; Qureshi, H.; Younus, M.; Hassan, M.W.; Sajid-Ur-Rehman, M.; Khalid, F.; Faiza; Zaman, W.; Soufan, W. Optimizing germination: Comparative assessment of various growth media on dragon fruit germination and early growth. BMC Plant Biol. 2024, 24, 533. [Google Scholar] [CrossRef]
- Santoso, V.R.; Pramitasari, R.; Anugrah, D.S.B. Development of Indicator Film Based on Cassava Starch-Chitosan Incorporated with Red Dragon Fruit Peel Anthocyanins-Gambier Catechins to Detect Banana Ripeness. Polymers 2023, 15, 3609. [Google Scholar] [CrossRef]
- Abirami, K.; Swain, S.; Baskaran, V.; Venkatesan, K.; Sakthivel, K.; Bommayasamy, N. Distinguishing three Dragon fruit (Hylocereus spp.) species grown in Andaman and Nicobar Islands of India using morphological, biochemical and molecular traits. Sci. Rep. 2011, 11, 2894. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Samokhin, A.S.; Matyushin, D.D. How searching against multiple libraries can lead to biased results in GC/MS-based metabolomics. Rapid Commun. Mass Spectrom. 2023, 37, e9437. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bacelar, E.; Pinto, T.; Anjos, R.; Morais, M.C.; Oliveira, I.; Vilela, A.; Cosme, F. Impacts of Climate Change and Mitigation Strategies for Some Abiotic and Biotic Constraints Influencing Fruit Growth and Quality. Plants 2004, 13, 1942. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, L.; Chen, J.; Zhang, N.; Zhou, W.; Song, Y. Altitudinal variation of dragon fruit metabolite profiles as revealed by UPLC-MS/MS-based widely targeted metabolomics analysis. BMC Plant Biol. 2024, 24, 344. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Han, X.; Qiao, G.; Yang, K.; Wen, Z.; Wen, X. Comparative Transcriptome Analysis Combining SMRT- and Illumina-Based RNA-Seq Identifies Potential Candidate Genes Involved in Betalain Biosynthesis in Pitaya Fruit. Int. J. Mol. Sci. 2020, 21, 3288. [Google Scholar] [CrossRef]
- Carmen, F.; Frances, C.; Barthe, L. Trends on valorization of pitaya fruit biomass through value-added and green extraction technology–A critical review of advancements and processes. Trends Food Sci. Technol. 2023, 138, 339–354. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, S.; Ma, D.; Liu, Z.; Qi, P.; Wang, Z.; Di, S.; Wang, X. Review of fruits flavor deterioration in postharvest storage: Odorants, formation mechanism and quality control. Food Res. Int. 2024, 182, 114077. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, Z.; Gao, Y.; Wang, K.; Sun, S.; Guo, H.; Tian, W.; Wang, L.; Li, Z.; Li, L.; et al. Analysis of Aroma Characteristics of ‘Binzi’ and ‘Xiangguo’ Apple-Ancient Cultivars in China. Foods 2024, 13, 2869. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Kuang, H.; Zhang, J.; Wang, B.; Wang, S. Transcriptomic and Physiological Analysis Reveals the Possible Mechanism of Inhibiting Strawberry Aroma Changes by Ultrasound after Harvest. Foods 2024, 13, 2231. [Google Scholar] [CrossRef]
- Abbas, F.; Zhou, Y.; O’Neill Rothenberg, D.; Alam, I.; Ke, Y.; Wang, H.C. Aroma Components in Horticultural Crops: Chemical Diversity and Usage of Metabolic Engineering for Industrial Applications. Plants 2023, 12, 1748. [Google Scholar] [CrossRef]
- Zhu, X.; Song, Z.; Li, Q.; Li, J.; Chen, W.; Li, X. Physiological and transcriptomic analysis reveals the roles of 1-MCP in the ripening and fruit aroma quality of banana fruit (Fenjiao). Food Res. Int. 2020, 130, 108968. [Google Scholar] [CrossRef]
- Ho, P.L.; Tran, D.T.; Hertog, M.L.; Nicolaï, B.M. Effect of controlled atmosphere storage on the quality attributes and volatile organic compounds profile of dragon fruit (Hylocereus undatus). Postharvest Biol. Technol. 2021, 173, 111406. [Google Scholar] [CrossRef]
- Ferreira, V.; de-la-Fuente-Blanco, A.; Sáenz-Navajas, M.P. A New Classification of Perceptual Interactions between Odorants to Interpret Complex Aroma Systems. Application to Model Wine Aroma. Foods 2021, 10, 1627. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Zhang, J.; Wang, Z.; Qi, K.; Li, H.; Tian, R.; Wu, X.; Qiao, X.; Zhang, S.; et al. Comparative analysis of volatile aromatic compounds from a wide range of pear (Pyrus L.) germplasm resources based on HS-SPME with GC-MS. Food Chem. 2023, 418, 135963. [Google Scholar] [CrossRef]
- Sheng, L.; Ni, Y.; Wang, J.; Chen, Y.; Gao, H. Characteristic-Aroma-Component-Based Evaluation and Classification of Strawberry Varieties by Aroma Type. Molecules 2021, 26, 6219. [Google Scholar] [CrossRef]
- Wu, H.; Xu, Y.; Wang, H.; Miao, Y.; Li, C.; Zhao, R.; Shi, X.; Wang, B. Physicochemical Characteristics, Antioxidant Activities, and Aroma Compound Analysis of Seven Peach Cultivars (Prunus persica L. Batsch) in Shihezi, Xinjiang. Foods 2022, 11, 2944. [Google Scholar] [CrossRef] [PubMed]
- Althiab-Almasaud, R.; Teyssier, E.; Chervin, C.; Johnson, M.A.; Mollet, J.C. Pollen viability, longevity, and function in angiosperms: Key drivers and prospects for improvement. Plant Reprod. 2024, 37, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Hong, W.; Liu, H.; Shi, X.; Liu, Y.; Liu, Z. The Pollen Donor Affects Seed Development, Taste, and Flavor Quality in ‘Hayward’ Kiwifruit. Int. J. Mol. Sci. 2023, 24, 8876. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, C.; Cheng, L.; Chang, Y.; He, P.; Li, L. Effect of metaxenia on volatile compounds in bagged apple fruit of Fuji. Agric. Sci. Technol. 2017, 18, 583–610. [Google Scholar]
- Durán-Soria, S.; Pott, D.M.; Osorio, S.; Vallarino, J.G. Sugar Signaling During Fruit Ripening. Front. Plant. Sci. 2020, 11, 564917. [Google Scholar] [CrossRef]
- Yang, S.; Meng, Z.; Li, Y.; Chen, R.; Yang, Y.; Zhao, Z. Evaluation of Physiological Characteristics, Soluble Sugars, Organic Acids and Volatile Compounds in ‘Orin’ Apples (Malus domestica) at Different Ripening Stages. Molecules 2021, 26, 807. [Google Scholar] [CrossRef]
- Zhang, Q.; Feng, C.; Li, W.; Qu, Z.; Zeng, M.; Xi, W. Transcriptional regulatory networks controlling taste and aroma quality of apricot (Prunus armeniaca L.) fruit during ripening. BMC Genom. 2019, 20, 45. [Google Scholar] [CrossRef]
- Chen, S.; Yang, Y.; Shi, W.; Ji, Q.; He, F.; Zhang, Z.; Cheng, Z.; Liu, X.; Xu, M. Badh2, encoding betaine aldehyde dehydrogenase, inhibits the biosynthesis of 2-acetyl-1-pyrroline, a major component in rice fragrance. Plant Cell 2008, 20, 1850–1861. [Google Scholar] [CrossRef]
- Imran, M.; Shafiq, S.; Tang, X. CRISPR-Cas9-mediated editing of BADH2 gene triggered fragrance revolution in rice. Physiol. Plant 2023, 175, e13871. [Google Scholar] [CrossRef]
- Singh, G.; Gopala Krishnan, S.; Kumar, A.; Vinod, K.K.; Bollinedi, H.; Ellur, R.K.; Nagarajan, M.; Bhowmick, P.K.; Madhav, S.M.; Singh, K.; et al. Molecular profiling of BADH2 locus reveals distinct functional allelic polymorphism associated with fragrance variation in Indian aromatic rice germplasm. Physiol. Mol. Biol. Plants 2022, 28, 1013–1027. [Google Scholar] [CrossRef]
- Shao, G.; Tang, S.; Chen, M.; Wei, X.; He, J.; Luo, J.; Jiao, G.; Hu, Y.; Xie, L.; Hu, P. Haplotype variation at Badh2, the gene determining fragrance in rice. Genomics 2013, 101, 157–162. [Google Scholar] [CrossRef]
- Addison, C.K.; Angira, B.; Kongchum, M.; Harrell, D.L.; Baisakh, N.; Linscombe, S.D.; Famoso, A.N. Characterization of Haplotype Diversity in the BADH2 Aroma Gene and Development of a KASP SNP Assay for Predicting Aroma in U.S. Rice. Rice 2020, 13, 47. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).