Abstract
Several strains of Bifidobacterium animalis subsp. lactis are blockbusters of commercial dietary supplement cocktails, widely recognized for their probiotic properties and found in various ecological niches. The present study aimed to perform an in-depth comparative genomic analysis on 71 B. animalis subsp. lactis strains isolated from diverse sources, including human and animal feces, breast milk, fermented foods, and commercial dietary supplements, to better elucidate the strain level diversity and biotechnological potential of this species. The average genome size was found to be 1.93 ± 0.05 Mb, with a GC content of 60.45% ± 0.2, an average of 1562 ± 41.3 coding sequences (CDS), and 53.4 ± 1.6 tRNA genes. A comparative genomic analysis revealed significant genetic diversity among the strains, with a core genome analysis showing that 34.7% of the total genes were conserved, while the pan-genome remained open, indicating ongoing gene acquisition. Functional annotation through EggNOG-Mapper and CAZYme clustering highlighted diverse metabolic capabilities, particularly in carbohydrate metabolism. Nearly all (70 of 71) Bifidobacterium animalis subsp. lactis strains were found to harbor CRISPR-Cas adaptive immune systems (predominantly of the Type I-E subtype), underscoring the ubiquity of this phage defense mechanism in the species. A comparative analysis of spacer sequences revealed distinct strain-specific CRISPR profiles, with certain strains sharing identical spacers that correlate with common phylogenetic clades or similar isolation sources—an indication of exposure to the same phage populations and shared selective pressures. These findings highlight a dynamic co-evolution between B. lactis and its bacteriophages across diverse ecological niches and point to the potential of leveraging its native CRISPR-Cas systems for future biotechnological applications. Our findings enhance our understanding of the genetic and functional diversity of B. animalis subsp. lactis, providing valuable insights for its use in probiotics and functional foods.
1. Introduction
The genus Bifidobacterium is a member of the family Bifidobacteriaceae and the order Bifidobacteriales and belongs to the phylum Actinobacteria, which represents one of the largest bacterial taxonomic units [1]. Bifidobacteria, a genus of lactic- and acetic-acid-producing, Gram-positive, non-spore-forming, non-motile, anaerobic bacteria, were first discovered and isolated from the feces of a breastfed infant in 1899 [2]. Bifidobacteria are found in different ecological niches represented by the human, insect, and animal gut (i.e., cow, rabbit, mouse, and chicken), oral cavity, sewage, blood, and food [3,4].
Bifidobacteria are the major symbiotics in the gut microbiome and provide beneficial effects to human health by maintaining a balanced gut microbiome via producing organic acids of lactic and acetic acids, which prohibit pathogen colonization in the gut [4]. The decline in the proportion of the genus Bifidobacterium in the gut has been linked to the progression of allergies, diabetes, and obesity [5]. Obesity, for example, has been shown to be associated with gut microbiota dysbiosis [6].
The health improvement effects and efficacy of Bifidobacteria are strain-specific; for instance, Bifidobacterium animalis subsp. lactis (B. lactis), one of the Bifidobacterium species, is generally found in the intestinal microbiome, and it has also been found to have probiotic properties [3]. Bifidobacterium lactis strains are usually isolated from the human intestinal tract or fecal samples of different age groups. While genotypic and phenotypic similarities were observed across B. lactis strains of animal and human intestinal origin, isolates obtained from German shepherd, mouflon, and ovine feces showed significant genetic and phenotypic diversity [7]. The B. lactis HN019 strain, which has been the subject of various studies, is thought to be capable of enhancing defense mechanisms, thereby increasing a person’s ability to fight against gastrointestinal pathogens and secondary-infection bacteria [8]. This defense mechanism in Bifidobacterium could also be attributed to the luxS gene, which is involved in the biosynthesis of AI-2, a fundamental quorum-sensing signaling molecule that triggers the anti-pathogenic potential of the cell [4]. Bifidobacterium animalis lactis BB-12 has multiple effects that are clinically proven, such as the prevention of gastrointestinal infections [9], reduction in allergic symptoms and eczema incidence [10], improvement in clinical conditions in children with Alzheimer’s disease [11], improvement in symptoms in lactose-intolerant patients [12], and improvement in immune function in resected colorectal cancer patients [13].
The species of Bifidobacteria, similar to other bacteria that exist in the gut, are saccharolytic and have a critical role in sugar fermentation [14]. The species of Bifidobacteria is known to hydrolyze a variety of different sugars, allowing them to inhabit various niches [15]. Understanding the carbohydrate metabolism of an organism is crucial as it explains the prevalence of a species in different niches. Moreover, the elucidation of the metabolic capacity of a species might uncover carbohydrate utilization characteristics unique to that particular species [15]. For bacteria to be assigned to the Bifidobacterium genus, the fructose-6-phosphate phosphoketolase (F6PPK) activity must be observed in cell extracts [16].
Bifidobacteria can metabolize complex carbohydrates derived from plants that function as prebiotics [15]. For a carbohydrate to be considered a prebiotic, a considerable amount of it must reach the colon undigested. Prebiotics offer a therapeutic capacity for the treatment of inflammatory diseases [17]. It has been discovered that Bifidobacteria can utilize starch, arabinoxylan, xylo-oligosaccharides (XOS), galacto-oligosaccharides (GOS), arabinogalactans, fructo-oligosaccharides (FOS), and inulin [15,16,18,19].
Glycoside hydrolases (GHs) encompass 113 protein families that facilitate the breakdown and/or transfer of glycosidic bonds [20]. It is estimated that Bifidobacterium genomes encode between 25 and 126 GHs, representing 12 to 38 GH families [15,21]. The glycoside hydrolase (GH) enzyme families and numbers in the genomes of B. lactis strains exhibit considerable diversity. For instance, several B. lactis strains were predicted to encode between 35 to 42 GH enzymes from around 16 GH families. This diversity indicates a wide range of carbohydrate-degrading capabilities among B. lactis strains [15].
The isolation sources of B. lactis were reportedly diverse such as human adult and infant feces, human adult and infant gut, human breast milk and breast-fed infant feces, Barbary macaque feces, Vervet monkey feces, chimpanzee feces, chicken feces, domestic dog feces, pheasant feces, pig feces, rabbit feces, sewage, commercial dietary supplements, culture tube, cultured food, dairy product, fermented food, microbial food, and probiotics. Since isolation sources of B. lactis strains are strongly diverse, comparative genomic studies are needed to explore genetic diversity at the species level and uncover their metabolic potentials and lifestyle adaptations. To our knowledge, a comprehensive evaluation of B. lactis genomes from different sources is limited in the literature. Thus, the present study aims to fill this gap by comparing whole-genome sequences of seventy-one B. lactis strains that were isolated from twenty-six different ecological niches.
2. Materials and Methods
2.1. Strains and Data Acquisition
Whole-genome sequences of a total of seventy-one B. lactis strains, both complete and draft, were acquired from NCBI GenBank and their accession numbers given in Supplementary Table S1.
2.2. Comparative Genomics of B. lactis
Whole-genome sequences of seventy-one B. lactis genomes were run through a BUSCO analysis to inspect the completeness of the genome assemblies using the bacteria_odb10 lineage dataset [22,23]. The genome assemblies showing above 90% completeness were annotated using Prokka (version 1.14.6) [24].
The core and pan-genome analysis was conducted by annotating genomes first with Prokka [24] with the following arguments: --kingdom Bacteria. Then, output files from Prokka were sent to Roary (version 3.13.0) [25] using arguments -e -n -v -r to carry out the analysis. Pan- and core genes were annotated as functional COG using EggNOG- Mapper [26]. The COG database [27] is publicly available for download with the following link: https://www.ncbi.nlm.nih.gov/COG/ (15 May 2024). Core and Pan COGs were visualized using R [28] and ggplot2 [29]. The SNP-based phylogenetic tree was created using TYGS and iTOL [30,31]. CAZy database (v10) in dbCAN server (https://bcb.unl.edu/dbCAN2/index.php, 15 May 2024) [32] and HMMER (version 3.3.2) [33] were utilized to identify Carbohydrate-active enzyme (CAZyme)-related genes according to suggested protocol dbCAN. Then, B. lactis strains were classified based on the number of CAZymes they harbored. The heatmaps were generated using ComplexHeatmap, circlize, and grid libraries available in R (version 4.1.1).
Identification, alignment, and visualization of CRISPR elements, such as repeats and spacers, were conducted with the CRISPRviz tool [34]. CRISPRCasFinder [35,36] was also utilized to confirm CRISPR types. Identification of plasmids in genomes of B. lactis was performed with PLSDB (version 2021_06_23_v2) using default settings [37,38]. Phage Search Tool Enhanced Release (PHASTEST) was utilized to identify the prophages [39,40]. To identify potential bacteriocins and bacteriocin-expressing regions, BAGEL4 web tool was utilized [41].
Identification, alignment, and visualization of CRISPR elements, such as repeats and spacers, were conducted with the CRISPRviz tool [34]. CRISPRCasFinder [35,36] was also utilized to confirm CRISPR types. Types of CRISPR loci were identified with CRISPRClassify [42]. Prodigal (v2.6.3) [43] was utilized to predict open reading frames (ORFs) within the +/− 20 kb of each CRISPR array. Annotation of Cas proteins in ORFs was conducted with hmmsearch using curated HMM profiles [44] with the following criteria: HMM E-value < 1 × 10−5, HMM alignment coverage >70%, and bitscore >= 40. Partially predicted proteins via Prodigal [31] were removed. Potential targets of spacers were predicted by PAMPredict (v1.0.2) [45], which utilizes blastn [46] against IMG/VR v4 [47] database.
3. Results
3.1. Genome Annotation
A comprehensive comparative genomic analysis of 71 Bifidobacterium animalis subsp. lactis strains was performed. The genome assemblies downloaded from NCBI GenBank were processed through BUSCO analysis, and above 90% completeness was achieved. The average genome size of 1.93 ± 0.05, the average GC (%) content of 60.45 ± 0.2, the average CDS of 1580 ± 39.0, and the average tRNA of 53.4 ± 1.6 were obtained. The complete genome statistics for all 71 strains are available in Supplementary Table S1.
3.2. Phylogenetic Analysis
To identify evolutionary relationships within the species of B. lactis, a SNP-based phylogenetic tree was constructed and shown in Figure 1. Eight main clades were identified. From the bottom up, the first clade contained 19 strains that were mainly isolated from animal feces (i.e., Barbary Macaque feces, Vervet monkey feces, chimpanzee feces, and domestic dog feces) and a few from cultured food in addition to sewage and adult feces. The second clade was composed of 8 strains that originated from adult feces, microbial food, infant feces, and intestines of healthy infants, as well as breast milk. The third clade contained 17 isolates from commercial dietary supplements, human feces, human gut, fermented foods, and rabbit feces. The fourth main clade was made of 2 strains isolated from either infant feces or chicken feces. The fifth clade included 3 isolates from adult feces, the human gut, and fermented food. The sixth clade had three members and only V9 had a known isolation source of intestines of healthy infants. The sixth clade was composed of 12 strains that were isolated from breast milk or infant feces or the human gut or human feces. The last two clades were composed of dairy products, breast milk, human feces, or fermented foods systems.
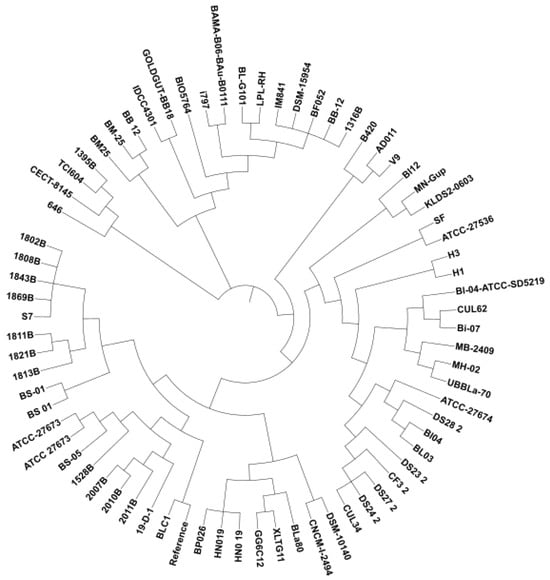
Figure 1.
Phylogenetic tree of 71 Bifidobacterium animalis subsp. lactis strains according to SNP sequences.
Core and Pan-Genome Analysis and eggNOG Mapper
The genomic conservation of 71 strains was analyzed with regard to pan- and core genomes. It was found that 34.7% of the total genes were conserved at 95% blastp identity. Among the total 3173 coding sequences, 1102 of them were shared among 71 genomes that represent the core genome (Figure 2A). The cloud genes comprised 57.4% of the total CDS; however, shell genes represented 7.8% of total coding sequences suggesting the phenotypic variations across B. lactis isolates. To elaborate further, random subsampling was performed to create trendlines of each isolate’s core genome and pan-genome (Figure 2B). Although the size of the core genome was close to a flat line, the pan-genome size never reached a plateau. Because the number of genomes continues to go up and novel genes are still in the process of discovery, the pan-genome stays open among B. lactis strains.
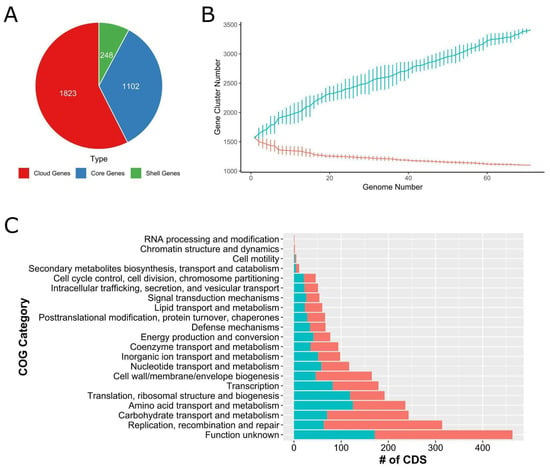
Figure 2.
(A) Coding sequence distributions in the seventy-one B. lactis pan-genome. Cloud genes (red), core genes (blue), and shell genes (green). (B) Estimation of core (red line) and pan-genomes (turquoise line) of the seventy-one B. lactis strains by including genomes one by one. (C) Functional coding sequences in the core genome are shown in red bars, whereas functional coding sequences in the pan-genome are shown in turquoise bars across the genome of 71 B. lactis strains.
The pan- and core genomes were annotated via eggNOG-Mapper [26]. The highest number of coding sequences were assigned to function unknown with regard to both pan- and coregenomes. The second largest number of CDS was achieved with the replication, recombination, and repair-related COG category for coregenomes. The carbohydrate transport and metabolism, along with amino acid transport and metabolism-related COG functions, ranked third and fourth, respectively. The number of CDS as it pertains to amino acid transport and metabolism, as well as translation, ribosomal structure, and biogenesis were the second largest function assigned to the pan-genome. Transcription, nucleotide transport and metabolism, inorganic ion transport and metabolism, energy production and conversion, signal transduction, and mechanism carried a similar number of CDS for both pan- and core genomes.
3.3. Analysis of Carbohydrate-Active Enzymes
Four main clades were identified according to the CAZyme clustering and distribution across the seventy-one B. lactis genomes analyzed [48]. The first clade from the very left was composed of 2010B, 2007B, and BS-05. The second clade was composed of 1821B, UBBLa-70, ATCC-27673, 19-D-1, and ATCC_27673. The third clade had the highest population and included fifty-eight genomes as shown in Figure 3A. The last clade members were 2011B, 1528B, BB_12, and BM-25. Due to being identified as an outlier in the heatmap, the genome Bl03 was excluded.
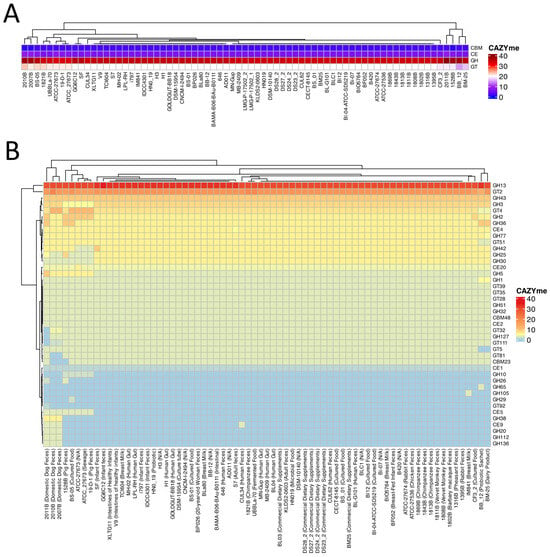
Figure 3.
(A) The color gradient from lighter to darker colors represents the abundance of CAZymes found in each genome. GH: Glycoside hydrolase, GT: Glycosyltransferase, CE: Carbohydrate esterase, AA: Auxiliary activity, and CBM: Carbohydrate binding module. R programming language (version 4.1.1; R Core Team, 2021) was used to draw the heatmap. (B) A heatmap of the detailed CAZyme distribution and clustering in the 71 Bifidobacterium animalis subsp. lactis genomes.
Twenty-three Glycoside Hydrolases were determined across 71 B. lactis strains being evaluated. Thirteen of GHs, including GH1, GH13, GH32, GH2, GH25, GH3, GH30, GH36, GH42, GH43, GH5, GH51, and GH77, existed in all strains. Some of the GHs, for example, GH112, GH136, GH20, and GH38, were unique to 2007B, 2010B, and 2011B, respectively. The GHs of GH105, GH29, and GH65 were only identified in IM841, BS-05, and BB_12, respectively. Across all B. lactis strains tested, eleven glycosyltransferases (GT111, GT2, GT28, GT32, GT35, GT39, GT4, GT5, GT51, GT81, and GT92) were detected. Eight GTs (GT111, GT2, GT28, GT32, GT35, GT39, GT4, and GT51) were identified in all seventy-one B. lactis strains. It was interesting to note that GT5 and GT81 did exist in all strains, with the exception of 2011B and BB_12. GT92 only existed in 2011B. Across two CBMs identified across the entire B. lactis genomes analyzed, CBM48 was found in all strains; however, CBM23 was found in all strains with the exception of 1528B, 2007B, and 2011B. Among six CEs identified, CE1, CE2, CE20, and CE4 were determined in all strains. On the other hand, CE5 and CE9 were found in 11% and 5% of B. lactis genomes analyzed, respectively.
3.4. CRISPR Characterization
All 71 B. animalis subsp. lactis genomes were analyzed to survey their CRISPR spacer and repeat content. We identified a total of 86 CRISPR loci across these genomes using CRISPRviz, with only one strain (646) lacking any detectable CRISPR array. The clustering of the loci based on spacer similarity revealed two major groups (containing 38 and 14 loci, respectively) and three minor groups (each containing 2 loci), while the remaining 28 loci displayed unique spacer compositions (Supplementary Figure S1). Using CRISPRCasFinder (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index, 23 October 2024) for system classification, we found that only seven strains (ATCC-27673, 2010B, 2011B, 1528B, 2007B, BS-05, and 19-D-1) could be assigned a CRISPR-Cas subtype—all of which were identified as Class 1, subtype I-E. Notably, strain ATCC-27673 harbored two distinct Type I-E CRISPR loci, whereas each of the other six subtype-assigned strains carried a single I-E locus.
Type I-E CRISPR-Cas systems carry out interference against invading genetic elements via a multi-protein cascade complex. In this subtype, the Cas8 protein recognizes the protospacer adjacent motif (PAM) on the target DNA and anchors the cascade complex for cleavage [49]. The alignment of the Cas8 sequences from the B. animalis subsp. lactis strains revealed four distinct clusters of variants (Supplementary Figure S2). An analysis of the PAM sequences derived from unique protospacers associated with each cluster showed a global consensus motif of 5′-AAC-3′ located at the upstream of the non-target strand of the protospacer, in alignment with previous reports identifying 5′-AAC-3′ as a noncanonical PAM in type I-E systems [50] (Figure 4A, Table S3).
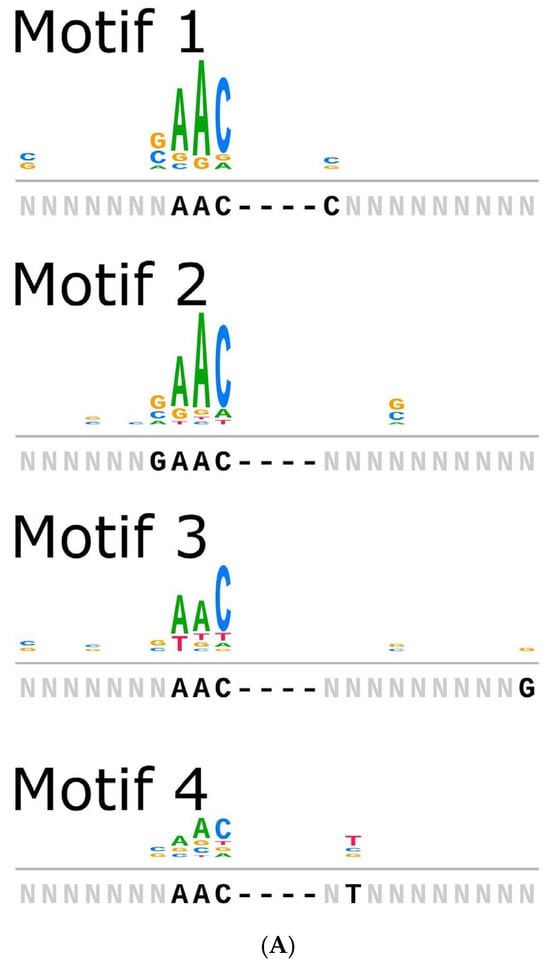
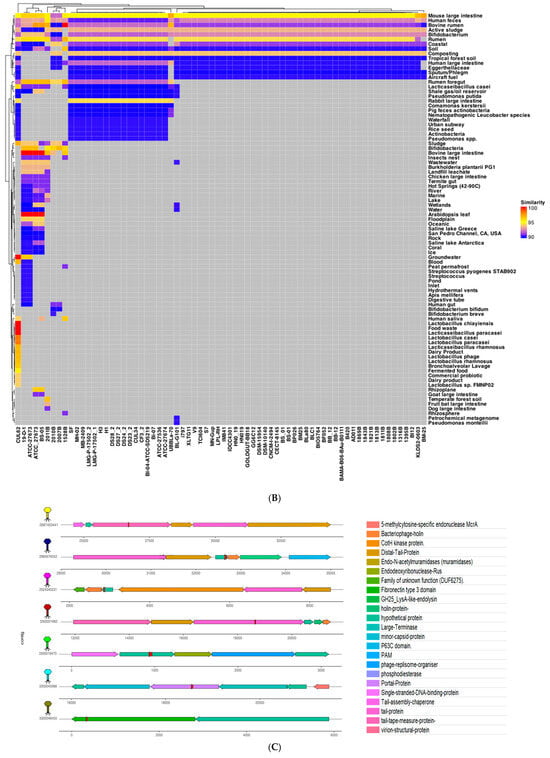
Figure 4.
(A) Strains predicted to encode cas8 were analyzed for PAM sequences under four motifs. Motifs carry 28, 43, 41, and 70 (total 182) spacer sequences. (B) Heatmap of all spacers with significant hits, identified by PAMPredict, when searched against IMGVR and IMGPR databases. Target samples are binned into categories instead of displaying the individual strain information. Dendrograms show the relationships between spacers and hits. (C) The genomic context of 7 distinct phages and prophages.
To investigate the putative origins of the CRISPR spacers, we compared all unique spacer sequences from the 86 loci against comprehensive viral and plasmid databases. Spacer searches against IMG/VR v4 (15,663,652 viral sequences) and IMG/PR (699,973 plasmid sequences) using the PAMPredict pipeline revealed that nearly all B. lactis strains have spacers matching the phage or plasmid sequences from mouse and human intestines, human feces, bovine serum, rumen fluid, coastal marine environments, soil, and even other Bifidobacterium species. The clustering of the spacer “resistome” based on these target profiles yielded two dominant clusters (Figure 4B) alongside several smaller unique clusters, reflecting differences in the phage/plasmid exposure history of each strain. A few strains—such as CUL62, 19-D-1, ATCC-27673, ATCC_27673, and BS-05—stood out by harboring spacers with an especially broad array of target origins, suggesting these particular strains have encountered a wider diversity of mobile genetic elements over their evolution.
To visualize representative phage–host interactions captured by the CRISPR spacers, we examined seven high-confidence spacer matches in detail (Figure 4C). These spacers were selected for having a high sequence identity to their protospacers (~93–100%) and for originating from distinct environmental niches. Phage discovery tools (Cenote-Taker3 and PHASTEST) were then used to identify and annotate the corresponding viral/proviral sequences. For example, strain CUL62 contained two spacers targeting prophages in lactic acid bacteria: one spacer showed 96.8% identity to a Lacticaseibacillus rhamnosus prophage from a human vaginal isolate (within a tail tape measure protein gene), and another showed 96.97% identity to a Lacticaseibacillus rhamnosus (formerly Lactobacillus rhamnosus) prophage from a human fecal sample (targeting a hypothetical gene flanked by a bacteriophage holin and a CotH kinase gene). The latter spacer from CUL62 also matched a prophage in Lactobacillus baoqingensis (a fermented-food isolate) with ~96.8% identity, indicating that a single spacer can target multiple related phages from different environments. Similarly, a spacer from strain 2011B matched a phage assembled from activated sludge (93.8% identity, hitting a tail protein gene region flanked by other tail genes), while a spacer from strain 2010B aligned with a phage from an animal feces metagenome (94.7% identity, within a region flanked by a DNA-binding protein gene and an endodeoxyribonuclease Rus gene). Furthermore, strain 2007B harbored a spacer that perfectly matched a human gut metagenomic phage (100% identity, targeting a portal protein gene flanked by minor capsid and large terminase subunit genes), and strain 1528B contained a spacer with a 100% match to a phage from a soil metagenomic sample (targeting a fibronectin type III domain protein gene). These examples illustrate the broad ecological spectrum of bacteriophages that B. animalis subsp. lactis has encountered, as reflected in its diverse CRISPR spacer repertoire.
4. Discussion
In the current work, an in-depth comparative genomic evaluation of Bifidobacterium animalis subsp. lactis was performed across seventy-one complete genomes available in NCBI Genbank. The strains were isolated from twenty-nine different isolation sources representing the human gut, human feces, infant gut, infant feces, breast milk, animal feces, dietary supplements, cultured food, and dairy food. The average GC (mol %) content achieved was 60.45 ± 0.2 which is typical for B. lactis genomes [51], although remarkably higher than lactic acid bacteria, which has a lower GC content compared to the genus Bifidobacterium. The genome size of Bifidobacterium animalis subsp. lactis was smaller compared to those identified in other Bifidobacterium species. This can be explained by the genome reduction process to decrease the biosynthesis capacity of the organism to retain and acquire genes related to the fermentation of a broad range of nitrogen and carbon sources [52,53].
When the SNP-based phylogenetic tree was constructed, it was not clear whether a close relationship existed between the isolation source and the phylogenetic location. For example, 1316B isolated from pheasant feces was clustered with the isolates from human feces, infant feces, breast milk, and the human gut. Likewise, 646, CECT-8145, TCI604, and 1395B were isolated from human feces, cultured food, breast milk, and rabbit feces, respectively, positioned in the same subclade. On the other hand, domestic dog feces isolates of 2011B, 202B, and 2007B were closely located to each other in the phylogenetic tree. In parallel, the Velvet Monkey feces isolates, Barbery Macau feces, and Chimpanzee feces isolates were clustered together with the exception of the S7 strain from adult feces that were located in between those animal-based isolates. Probiotic dietary supplements of BL-04, HN019, Bi-07, and B420 were located not too far from each other, although the isolation sources of the latter two strains were not reported. We speculate that, since no clear relationship between isolation source and phylogenomic locations was evident across all genomes analyzed, B. lactis strains exhibit a free-living lifestyle.
It was reported that Bifidobacterium species carry large reservoirs of glycoside hydrolase that ease up the metabolism and digestion of glycans [54]. A total of thirteen different GHs were found across all B. lactis strains analyzed. The predominant glycoside hydrolase found among all genomes was GH13 which is functional in the hydrolysis of alpha glycosidic linkages in disaccharides, oligosaccharides, and polysaccharides [55]. It is noteworthy that the highest number of GH13-enzyme-encoding genes were found in those strains isolated from breast milk, infant gut, infant feces, probiotic sachet, adult feces, and cultured food. The second largest GH family found in B. lactis genomes was GH43, which carries enzymes like beta-xylosidase that participate in xylan fermentation. The absence of the GH33 gene cluster that encodes exo-sialidases might suggest that B. lactis is not capable of directly fermenting host mucins to generate sialic acids, and utilize sialic acids in the gut for cross-feeding [56]. However, it was reported that the lack of the GH gene in some Bifidobacterium species does not necessarily reveal a missing sialic acid cross-feeding event [57].
The pan-genome analysis indicated that the genome of B. lactis remains open, suggesting the ongoing acquisition of genetic elements from diverse ecological niches [58]. This open pan-genome suggests that B. lactis has a high capacity for functional diversity, allowing it to adapt to various environments. All 71 strains shared a mere 63.1% of Cluster of Orthologous Groups (COGs) in the core genome, revealing that the genotypic differences in B. lactis were primarily determined by the accessory genome. The core genome, which is shared across all investigated strains, comprises 34.7% of the total genes and includes the essential genetic components necessary for the fundamental cellular functions that are critical to the survival and propagation of this species. We defined a core orthogroup in which all studied genomes were present. The core genome of B. lactis is composed of 1012 shared orthogroups, containing genes associated with the fundamental cellular processes essential for the proliferation and survival of this species [57]. A COG analysis revealed that the majority of the COGs are predicted to participate in diverse housekeeping functions, particularly those associated with carbohydrate and amino acid metabolism, as well as related transport activities [57,59]. A substantial portion of the coding sequences in both the pan-genome and core genome were categorized as having unknown functions. Although 71 B. lactis strains were also examined for the presence of prophages and plasmids, neither prophages nor plasmids were found in the strains [37,60].
CRISPR-Cas adaptive immune systems—beyond serving as bacterial defenses—have also been harnessed as powerful genome-editing tools [34]. In our comparative analysis, these systems proved to be pervasive in B. animalis subsp. lactis, underlining the species’ ongoing arms race with bacteriophages. We detected CRISPR-Cas loci in 70 of the 71 genomes examined, a prevalence much higher than the typical incidence across prokaryotes and indicative of strong selective pressure from phage predation. Moreover, all the identified CRISPR-Cas loci in B. lactis belonged to the Type I-E subtype, suggesting a dominant and conserved mode of immunity in this subspecies.
Intriguingly, we also observed instances where different B. lactis strains carried identical CRISPR spacers, implying that those strains have encountered similar viruses or perhaps exchanged CRISPR cassettes through horizontal gene transfer. For example, strains 2007B and 2010B (isolated from the same source) clustered together in the phylogenetic tree and shared identical spacer sequences, pointing to common phage challenges in their history. Likewise, strain BS-05 and ATCC-27673, which reside in the same clade, displayed a very similar spacer profile, and the pig-feces isolate 19-D-1 harbored a spacer identical to one found in ATCC-27673 (a strain of unknown source). Similarly, the isolates BIO5764, IDCC4301, and GG6C12—recovered from breast milk and infant feces—exhibited matching spacer sequences along with conserved repeat regions. These patterns suggest that strains occupying similar niches or having close lineage relationships often acquire and retain the same phage-derived spacers. In essence, exposure to a shared pool of viruses (or the direct transfer of CRISPR elements between strains) has led to overlapping CRISPR “memories” in certain B. lactis lineages, even as the overall spacer content remains unique to each strain.
Analyzing the CRISPR spacer content at a single-strain resolution provides valuable insight into how B. lactis has adapted to its virus-rich environments. Bacteria archive snippets of viral DNA in their CRISPR arrays as a record of past infections; thus, the spacers within a strain’s genome serve as a chronological log of the phage threats it has survived. Our findings reinforce that the CRISPR-Cas system plays a critical role in strain-specific immunity, allowing B. lactis to endure and thrive under constant phage predation. The high abundance of CRISPR loci in these genomes (far above the prokaryotic average) underscores the importance of this defense mechanism in the species’ lifestyle. The recurrence of identical spacers within and between strains may indicate particularly prevalent phages exerting common selective pressures. Such spacer conservation could arise from different strains independently encountering the same ubiquitous phage (leading to convergent spacer acquisition) or from the sharing of CRISPR loci via genetic duplication or horizontal gene transfer. In either case, the retention of these specific spacers suggests they confer a tangible advantage—likely immunity against phages that B. lactis frequently faces in its habitats.
Linking spacer sequences to their protospacers in viral genomes further revealed how CRISPR profiles correlate with the ecological niches of B. lactis (Figure 5). Shared spacer matches between strains imply overlapping environmental exposures and help map out the bacterium’s natural lifecycle. The evidence points to a broad-spectrum adaptive immunity against phages, spanning the diverse settings where B. lactis is found. For instance, B. animalis subsp. lactis is commonly associated with fermented dairy products [54] and is known to survive transit through the human gastrointestinal tract while contributing to gut health [55]. Consistent with these primary niches, many spacers in our study correspond to phages or prophages derived from human intestinal and fecal samples, reflecting the frequent exposure to gut-associated viruses. Although B. lactis is not typically considered a resident of the vaginal microbiome, oral supplementation or dietary intake can introduce this bacterium into the vaginal environment. Such transient colonization—explored for its potential to prevent urogenital infections and alleviate menopausal symptoms [56]—could explain why we observed spacers targeting phages isolated from vaginal samples. It also raises the possibility of maternal–infant transfer: a mother ingesting B. lactis (via probiotic foods or supplements) might harbor the bacterium in the gut and occasionally in the birth canal or breast milk, seeding her infant’s microbiome during birth and nursing. This scenario would be consistent with our finding that certain breast milk and infant feces isolates share identical CRISPR spacers.
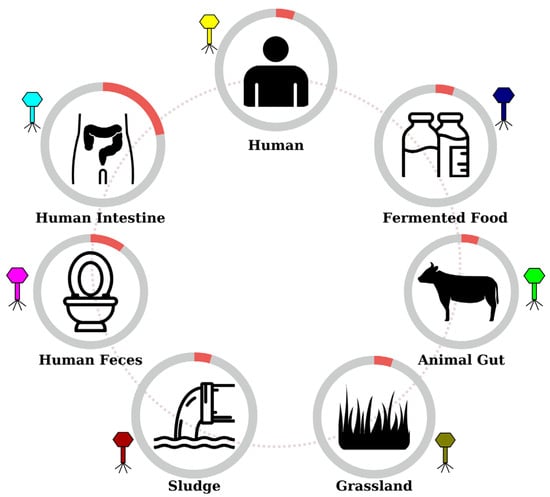
Figure 5.
Representative schematic of the potential life cycle of B. lactis strains. The detailed table with retrieved ecosystems and IMGs is available in Supplementary File Table S2.
Interestingly, some spacers in B. lactis even matched viruses from unexpected sources such as sewage sludge and soil. The direct isolation of B. animalis subsp. lactis from these environments has not been reported, but indirect transmission routes can be proposed. For example, bacteria shed in animal or human waste could enter sewage systems and persist in sludge; the subsequent use of sludge in agriculture or environmental runoff might carry B. lactis (or its phages) into soils and water, which then come in contact with plants and grazing animals. Such a cycle would allow B. lactis or its genetic material to disseminate into grassland ecosystems, providing a plausible explanation for the spacer matches to phages from sludge and grassland habitats [57]. Ultimately, the diverse array of protospacer sources captured in the CRISPR loci of B. lactis highlights the extensive co-adaptation, both evolutionary and ecological, between B. animalis subsp. lactis and its viral predators across the species’ natural lifecycle and a broad spectrum of environments.
5. Conclusions
Overall, the present study provides a comprehensive basis for the genomic analysis of Bifidobacterium animalis subsp. lactis strains isolated from diverse ecological niches. The whole-genome sequencing and comparative genomic analysis of 71 strains revealed significant genetic variability, enriched with CRISPR/Cas systems, diverse carbohydrate-active enzymes (CAZymes), and mobile genetic elements. The analysis identified that all strains lacked plasmids and prophages but displayed a robust presence of CRISPR/Cas systems. A Comparative analysis of spacer sequences revealed distinct strain-specific CRISPR profiles, with certain strains sharing identical spacers that correlate with common phylogenetic clades or similar isolation sources—an indication of exposure to the same phage populations and shared selective pressures. These findings highlight a dynamic co-evolution between B. lactis and its bacteriophages across diverse ecological niches and point to the potential of leveraging its native CRISPR-Cas systems for future biotechnological applications. This study also highlights the metabolic versatility of B. lactis, particularly in carbohydrate metabolism, which is crucial for its probiotic functionality and adaptability to different environments. These findings pave the way for further functional evaluations and biotechnological applications of B. lactis, emphasizing its role in enhancing gut health and its potential use in probiotic and functional food industries. The insights gained from this study contribute significantly to the understanding of B. lactis and support the development of targeted strategies for utilizing its probiotic and industrially relevant traits.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/fermentation11040179/s1, Figure S1: Alignment of spacers of each detected CRISPR locus. Each colored diamond represents a unique repeat, and each colored square represents a unique spacer in the CRISPR-Cas system. Grey “x” boxes showed missing spacer.; Figure S2: Multiple sequence alignment (mafft) and phylogenetic analyis (fasttree) of cas8 encoding B. lactis strains.; Table S1: Whole-Genome assembly statistics of each of 71 B. lactis genomes; Table S2: Isolation sources and IMG Taxon IDs of 7 different phages.; Table S3: Flanked protospacer sequences targeted by cas8 encoding B. lactis species.
Author Contributions
Conceptualization, F.O. and I.C.K.; methodology, F.O. and I.C.K.; software, O.C., I.G. (Ismail Gumustop), and I.G. (Ibrahim Genel); validation, F.O., I.C.K., O.C., I.G. (Ismail Gumustop), and I.G. (Ibrahim Genel); formal analysis, O.C., I.G. (Ismail Gumustop), I.G. (Ibrahim Genel), and H.U.; investigation, O.C., I.G. (Ismail Gumustop), H.U., E.D. and I.G. (Ibrahim Genel); resources, F.O., I.C.K., H.U., O.C., I.G. (Ismail Gumustop), I.G. (Ibrahim Genel), and H.U.; writing—original draft preparation, F.O., I.C.K., O.C., I.G. (Ismail Gumustop), and I.G. (Ibrahim Genel); writing—review and editing, F.O., I.C.K. and E.D., visualization, O.C., I.G. (Ismail Gumustop), and I.G. (Ibrahim Genel); supervision, F.O. and I.C.K.; project administration, F.O. and I.C.K.; funding acquisition, F.O., I.C.K. and H.U. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by the Istanbul Technical University Scientific Research Projects Unit with grant number TGA-2025-46583.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Whole-genome sequences of a total of seventy-two B. lactis strains are available in NCBI GenBank with the following accession numbers of GCA_004154565.1 (1316B), GCA_004154695.1 (1395B), GCA_004154555.1 (1528B), GCA_004154655.1 (1802B), GCA_004154545.1 (1808B), GCA_004154535.1 (1811B), GCA_004154525.1 (1813B), GCA_004154435.1 (1821B), GCA_004154475.1 (1843B), GCA_004154445.1 (1869B), GCA_020309945.1 (19-D-1), GCA_004154645.1 (2007B), GCA_004154425.1 (2010B), GCA_004154455.1 (2011B), GCA_001892925.1 (646), GCA_000021425.1 (AD011), GCA_001263975.1 (ATCC-27536), GCA_000471945.1 (ATCC-27673), GCA_001264015.1 (ATCC_27673), GCA_001263985.1 (ATCC-27674), GCA_000277325.1 (B420), GCA_028463865.1 (BAMA-B06/BAu-B0111), GCA_000025245.2 (BB-12), GCA_002762435.1 (BB-12), GCA_000818055.1 (BF052), GCA_008868455.1 (BIO5764), GCA_000277345.1 (Bi-07), GCA_000022705.1 (Bl-04-ATCC-SD5219), GCA_000414215.1 (Bl12), GCA_023375105.1 (BLa80), GCA_000224965.2 (BLC1), GCA_017963615.1 (BL-G101), GCA_002803775.1 (BM 25), GCA_035282465.1 (BM25) GCA_026683895.1 (BP026), GCA_000240765.1 (BS 01), GCA_018408975.1 (BS 01), GCA_018408985.1 (BS 05), GCA_000612705.1 (CECT 8145), GCA_003094915.1 (CF3_2), GCA_000220885.1 (CNCM I-2494), GCA_025506255.1 (CUL34), GCA_036281465.1 (CUL62) GCA_003094835.1 (DS23_2), GCA_003094815.1 (DS24_2), GCA_003094775.1 (DS27_2), GCA_003095015.1 (DS28_2), GCA_000022965.1 (DSM-10140), GCA_021018785.1 (DSM-15954), GCA_030253625.1 (GG6C12), GCA_034561975.1 (GOLDGUT-BB18), GCA_016835115.1 (H1), GCA_016835135.1 (H3), GCA_000172535.1 (HN019), GCA_003606305.1 (HN0_19), GCA_003428375.1 (IDCC4301), GCA_925048565.1 (IM841), GCA_019576095.1 (i797), GCA_000816205.1 (KLDS2.0603), GCA_002914815.1 (BL03), GCA_002914895.1 (Bl04), GCA_030284625.1 (LPL-RH), GCA_018409015.1 (MB 2409), GCA_029167605.1 (MH-02), GCA_009834975.1 (MN-Gup), GCA_003390755.1 (S7), GCA_029542645.1 (SF) GCA_036923305.1 (TCI604), GCA_003970855.1 (UBBLa-70), GCA_000092765.1 (V9), and GCA_030552855.1 (XLTG11).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Turroni, F.; van Sinderen, D.; Ventura, M. Genomics and Ecological Overview of the Genus Bifidobacterium. Int. J. Food Microbiol. 2011, 149, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Jungersen, M.; Wind, A.; Johansen, E.; Christensen, J.E.; Stuer-Lauridsen, B.; Eskesen, D. The Science behind the Probiotic Strain Bifidobacterium animalis subsp. lactis BB-12®. Microorganisms 2014, 2, 92–110. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G. The Lactobacillus and Bifidobacterium Microflora of the Human Intestine: Composition and Succession. Curr. Issues Intest. Microbiol. 2001, 2, 43–53. [Google Scholar]
- Li, H.; He, B.; Ma, N.; Liu, C.; Cai, K.; Zhang, X.; Ma, X. Quorum Sensing of Bifidobacteria: Research and Progress. Microbiol. Res. 2025, 294, 128102. [Google Scholar] [CrossRef]
- Da Silva, C.C.; Monteil, M.A.; Davis, E.M. Overweight and Obesity in Children Are Associated with an Abundance of Firmicutes and Reduction of Bifidobacterium in Their Gastrointestinal Microbiota. Child. Obes. 2020, 16, 204–210. [Google Scholar] [CrossRef]
- Liu, Y.-M.; Liu, C.; Deng, Y.-S.; Chen, Y.; Qiu, Q.-W.; Shang, X.-X.; Wang, C.-R.; Han, L.-J.; Huang, L.; Yang, Z.-M.; et al. Beneficial Effects of Dietary Herbs on High-Fat Diet-Induced Obesity Linking with Modulation of Gut Microbiota. Food Med. Homol. 2025, 2, 9420034. [Google Scholar] [CrossRef]
- Bunesova, V.; Killer, J.; Javurkova, B.; Vlkova, E.; Tejnecky, V.; Musilova, S.; Rada, V. Diversity of the Subspecies Bifidobacterium animalis Subsp. lactis. Anaerobe 2017, 44, 40–47. [Google Scholar] [CrossRef]
- Arunachalam, K.; Gill, H.; Chandra, R. Enhancement of Natural Immune Function by Dietary Consumption of Bifidobacterium lactis (HN019). Eur. J. Clin. Nutr. 2000, 54, 263–267. [Google Scholar] [CrossRef]
- Nocerino, R.; De Filippis, F.; Cecere, G.; Marino, A.; Micillo, M.; Di Scala, C.; de Caro, C.; Calignano, A.; Bruno, C.; Paparo, L.; et al. The Therapeutic Efficacy of Bifidobacterium animalis Subsp. lactis BB-12® in Infant Colic: A Randomised, Double Blind, Placebo-Controlled Trial. Aliment. Pharmacol. Ther. 2020, 51, 110–120. [Google Scholar] [CrossRef]
- Miraglia Del Giudice, M.; Indolfi, C.; Capasso, M.; Maiello, N.; Decimo, F.; Ciprandi, G. Bifidobacterium Mixture (B. longum BB536, B. infantis M-63, B. breve M-16V) Treatment in Children with Seasonal Allergic Rhinitis and Intermittent Asthma. Ital. J. Pediatr. 2017, 43, 25. [Google Scholar] [CrossRef]
- Huidrom, S. Therapeutic Approach of Probiotics in Children with Atopic Dermatitis. Former. Curr. Med. Chem.—Anti-Inflamm. Anti-Allergy Agents 2021, 20, 2–9. [Google Scholar] [CrossRef]
- Leis, R.; de Castro, M.-J.; de Lamas, C.; Picáns, R.; Couce, M.L. Effects of Prebiotic and Probiotic Supplementation on Lactase Deficiency and Lactose Intolerance: A Systematic Review of Controlled Trials. Nutrients 2020, 12, 1487. [Google Scholar] [CrossRef] [PubMed]
- Sivamaruthi, B.S.; Kesika, P.; Chaiyasut, C. The Role of Probiotics in Colorectal Cancer Management. Evid. Based Complement. Alternat. Med. 2020, 2020, 3535982. [Google Scholar] [CrossRef] [PubMed]
- Pokusaeva, K.; Fitzgerald, G.F.; van Sinderen, D. Carbohydrate Metabolism in Bifidobacteria. Genes Nutr. 2011, 6, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.; Sinderen, D.V. Chapter 8—Carbohydrate Metabolism in Bifidobacteria. In The Bifidobacteria and Related Organisms; Mattarelli, P., Biavati, B., Holzapfel, W.H., Wood, B.J.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 145–164. ISBN 978-0-12-805060-6. [Google Scholar]
- Gavini, F.; Esbroeck, M.V.; Touzel, J.P.; Fourment, A.; Goossens, H. Detection of Fructose-6-Phosphate Phosphoketolase (F6PPK), a Key Enzyme of the Bifid-Shunt, inGardnerella Vaginalis. Anaerobe 1996, 2, 191–193. [Google Scholar] [CrossRef]
- Shi, D.-C.; Wang, P.-Y.; Xu, L.; Zhu, H.; Zhang, W.-Y.; Wu, Q.-Y.; Bu, T.-T.; Tian, B.-M.; Sun, P.-L.; Cai, M. Potential of Dendrobium Officinale Oligosaccharides to Alleviate Chronic Colitis by Modulating Inflammation and Gut Microbiota. Food Med. Homol. 2025, 2, 9420077. [Google Scholar] [CrossRef]
- Rossi, M.; Corradini, C.; Amaretti, A.; Nicolini, M.; Pompei, A.; Zanoni, S.; Matteuzzi, D. Fermentation of Fructooligosaccharides and Inulin by Bifidobacteria: A Comparative Study of Pure and Fecal Cultures. Appl. Environ. Microbiol. 2005, 71, 6150–6158. [Google Scholar] [CrossRef]
- Davis, L.M.G.; Martínez, I.; Walter, J.; Hutkins, R. A Dose Dependent Impact of Prebiotic Galactooligosaccharides on the Intestinal Microbiota of Healthy Adults. Int. J. Food Microbiol. 2010, 144, 285–292. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes Database (CAZy): An Expert Resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Mancabelli, L.; Ferrario, C.; Mangifesta, M.; Hevia, A.; Viappiani, A.; Scholz, M.; et al. Bifidobacteria Exhibit Social Behavior through Carbohydrate Resource Sharing in the Gut. Sci. Rep. 2015, 5, 15782. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Tatusov, R.L. The COG Database: A Tool for Genome-Scale Analysis of Protein Functions and Evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER Web Server: 2018 Update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Nethery, M.A.; Barrangou, R. CRISPR Visualizer: Rapid Identification and Visualization of CRISPR Loci via an Automated High-Throughput Processing Pipeline. RNA Biol. 2019, 16, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A Web Tool to Identify Clustered Regularly Interspaced Short Palindromic Repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an Update of CRISRFinder, Includes a Portable Version, Enhanced Performance and Integrates Search for Cas Proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Schmartz, G.P.; Hartung, A.; Hirsch, P.; Kern, F.; Fehlmann, T.; Müller, R.; Keller, A. PLSDB: Advancing a Comprehensive Database of Bacterial Plasmids. Nucleic Acids Res. 2022, 50, D273–D278. [Google Scholar] [CrossRef]
- Galata, V.; Fehlmann, T.; Backes, C.; Keller, A. PLSDB: A Resource of Complete Bacterial Plasmids. Nucleic Acids Res. 2019, 47, D195–D202. [Google Scholar] [CrossRef]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, Better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A User-Friendly Web Server to Thoroughly Mine RiPPs and Bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]
- Nethery, M.A.; Korvink, M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V.; Barrangou, R. CRISPRclassify: Repeat-Based Classification of CRISPR Loci. CRISPR J. 2021, 4, 558–574. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An Updated Evolutionary Classification of CRISPR–Cas Systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Ciciani, M.; Demozzi, M.; Pedrazzoli, E.; Visentin, E.; Pezzè, L.; Signorini, L.F.; Blanco-Miguez, A.; Zolfo, M.; Asnicar, F.; Casini, A.; et al. Automated Identification of Sequence-Tailored Cas9 Proteins Using Massive Metagenomic Data. Nat. Commun. 2022, 13, 6474. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Camargo, A.P.; Nayfach, S.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Ritter, S.J.; Reddy, T.B.K.; Mukherjee, S.; Schulz, F.; et al. IMG/VR v4: An Expanded Database of Uncultivated Virus Genomes within a Framework of Extensive Functional, Taxonomic, and Ecological Metadata. Nucleic Acids Res. 2022, 51, D733–D743. [Google Scholar] [CrossRef]
- Gavande, P.V.; Goyal, A.; Fontes, C.M.G.A. Chapter 1—Carbohydrates and Carbohydrate-Active enZymes (CAZyme): An Overview. In Glycoside Hydrolases; Foundations and Frontiers in Enzymology; Goyal, A., Sharma, K., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 1–23. ISBN 978-0-323-91805-3. [Google Scholar]
- Zheng, Y.; Li, J.; Wang, B.; Han, J.; Hao, Y.; Wang, S.; Ma, X.; Yang, S.; Ma, L.; Yi, L.; et al. Endogenous Type I CRISPR-Cas: From Foreign DNA Defense to Prokaryotic Engineering. Front. Bioeng. Biotechnol. 2020, 8, 62. [Google Scholar] [CrossRef]
- Xu Hua Fu, B.; Wainberg, M.; Kundaje, A.; Fire, A.Z. High-Throughput Characterization of Cascade Type I-E CRISPR Guide Efficacy Reveals Unexpected PAM Diversity and Target Sequence Preferences. Genetics 2017, 206, 1727–1738. [Google Scholar] [CrossRef]
- Barrangou, R.; Briczinski, E.P.; Traeger, L.L.; Loquasto, J.R.; Richards, M.; Horvath, P.; Coûté-Monvoisin, A.-C.; Leyer, G.; Rendulic, S.; Steele, J.L.; et al. Comparison of the Complete Genome Sequences of Bifidobacterium snimalis subsp. Lactis DSM 10140 and Bl-04. J. Bacteriol. 2009, 191, 4144–4151. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Evolutionary Genomics of Lactic Acid Bacteria. J. Bacteriol. 2007, 189, 1199–1208. [Google Scholar] [CrossRef]
- Makarova, K.; Slesarev, A.; Wolf, Y.; Sorokin, A.; Mirkin, B.; Koonin, E.; Pavlov, A.; Pavlova, N.; Karamychev, V.; Polouchine, N.; et al. Comparative Genomics of the Lactic Acid Bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 15611–15616. [Google Scholar] [CrossRef]
- Lawson, M.A.E.; O’Neill, I.J.; Kujawska, M.; Gowrinadh Javvadi, S.; Wijeyesekera, A.; Flegg, Z.; Chalklen, L.; Hall, L.J. Breast Milk-Derived Human Milk Oligosaccharides Promote Bifidobacterium Interactions within a Single Ecosystem. ISME J. 2019, 14, 635–648. [Google Scholar] [CrossRef]
- Lordan, C.; Roche, A.K.; Delsing, D.; Nauta, A.; Groeneveld, A.; MacSharry, J.; Cotter, P.D.; van Sinderen, D. Linking Human Milk Oligosaccharide Metabolism and Early Life Gut Microbiota: Bifidobacteria and Beyond. Microbiol. Mol. Biol. Rev. 2024, 88, e00094-23. [Google Scholar] [CrossRef] [PubMed]
- Juge, N.; Tailford, L.; Owen, C.D. Sialidases from Gut Bacteria: A Mini-Review. Biochem. Soc. Trans. 2016, 44, 166–175. [Google Scholar] [CrossRef]
- Bottacini, F.; O’Connell Motherway, M.; Kuczynski, J.; O’Connell, K.J.; Serafini, F.; Duranti, S.; Milani, C.; Turroni, F.; Lugli, G.A.; Zomer, A.; et al. Comparative Genomics of the Bifidobacterium brevetaxon. BMC Genom. 2014, 15, 170. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Ferrario, C.; Modesto, M.; Mattarelli, P.; Jiří, K.; et al. Comparative Genomic and Phylogenomic Analyses of the Bifidobacteriaceae Family. BMC Genom. 2017, 18, 568. [Google Scholar] [CrossRef]
- Turroni, F.; Bottacini, F.; Foroni, E.; Mulder, I.; Kim, J.-H.; Zomer, A.; Sánchez, B.; Bidossi, A.; Ferrarini, A.; Giubellini, V.; et al. Genome Analysis of Bifidobacterium bifidum PRL2010 Reveals Metabolic Pathways for Host-Derived Glycan Foraging. Proc. Natl. Acad. Sci. USA 2010, 107, 19514–19519. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid Identification, Annotation and Analysis of Secondary Metabolite Biosynthesis Gene Clusters in Bacterial and Fungal Genome Sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).