Abstract
Unexpected activation of the tetrabutylammonium cation in the presence of hexachloroplatinate(IV) under light to give a dinuclear complex of trans-μ2:η2,η2-1,3-butadiene-bis(trichloroplatinate(II)) along with a proposed mechanism of the activation has been reported. The mechanism has been investigated using a combination of photodissociation photodetachment mass spectrometry, and frozen-matrix EPR spectroscopy, in addition to 1D and 2D NMR spectroscopy. In addition to the Bu4N+ salts of [PtCl6]2− that were part of the original observations, the reactivity of Bu4P+, Pr4N+, and Pe4N+ (Pe = pentyl) salts has also been investigated, and, in addition, the possible involvement of η2-butene complex intermediates has been investigated. The combined results provide additional evidence and support for the originally proposed mechanism of activation of the Bu4N+ cation.
1. Introduction
As part of a study on possible syntheses of chloro-bridged, mixed-metal complexes of platinum, palladium and gold, we previously reported that it was possible to prepare the anion [Cl2Pd(μ-Cl)2PtCl2]2− [1]. It was obtained as its [K(18-crown-6)]+ salt as a statistical, ca 25% part of a mixture that also contained [Pd2Cl6]2− and [Pt2Cl6]2−, following a purely thermal reaction of K2[PtCl4] and K2[PdCl4] in the presence of [K(18-crown-6)]+. In then attempting to prepare an analogous anion containing both platinum and gold, K[AuCl4] and K2[PtCl4] were reacted together in water, leading to a redox reaction and formation of [PtCl6]2− and either [AuCl2]− or Au0 depending on the ratio of salts used. In determining the course of these reactions and the products formed, [Bu4N]+ was added to the solution post reaction, which resulted in the formation of [Bu4N]2[PtCl6]. On one occasion in the course of this work, a sample was left in an NMR tube from which crystals were recovered that turned out to be of the [Bu4N]+ salt of the dinuclear PtII complex [Cl3Pt(μ2:μ2-1,3-butadiene)PtCl3]2− (Figure 1, 1) [2]. This complex exists in up to four isomeric forms, and we have reported on the dynamics of these complexes in solution as studied by NMR spectroscopy [3].
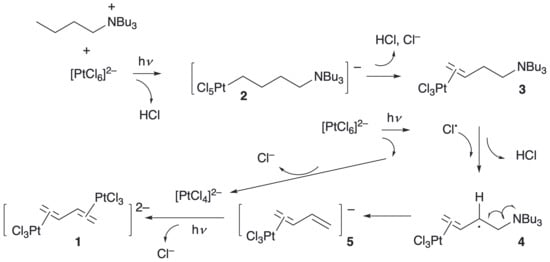
Figure 1.
Proposed mechanism for the formation of the dinuclear butadiene complex.
We reported on this observation and our finding that the reaction was mediated photochemically through excitation of the [PtCl6]2− anion [2], even though our first isolation of the complex arose from serendipitous ambient irradiation. Understanding that the reaction was driven photochemically and using calculations of excited states and in situ EPR measurements, we were able to propose a mechanism consistent with both the observed products and with literature precedent that invoked two different C–H activation steps, each with concomitant reduction to PtII. We now report the results of further experiments to investigate aspects of the mechanism of this somewhat unprecedented reaction and to understand something of its scope.
2. Results
To probe the scope and hence mechanism of this reaction further, complementary experiments were identified that would provide additional support and insight, and it is these that form the basis of this paper. First, revisiting the mechanism (Figure 1) proposed originally, the photochemically driven formation of the σ-alkyl complex 2 is uncontentious, being based upon the extensive work of Shilov and Shul’pin [4,5,6,7,8,9,10,11,12], and β-hydrogen elimination from this complex to form 3 would be anticipated. Complex 3 then undergoes hydrogen abstraction followed by Hofmann elimination [7] to extrude the monoplatinum η2-butadiene complex 5, which then complexes with PtII to give 1. Both the formation of the σ-alkyl complex and the generation of Cl• occur via photoexcitation that generates PtIII and leads ultimately to PtII.
The lower part of Figure 1 shows photoexcitation of [PtCl6]2− leading to Cl• (and PtIII), which can then abstract a hydrogen from the β-position of the now functionalised alkyl group to give radical 4. In turn, this extrudes alkene 5, which reacts photochemically with [PtCl4]2− (from reduction of PtIII) to give product 1; proposing Bu3N• is consistent with the observation of Bu3NH+ by liquid injection field desorption/ionisation mass spectrometry reported earlier (requiring only proton abstraction in the medium) [2]. We also note that ‘consumption’ of two equivalents of PtIV requires the formation of a single equivalent of butadiene, and so with Bu4N+ in excess, it is statistically most likely that each butadiene will result from activation of Bu4N+, which would require Bu3NH+ as the major nitrogen-containing product.
2.1. Photodissociation and Photodetachment Mass Spectroscopy of [Bu4N]2[PtCl6]
Photodissociation and photodetachment mass spectrometry was exploited to identify species resulting from the irradiation of [PtCl6]2−, offering the advantage of being able to probe the very reaction solutions used in the bulk experiments. The technique subjects the compound of interest to photoexcitation using energies in excess of those required for fragmentation and then detects the products using mass spectrometry, accessing both positive and negative ion modes. Solutions are introduced into the ion-trap mass spectrometer by electrospray methods.
Details of how the mass spectrometer is configured and the general way in which the experiments are carried out are found in reference [13]. To give an example, previous studies using aqueous solutions of Na2[PtCl6] in the presence of nucleobases showed both the formation of clusters of one or more of the bases with [PtCl6]2− upon ESI, with the cluster UV photochemistry then being studied in the mass spectrometer via laser spectroscopy [8]. Previous work using a similar instrument to investigate the photochemistry of [PtCl6]2− provided evidence for photochemically activated redox processes generating chloroplatinate anions of PtV, PtIII, and PtII, accompanied by the formation of both chlorine radicals and chloride anions via dissociation, leading to chlorometallate(II) anions [14].
Given that in the present system, both calculation and experiment show photogeneration of PtIII on irradiation of solutions of [PtCl6]2−, which can subsequently lead to PtII, there was confidence that these experiments would mimic conditions of the bulk photolysis preparations that had been carried out. Indeed, similar evidence has been observed for Ru carbonyl complexes with gas-phase photolysis mirroring solution-phase photolysis [15].
The positive-ion ESI mass spectrum of an unirradiated solution of [Bu4N]2[PtIVCl6] in acetone shows the precursor ion at m/z 1135 associated with the cluster ion {[Bu4N]3[PtIVCl6]}+ and a fragment at m/z 242 corresponding to {Bu4N}+ (Figure S1). The negative-ion ESI mass spectrum (Figure S2) depicts the precursor ion at m/z 926 associated with {[Bu4N]2[PtIVCl6]Cl}− with its main fragment at m/z 649—{[Bu4N][PtCl6]}−.
Irradiation was carried out at wavelengths in the range of 220 to 270 nm, and evidence from the resulting mass spectra in both the positive and negative ion modes showed the formation of platinum(II); an example from the positive mode is shown in Figure 2 (other examples in Figures S3–S5). Thus, {[Bu4N]3[PtCl6]}+ was found to photofragment giving {[Bu4N]3[PtIICl4]}+ (with the corresponding ion {[Bu4N][PtIICl4]}− being seen in the negative ion spectrum). Note also that PtIII was observed directly both as the {[PtCl4]}− anion and as {[Bu4N]3[PtIIICl5]}+. These data are consistent with observations from the previous experiments using aqueous solutions of Na2[PtCl6], which photolysed [PtCl6]2− directly [8]. However, crucially, it was possible to detect two complexes invoked in the mechanistic proposal. Thus, zwitterionic complex 3 was observed as part of a cluster with m/z = 1061 that corresponded to {(Bu4N)2Cl[(Bu3N)(C4H7)PtIICl3]}+, while σ-bonded complex 1 was observed directly as {Bu3N-C4H8PtIVCl5}− at m/z 372 (Figure S4). Finally, here, production of complex 5 from 4 produces the Bu3N+• radical cation, which would be expected to abstract a hydrogen in the medium to give [Bu3NH]+, which was observed at m/z 186 (Figure S5).
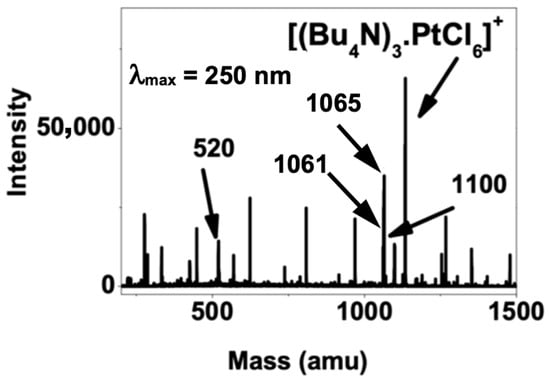
Figure 2.
The UV photodepletion spectrum of [Bu4N]3[PtCl6]+ ion (λ = 250 nm) showing signals at m/z 520 = [(Bu4N)2Cl]+, 1061 = [(Bu4N)2Cl[(Bu3N)(C4H7)PtIICl3]]+, 1065 = [(Bu4N)3PtIICl4]+ and 1100 = [(Bu4N)3PtIIICl5]+.
With the ability to mimic conditions of the photolytic preparations carried out, these experiments provide valuable and compelling support for the mechanism proposed previously for this quite unique reaction.
2.2. Possible Reactivity of [PtCl3(1-butene)]− and [PtCl3(2-butene)]−
One of the key ideas in the proposed mechanism is that the butyl fragment is functionalised in two steps and that in both of these it remains attached to a Bu3N fragment—i.e., the formation of complex 2 and then 3, and also the formation and reaction of complex 4 (Figure 3). This pathway assumes that the initial C–H activation of Bu4N+ occurs at a terminal carbon, leaving the modified Bu4N+ with four C–N bonds. However, activation at a carbon closer to the nitrogen could conceivably have led to elimination of the alkene through the breaking of a C–N bond and the formation of [PtCl3(η2-butene)]−. If this were the case, then further reaction would require activation of [PtCl3(η2-butene)]− by [PtCl6]2− in a second photochemical step to generate the product. So, depending on the way the elimination occurred, either [PtCl3(η2-1-butene)]− or [PtCl3(η2-2-butene)]− could be formed.
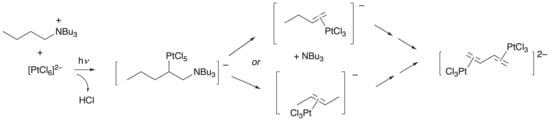
Figure 3.
Possible alternative products following first metalation on a non-terminal carbon atom.
To investigate the viability of such a pathway, trichloroplatinate(II) complexes of 1-butene and of 2-butene were prepared as their PPN+ ((PPN is bis(triphenylphosphino)iminium − [N(PPh3)2]+)) salts in order to remove any reactivity associated with Bu4N+. The salts were then dissolved in CD2Cl2 in a gas-tight NMR tube and irradiated before the 1H NMR spectrum was recorded (CD2Cl2 was used owing to the much better solubility of the salt when compared with acetone). In both cases, after 5 min of irradiation, there was a small decrease in the intensity of the resonances associated with the coordinated alkene and the appearance of weak signals consistent with the free alkene (comparison with the chemical shifts in a solution of the free alkene). Interestingly, on standing after irradiation, the signals associated with the free alkene disappeared, and the intensity of the complex resonances was restored, implying re-complexation.
In the second experiment, [PPN]2[PtCl6] was added to a fresh solution of the alkene complex, and irradiation was carried out once more. In both cases, the resonances corresponding to the free alkene were more intense, and indeed, after 60 min, there were no signals from the starting alkene complex. However, after standing for 24 h, most of the intensity of the resonances associated with the starting complex was restored. The 1H NMR spectrum also showed new, weak resonances in the region 3.5–3.8 ppm, but in the absence of any evidence of coupling to 195Pt in the 2D spectra, it is assumed that these are some by-products, perhaps formed by the reaction of decomplexed alkene with [PtCl6]2−.
Finally, here, a dichloromethane solution was prepared containing [PPN]2[PtCl6] and [PPN]2[PtCl3(1-butene)], which was then frozen, placed in an EPR cavity, and irradiated. The observed spectrum was compared with that obtained on performing an equivalent experiment irradiating [Bu4N]2[PtCl6]. In the Pt region, the two spectra (Figure 4a) are in effect superimposable, which is consistent in both cases with the formation of [PtIIICl5]2− as identified previously (g = 2.408, aPt = 425 G). In addition, there is also a signal at g = 2.008 (Figure 4b), which is again overlaid with the signal from a photolysis of [Bu4N]2[PtCl6]. While there are similarities, the two signals are evidently not the same, although that originating from the PPN+ salt does have a structure not unlike that arising from an N-based radical. While there appears to be little radical chemistry of the PPN cation (it tends to be used normally as an ‘innocent’ cation in conjunction with much more redox-labile metalate salts), there is a report [16] of a radical derived from an iminophosphorane dimer (Figure 5) with aN = 2.6 G and aP = −5.4 G. While 31P hyperfine is larger than that of 14N, it is nonetheless relatively small, and so it is not impossible that for two equivalent 31P, it will all merge to a single line, and a nitrogen radical would be observed, leaving open the possibility of a signal arising from a PPN-based radical.
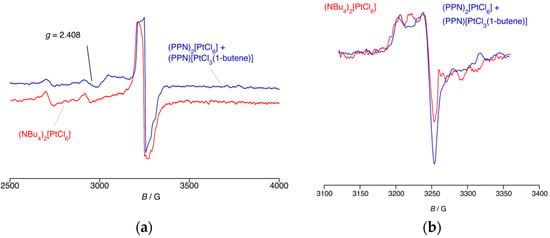
Figure 4.
(a) The X-band EPR spectrum of the mixture [PPN]2[PtCl6] and [PPN]2[PtCl3(1-butene)] in dichloromethane at 120 K under UV irradiation for 25 min; the spectrum obtained from treating [Bu4N]2[PtCl6] under identical conditions is superimposed. (b) The organic region of the spectrum.
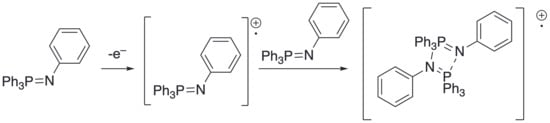
Figure 5.
Dimeric iminophosphorane radical and its proposed mechanism of formation.
While these reactions were carried out in dichloromethane on account of the better solubility of the reagents, some early experiments used acetone, and on one occasion, crystals of [PPN]2[PtCl4] grew from the solution where the reaction was not under constant volume (i.e., any decomplexing alkene could escape the reaction medium). The structure was solved using X-ray methods and is described below.
2.3. Possible Activation of Other Cations
2.3.1. Tetrabutylphosphonium—[Bu4P]+
In common with tetraalkylammonium cations, the tetrabutylphosphonium cation can be deployed as a counter cation to anionic inorganic complexes to engender solubility in organic solvents. With (Bu4N)+ identified as the source of the butadiene on photolysis of [Bu4N][PtCl6], it was of interest to determine whether the same chemistry might occur on irradiation of [Bu4P][PtCl6]. Thus, [Bu4P]2[PtCl6] was prepared and photolysed in acetone under the conditions used with [Bu4N]2[PtCl6] and, as a control, Bu4PCl and [Bu3PH][BF4] were also irradiated separately under the same conditions.
The 1H and 31P{1H} NMR spectra of the post-irradiation solutions of Bu4PCl and [Bu3PH][BF4] showed no change, whereas on irradiation of [Bu4P]2[PtCl6], [PtCl6]2− was consumed, giving [PtCl4]2− (195Pt NMR chemical shift), but no platinum(II)-olefin signals were observed. Furthermore, the post-photolysis 31P{1H} NMR spectrum showed a signal at δ = 46 ppm corresponding to (Bu4P)+. Unsuccessful attempts to isolate a product from the reaction mixture were made through crystallisation. This observation indicated the generation of active species from [PtCl6]2− under light to form the reduced species [PtCl4]2−, however, the activation of the butyl going in the same manner as for the tetrabutylammonium cation did not occur.
This can be rationalised by reference to work by Oh et al., who studied reactions of tetraalkylphosphonium ionic liquids in CO2 capture, in which the ionic liquid anion was a strongly basic amide (e.g., 2-cyanopyrrolide or 3-trifluoromethylpyrazolide) [17]. Under such conditions, it might reasonably be expected that a tetraalkylammonium cation would be unstable to deprotonation (recall that anhydrous Bu4N+F− is unstable with respect to elimination), but in these materials, even H/D exchange reactions were slow, and such exchange that did occur was exclusively at the hydrogens on the α-carbon. The liability of this hydrogen (compared to the β-hydrogen in alkylammonium cations) is related to the larger size of P compared to N, which then reflects a generally lower acidity for those hydrogens. Indeed, this last point is also shown by the chemical shift in the α-hydrogens, which is ca 3.7 ppm in [Bu4N]+ and 1.65 ppm in [Bu4P]+ [18].
2.3.2. Alkylammonium Cations—[Pr4N]2[PtCl6], [Bu4N]2[PtCl6] and [Pe4N]2[PtCl6]
Having determined that the Bu4P+ cation is unreactive under irradiation with [PtCl6]2−, the question arises concerning the reactivity of other tetraalkylammonium homologues, as it seems clear that the reactivity depends on the use of an ammonium derivative. To this end, [PtCl6]2− salts of both Pr4N+ and Pe4N+ (Pe = pentyl) have been prepared and studied.
Preparative Study: Both [Pr4N]2[PtCl6] and [Pe4N]2[PtCl6] are readily prepared from the chloride salt of the cation and K2[PtCl6] in water, being isolated in quantitative yield. While [Pe4N]2[PtCl6] showed high solubility in acetone in common with [Bu4N]2[PtCl6], the solubility of [Pr4N]2[PtCl6] was significantly lower, meaning that more dilute conditions were required in order to perform reactions with homogeneous solutions. Thus, in each case, an acetone solution of the salt was irradiated for 18 h, after which the acetone was removed, and the sticky solid that remained was treated with diethyl ether. However, in neither case was it possible to isolate a tractable product.
Solution NMR Studies: Given that no isolable product was obtained from irradiation of either [Pr4N]2[PtCl6] or [Pe4N]2[PtCl6], a solution NMR study was conducted on these two salts along with [Bu4N]2[PtCl6]. In each case, an anaerobic solution of the PtIV salt in an NMR tube in d6-acetone was irradiated, and NMR spectra were recorded either at intervals during the 18 h photolysis or at the end. Of necessity, for reasons of solubility, the concentrations of the solutions containing [Pr4N]2[PtCl6] were some ten times more dilute than those of the other salts. [Bu4N]2[PtCl6] and [Pe4N]2[PtCl6] are considered first and then compared with [Pr4N]2[PtCl6].
For both salts, following the reaction by 195Pt NMR spectroscopy (Figure 6) showed an immediate decrease in the intensity of the resonance associated with the [PtCl6]2− anion at δ = 377 ppm, which was accompanied by the formation of [PtCl4]2− at δ = −1420 ppm. All [PtCl6]2− was consumed by the end of the reaction. Trace amounts of [Pt2Cl6]2− (δ = −1164 ppm) were also observed, although this also tended to have disappeared by the end of the reaction. In addition to these chloroplatinate species, resonances were also observed in the region of the spectrum where Pt-alkene complexes are found, and it is interesting that their formation seemed to lag behind loss of PtIV and formation of PtII. Attempts to quantify these changes were frustrated by the differing T1 values for the different species.
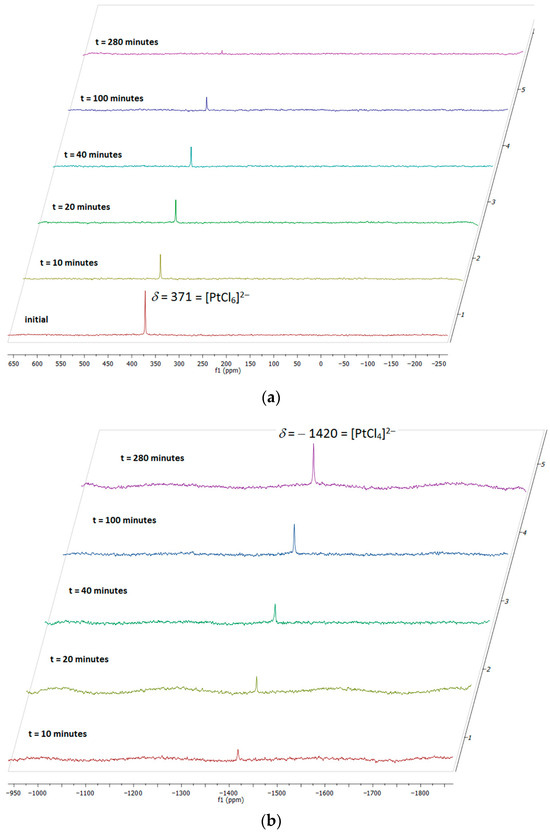
Figure 6.
Evolution of the 195Pt resonance from (a) [PtCl6]2− and (b) [PtCl4]2− on irradiation of [Bu4N]2[PtCl6] in acetone.
Then, to probe the organoplatinum complexes, 2D-(195Pt-1H) NMR spectra of the photoreaction mixture were obtained. After investigating a range of 10–135 Hz, a 195Pt-1H coupling constant of 60 Hz was found to give the best results in these experiments, and all data (Table 1) are collected using this value. Beginning with [Bu4N]2[PtCl6], the 2D-HMQC-195Pt-1H NMR spectrum of a photoreaction mixture obtained in acetone is presented in Figure S11. Four platinum resonances correlated to protons are observed, and two of the signals were also observed in the 1D-195Pt{1H} NMR spectrum. None of the signals corresponds to those of the isolated butadiene-platinum(II) complexes.

Table 1.
195Pt chemical shifts in the PtII–alkene region with correlated hydrogens observed in 2D-HMQC-195Pt-1H NMR spectra following photolysis of [Pr4N]2[PtCl6], [Bu4N]2[PtCl6] and [Pe4N]2[PtCl6] in acetone.
The signal at −2570 ppm is seen in the 1D-195Pt{1H} and in the 2D-195Pt-1H spectra and correlates with the hydrogen resonances at 3.4 and 1.56 ppm, noting that they could not be analysed in the 1D 1H spectrum as they were weak and poorly resolved. The signal at 3.4 ppm is likely related to −NCH2, indicating an olefin compound consisting of alkylammonium bound to a platinum(II) species, but the presence of 1H-195Pt coupling might suggest that this arises from an internal rather than a terminal alkene. In considering this possibility, the literature precedent for the formation of 3 is very strong, and so were an internal alkene to be formed, then it may well be via an isomerisation reaction. This is not at odds with the observation of the (Bu3N)(C4H7)PtIICl3 fragment found using photodissociation mass spectroscopy, as this technique would not discriminate between isomers. Of course, the possibility does exist that under the conditions employed, there is a pathway that would see C–H activation at other than the terminal carbon, which could in turn more easily lead to the formation of an internal alkene.
The remaining signals show 1H-195Pt coupling only into the coordinated alkene region (ca 4.0–4.5 ppm) of the 1H spectrum, with all Pt resonances consistent with coordinated alkene, but none corresponds with chemical shifts observed for either the 1-butene or 2-butene complex. One resonance could, however, correspond to the monoplatinated diene complex 5.
To see if there was a dynamic process, such as an exchange occurring during spectral acquisition, a variable-temperature 195Pt{1H} NMR experiment was conducted, recording in the Pt-olefin area between 200 and 298 K (Figure S12). As predicted [19,20], the chemical shifts decreased by 40 ppm when the temperature lowered from 298 K to 200 K. Moreover, the peaks were broader and less intense on cooling, but the pattern remained the same, implying an absence of a dynamic process; the broad peaks perhaps being attributed to a decrease in solubility at lower temperature. When the temperature increased back to 298 K, the spectrum reverted to the original intensities and resolution.
Now considering [Pe4N]2[PtCl6], the 195Pt{1H} NMR spectra (Figure S13) of the reaction mixture also showed a signal corresponding to [PtCl4]2− at −1425 ppm, indicating the reduction of [PtCl6]2− as found in the photolysis of [Bu4N]2[PtCl6]. In addition, the 1D-195Pt{1H}-NMR spectrum showed resonances associated with olefin-platinum(II) complexes at −2573 and −2569 ppm, while the 2D-(195Pt-1H) spectra (Figure S14) showed additional signals at −2559, −2555, −2543 and −2521 ppm. In common with the spectra obtained on irradiation of [Bu4N]2[PtCl6], one resonance (–2573 ppm) shows coupling between Pt and a signal at 3.4 ppm, which we have tentatively assigned as belonging to Pt bound to an internal alkene that remains bound to nitrogen. As for the other Pt signals, they are all coupled to 1H resonances in the range 4.0–4.5 ppm—indicative of coordinated alkene hydrogens—as well as a resonance at ca 1.6 ppm, which, by comparison with the spectra obtained for the complexes of 1-butene and 2-butene, could arise from either a CH2 or CH3 hydrogen adjacent to a coordinated double bond. Given the extra carbon in the chain in the [Pe4N]+ cation, then the possibility exists for more isomeric forms, either of a coordinated N-bound or free alkene, consistent with the similarities in the chemical shifts in the species observed after reaction of the two different cations. Indeed, the Supporting Information shows the results of quantum chemical calculations on the relative stability of the different possible dinuclear diene products.
[Pr4N]2[PtCl6] turned out to be much less soluble in acetone than the related Bu4N+ or Pe4N+ salts, so that only a fraction of the material was in solution at the beginning of the reaction. If, however, the photolysis was continued using what was in effect a solution and a suspension of the complex, then a homogeneous solution did, in due course, result. However, this led to rather long reaction times, and so experiments were instead conducted at concentrations some ten times lower. This presented challenges in terms of sensitivity in NMR experiments and, as a result, many signals present in the 1H NMR spectra were simply too weak to allow interpretation. Nonetheless, useful data could be obtained from both 1D 195Pt{1H} and 2D 195Pt–1H spectra.
Thus, post-irradiation 1D 195Pt{1H} spectra showed the presence of [PtCl4]2− (δ = −1425 ppm), an unidentified signal at −1631 ppm, and two signals in the PtII-alkene region (δ = −2430 and −2567 ppm). The 2D 195Pt–1H spectra (Figure S16) showed correlation of the resonance at −2567 ppm with 1H resonances in the region of coordinated alkene (δ ≈ 4.4 ppm), next to nitrogen (δ ≈ 3.4 ppm), as well as those from hydrogen atoms attached to a saturated carbon (δ ≈1.6 ppm). This implies the zwitterionic complex [Cl3Pt−(H3CCH=CHN+Pr3)] with internal alkene. This is consistent with the formation of a σ-alkyl complex analogous to 2, followed by β-hydrogen elimination to generate the analogue of 3.
The resonance at −2430 ppm correlated weakly with a 1H resonance at δ ≈ 5.7 ppm and again with one at δ ≈ 1.7 ppm; its origin is currently less clear.
2.4. EPR Study of Frozen Solutions
The EPR spectra of photolysed, glassy solutions of each of the three salts dissolved in CH2Cl2 at 120 K are given in Figure 7. The signals in the organic region with g = 2.013 and arising from a N-based cation are to all intents and purposes the same, while at lower field a signal is seen corresponding to paramagnetic platinum(III). For the Pe4N+ and Bu4N+ salts g = 2.402 with a(195Pt) = 1560 and 1590 MHz, while for the Pr4N+ salt, there are two signals with g = 2.393 and 2.463 and a(195Pt) = 1500 and 1430 MHz, respectively. The former would seem to be the same species as found for the other two salts, while the latter is new and of unclear origin.
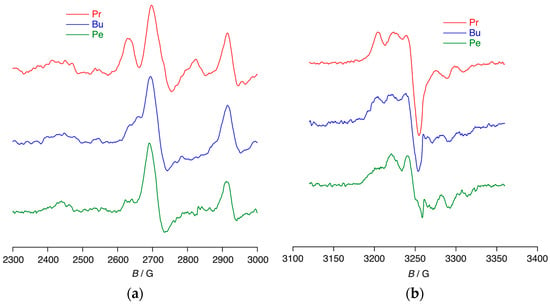
Figure 7.
The in situ EPR spectra of [Pr4N]2[PtCl6], [Bu4N]2[PtCl6], and [Pe4N]2[PtCl6] recorded in dichloromethane at 120 K, showing (a) platinum(III) radical and (b) organic N-based radical.
2.5. Molecular Structures of [PPN]2[PtCl4] and of [Pr4N]2[PtCl4]
Crystals of the PPN+ salt were obtained as an acetone solvate and were twinned, crystallising in the P–1 space group and refined to an R-factor of 5.3% (CCDC: 2477527), while those of the Pr4N+ salt in the monoclinic space group C2/c were refined to an R-factor of 3.1% and were solvent-free (CCDC: 2477528). In [PPN]2[PtCl4] there are two 180° trans-Cl–Pt–Cl angles and a very small deviation from orthogonality (cis-Cl–Pt–Cl angles = 89.79(4)° and 90.21(4)°), while for [Pr4N]2[PtCl4] the trans Cl–Pt–Cl angles are 180.0° and 179.61(4)° with cis angles of 89.805(19)° and 90.195(19)°. Curiously, while for [PPN]2[PtCl4] the Pt–Cl distances are very similar at 2.3096(11) Å and 2.3020(11) Å (symmetry-related pairs) and comparable with the three other structures of the [PtCl4]2− anion held in the Cambridge Crystallographic Database (846887, 1298726, 126477), [21,22,23] in [Pr4N]2[PtCl4] they are appreciably longer at 2.3459(10) Å, 2.3669(10) Å and 2.3211(8) Å (Cl1–Pt–Cl3 lies on a two-fold axis).
Implementing Olex2 [24], the structure was solved with the Superflip [25,26,27] structure solution program using Charge Flipping and refined with the ShelXL [28] refinement package using Least Squares minimisation. Crystallographic data are found in Table S1 in the Supporting Information.
In both structures, the [PtCl4]2− sits in a ‘pocket’ (Figure 8) defined by six cations (plus two molecules of acetone for [PPN]2[PtCl4]). A cation pocket in [Pr4N]2[PtCl4] is defined by three tetrachloroplatinate(II) anions, whereas for [PPN]2[PtCl4], two cations are located in a pocket formed of six tetrachloroplatinate(II) anions and four acetones of solvation. In both cases, there are contacts at ca 2.8 Å with hydrogen atoms of the respective cation.
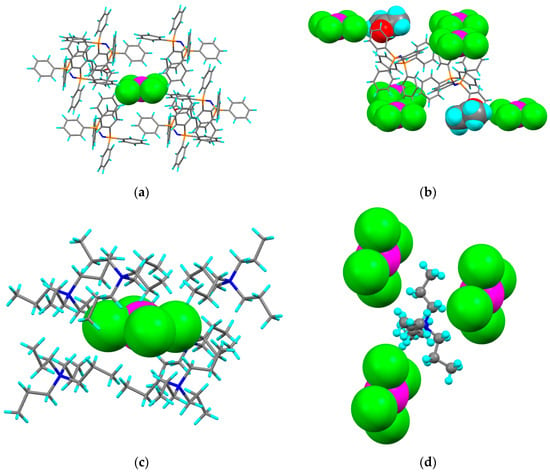
Figure 8.
Structure of: [PPN]2[PtCl4] showing (a) a [PtCl4]2− anion in a pocket of [PPN]+ cations and (b) two [PPN]+ cations in a pocket of [PtCl4]2− anions (showing acetone of solvation) and of [Pr4N]2[PtCl4] showing (c) a [PtCl4]2− anion in a pocket of [Pr4N]+ cations and (d) a [Pr4N]+ cation in a pocket of [PtCl4]2− anions.
3. Materials and Methods
3.1. Instrumentation
3.1.1. NMR Spectroscopy
1H NMR spectra were recorded using Jeol (Tokyo, Japan) ECS400 and Jeol ECX400 spectrometers operating at 400 MHz (1H). 1H (500 MHz), 31P{1H} (202.4 MHz), 2D NMR spectra (76.8 MHz) and 195Pt{1H} (107 MHz) spectra were recorded on Bruker (Billerica, MA, USA) Avance 500 spectrometer. Two-dimensional 195Pt-1H and 15N-1H NMR spectra were acquired via heteronuclear zero and double quantum coherence with decoupling during acquisition using gradient pulses for selection. The processing of all spectra was carried out using MestreNova software. For 1H-NMR, the residual protic solvent was used as the internal standard (CDCl3: 7.26 ppm, CD2Cl2: 5.32 ppm, CD3COCD3: 2.04 ppm), for the 195Pt-NMR spectra were referenced to K2[PtCl6] in D2O.
3.1.2. Electron Paramagnetic Resonance Spectroscopy
EPR spectroscopy was performed using a JEOL JES-RE1X-ESR spectrometer equipped with a 100 W Mercury lamps without a filter (λ > 250 nm). A sample of a solution of the analyte was loaded into a flat, quartz cell. A background spectrum was recorded at room temperature followed by the spectrum of sample at 120 K with a sweep time of 120 s and time constant of 0.3 s. At 176 K or above, no signals were observed. Before sample analysis, a background was recorded at an identical temperature over a wide range where the sweep width was 750 G and centre of the field was 3254 G. The internal standard was di(phenyl)(2,4,6-trinitrophenyl)iminoazanium (DPPH) with a g value of 2.0036. Parameters of the analysis and observed g value of samples are showed in Table S3. The spectra were processed using the EasySpin toolbox [29].
3.1.3. UV-Mass Spectrometry
Experiments were performed using a modified Bruker Esquire 6000 quadrupole ion trap mass spectrometer. The mass spectrometer was modified for the laser experiments by drilling holes (1 mm) in the ion trap ring electrode to allow laser access [13]. The source of UV photons for the laser spectroscopy experiments was from an Nd:YAG (Powerlite) pumped OPO (Panther Ex), producing ~2 mJ across the range 220–280 nm. (Bu4N)2[PtCl6] dissolved in water was prepared for the electrospraying.
3.2. 1H NMR Spectra from the Reaction of [PPN]2[PtCl6] with [PPN][PtCl3(1-butene)]− or [PPN][PtCl3(2-butene)]
To demonstrate the occurrence of the decomplexation, the mixture of the 1-butene-platinum(II) and hexachloroplatinate(IV) complexes in dichloromethane solution was irradiated in a Young’s NMR tube and the reaction was monitored using 1H NMR spectroscopy. The free 1-butene was removed using the freeze-pump-thaw method −116 °C, at which temperature, dichloromethane freezes whereas 1-butene is in the liquid phase. The 1H-NMR spectrum of the solution was then recorded and showed roughly about 50% reduction of the integration corresponding to the hydrogen signals of free 1-butene. The observation demonstrated the removal of 1-butene through freeze-pump-thaw and implied the decomplexation of 1-butene-platinum(II) to free 1-butene. On heating of the mixture of the [PtCl3(1-butene)]2− and [PtCl6]2− in dichloromethane at 40 °C for 48 h under the dark conditions, the decomplexation was absent implying the important role of light in the process. Note that (Figure S7) there is no change in the 1H NMR spectrum on simply applying vacuum in the absence of irradiation.
3.3. Quantum Chemical Calculations
In order better to understand the complexes that may result from the reactions using the tetrapentylammonium cation, DFT calculations were undertaken to find the conformers and isomers that may form preferentially. In principle, six such structures are possible (Figure S17), although as we have shown previously with closely related complexes of butadiene, not all of the possible structures exist separately.
The geometries were optimised at the M06/def2-TZVPP [30,31] level of theory with an effective core potentials (ECP) used for platinum atoms using Gaussian 16 package [32]. Calculated vibrational frequencies contained no imaginary values showing that each optimised geometry corresponds to a minimum on the respective potential energy surface. The calculated structures (S1, S3, S5 and S6) are shown in Figure S18 and the corresponding geometries with Cartesian coordinates of atomic positions are given in the separate coordinates file.
3.4. Synthesis
Salts with a tetrabutylammonium cation were prepared as described earlier.[2]
3.4.1. Tetrapropylammonium, Tetrapentylammonium and Tetrabutylphosphonium Hexachloroplatinate(IV)
These salts are prepared by stirring at room temperature and in the dark a suspension of potassium hexachloroplatinate (2.37 mmol) in acetone (15 cm3) and then adding the relevant cation as its chloride salt (4.74 mmol) as a solid in small batches. Once addition was complete, stirring continued overnight to afford an orange solution and a colourless precipitate. The solution was filtered in vacuo to remove the precipitate and then the solvent was removed resulting in an orange precipitate, which was dried in vacuo to give a crystalline orange solid. Isolated yields were effectively quantitative.
Tetrapropylammonium hexachloroplatinate(IV). Anal. calc. for C24H56N2Cl6Pt: Calc.: C = 36.93, H = 7.23, N = 3.59%. Found: C = 36.75, H = 7.20, N = 3.47%. 1H NMR, δ (400 MHz, d6-acetone): 1.02 (24 H, t, -CH3), 1.89 (16 H, m, -CH2-), 3.48 (16 H, m, -NCH2-). IR (νPt-Cl): 316 cm−1. 195Pt NMR: δ = 236 ppm.
Tetrapentylammonium hexachloroplatinate(IV). Anal. calc. for C40H88N2Cl6Pt: Calc.: C = 47.81, H = 8.83, N = 2.79%. Found: C = 47.57, H = 8.75, N = 2.71%. 1H NMR, δ (500 MHz, d6-acetone): 0.90 (24 H, t, -CH3), 1.39 (32 H, m, -CH2-CH2), 1.84 (16 H, m, -CH2), 3.49 (16 H, m, -NCH2-). 195Pt NMR: δ = 375 ppm.
Tetrabutylphosphonium Hexachloroplatinate(IV). Anal. calc. for C32H72P2Cl6Pt: Calc.: C = 41.48, H = 7.83%. Found: C = 41.92, H = 7.75%. 1H NMR, δ (500 MHz, d6-acetone): 0.95 (24 H, t, -CH3), 1.54 (16 H, m, -CH2), 1.66 (16 H, m, -CH2), 2.51 (16 H, m, -PCH2-). 195Pt{1H} NMR: δ = 373 ppm. 31P{1H} NMR: δ = 33.6 ppm.
3.4.2. Photolysis of Analogous Hexachloroplatinate(IV) Complexes
The method and equipment used for the irradiation of the analogous complexes was identical to the one employed to irradiate (NBu4)2[PtCl6] complex described previously. The photolysis experiments were carried out in Young’s NMR tubes for followed by 1H and 195Pt{1H} NMR spectroscopy. Quantities of the starting materials used are tabulated below (Table 2).
Table 2.
Amount of starting materials for the photolysis of analogous hexachloroplatinate(IV) complexes.
3.4.3. Irradiation of [PtCl3(butene)]− Complexes
The method and equipment used for the irradiation of the [PtCl3(butene)]− complexes were identical to those employed to irradiate (Bu4N)2[PtCl6] described previously. The photolysis experiments were carried out in Young’s NMR tubes and followed by 1H and 195Pt{1H} NMR spectroscopy. Quantities of the starting materials used are tabulated below (Table 3). Control reactions in the dark were also carried out.
Table 3.
Descriptions quantities of starting materials used and conditions of reactions.
4. Discussion and Conclusions
Key to this work is the photoactivity of [PtCl6]2−, which generates PtIII species and Cl• and leads to reduction to PtII as shown by studies in the 1970s [33,34]. Among more recent studies on the photochemistry of PtIV complexes by Gelbov and co-workers, [35,36,37,38] they reported studies of the stationary photolysis and flash photolysis of both [Bu4N]2[Pt(NO3)6] and [Bu4N]2[PtCl6] dissolved in acetonitrile or chloroform, respectively. In both cases and by analogy with the earlier work, PtIII is generated along with a radical derived from the ligand (NO3• or Cl•, respectively), ultimately generating PtII, yet despite both complexes having Bu4N+ as the counter cation, no activation was reported [39,40].
Therefore, to investigate this reactivity further and, in so doing, attempt to find further evidence supporting the proposed mechanism, photodissociation photodetachment mass spectrometry proved invaluable as it was able to identify solution species that had been proposed mechanistically, some of which were also identified by NMR spectroscopy.
Likewise, another crucial observation was that of the lack of reactivity of trichloroplatinate(II) complexes of 1-butene and 2-butene, which also provides support for the regiospecificity suggested by the mechanism.
Against these observations, perhaps the most intriguing aspect of the study centres around the photoreactivity of the tetraalkylammonium salts as studied by 1H and 195Pt NMR spectroscopy and by EPR spectroscopy. These studies showed that there is commonality between the signals observed on irradiation of the different salts and, where there are differences, then at a qualitative level they can be rationalised according to the different lengths of the alkyl chain bound to nitrogen.
Pulling all of these observations together allows for a more coherent understanding to emerge. Thus, the photodissociation mass spectrometry studies of [Bu4N]2[PtCl6] are consistent with EPR spectra in showing the presence of (photogenerated) platinum(III) in addition to platinum(II), and this is consistent with the known photochemistry of [PtCl6]2−. The mass spectrometry studies also show the presence of both unreacted platinum(IV) and of platinum(IV) σ-bound to the functionalised alkylammonium (2). Further, the results show the presence of zwitterionic η2-alkene complexes akin to 3 or an isomer thereof.
Curiously, complex 2 is not seen in any of the NMR spectra (signal would be expected at ca −700 ppm in the 195Pt spectrum [41]), but resonances in the alkene region of the spectra viewed in conjunction with 2D 1H-195Pt data are consistent with Pt-alkene complexes. Furthermore, for each salt studied, there is a resonance that shows evidence of Pt coupling to a methylene hydrogen that is bound to nitrogen (δ ≈ 3.4 ppm in the 1H spectra), which suggests a Pt complex of an isomer of 3 (or related species where the cation is [Pr4N]+ or [Pe4N]+). Other resonances are consistent with coordination to an alkene as found in 3, although it is not possible unequivocally to rule out that one or more species arise from complexation to free alkene. Another important observation is that there are more Pt-alkene species as the cation chain length increases from Pr to Pe, which speaks both to the possibility of the internal alkene isomerisation mentioned, plus the possibility of ‘free alkene’ complexes where the hydrocarbon fragment has broken its link to the nitrogen centre. Therefore, while unable to provide support for each step individually, when taken together, the data above are entirely consistent with the proposed mechanism.
However, one rather key observation is absent. Thus, where the starting material is [Bu4N]2[PtCl6], chemical shift and coupling constant data for the isolated butadiene-containing product (1, Figure 1), including its isomeric forms, have been recorded and reported previously [3]. However, in none of the post-irradiation spectra obtained using [Bu4N]2[PtCl6] is there any evidence of the product having formed and simply waiting to be precipitated out of solution. Indeed, this is true for experiments conducted by photolysis in NMR tubes, photolysis experiments conducted on a more preparative scale in larger photolysis tubes, and even the very early experiments conducted in more conventional glassware with adventitious ambient illumination and stirring, both with and without heating. It seems, therefore, that on the addition of diethyl ether to post-reaction solutions using [Bu4N]2[PtCl6] as starting material, the product forms. This is not to imply chemical reactivity on the part of the ether anti-solvent, rather simply to observe that its addition and the presence of the different species in solution lead to the formation of the bimetallic butadiene complex. It is also important to recall here that isolated yields of the product are ca 70%, so that this is not a minor side-product of some other reaction. What this suggests is that, at least for [Bu4N]2[PtCl6], whatever is present in solution, the bimetallic product complex 1 represents a thermodynamic minimum and so during crystallisation, this product forms preferentially from the solution species.
Supplementary Materials
Supporting information can be downloaded at https://www.mdpi.com/article/10.3390/inorganics13110362/s1. Figure S1: The positive-ESI-mass spectrum of [Bu4N]2[PtCl6] photodissociation showing the precursor ion at m/z 1135 associated with [Bu4N]3[PtCl6]+ ion and a fragment at m/z 242 assigned for (Bu4N)+ ion; Figure S2: The negative-ESI-mass spectrum of [Bu4N]2[PtCl6] showing the parent ion at m/z 926 associated with [Bu4N]2[PtCl7]− with main fragments at m/z 649 assigned for [Bu4N] [PtCl6]− along with other fragments at m/z 684 associated with [Bu4N] [PtCl7]−, m/z 579 assigned for [Bu4N] [PtCl4]−, m/z 312 associated with [Bu4NCl2]− and m/z 300 assigned for [PtCl3]−; Figure S3: The positive ion UV photodepletion spectra of [Bu4N]3[PtCl6]+ ion showing signals at m/z 520 corresponding to [(Bu4N)2Cl]+, m/z 1061 associated with [(Bu4N)2Cl[(nBu3N)(C4H7)PtIICl3]]+, m/z 1065 assigned for [(Bu4N)3PtIICl4]+ and m/z 1100 assigned for [(Bu4N)3PtIIICl5]+; Figure S4: The negative ion UV photodepletion spectra of [Bu4N.PtCl6]− ion showing signals at m/z 650 assigned for [Bu4N.PtIVCl6]− and m/z 337 assigned for [PtIIICl4]−, m/z 372 associated with [PtIVCl5]− and m/z 300 assigned for [PtIICl3]−; Figure S5: The positive ion UV photodepletion spectra of [Bu4N]3[PtCl6]+ ion in low mass showing signals at m/z 186 assigned for [Bu3NH]+ and m/z 242 assigned for [Bu4N]+; Figure S6: The 1H NMR spectra (500 MHz) of the mixture of [PPN][PtCl3(1-butene)], and [PPN]2[PtCl6] in CD2Cl2 on the irradiation under UV light (signals at ≈2 ppm and ≈2.2 ppm from water and acetone, respectively, from the work up; Figure S7: The 1H NMR spectra (500 MHz) of the mixture of [PPN][PtCl3(1-butene)], and [PPN]2[PtCl6] in CD2Cl2 under dark conditions; Figure S8: The 1H NMR spectra (500 MHz) of the mixture of [PPN][PtCl3(2-butene)], and [PPN]2[PtCl6] in CD2Cl2 during the irradiation. The assignment of free 2-butene was made by comparing the signals to the proton signal of 2-butene from a solution that was prepared separately; Figure S9: The 1H NMR spectra (500 MHz) of the mixture of [PPN][PtCl3(2-butene)], and [PPN]2[PtCl6] in CD2Cl2 after 60 min irradiation and after it stored in the dark showing re-complexation and reduction in intensity of the signals from free 2-butene; Figure S10: The 31P{1H} NMR spectrum of (PBu4)2[PtCl6] recorded in d6-acetone recorded before (lower) and after (upper) irradiation; Figure S11: Left (for comparison purposes): the 2D-(1H-195Pt)-NMR spectra (500 MHz for 1H) of the reaction mixture arising from a solution of [Bu4N]2[PtCl6] heated under reflux in acetone in ambient light recorded in d6-acetone at 298 K; right: the 1D-195Pt{1H} NMR spectrum showing resonances at −2570 and −2567 ppm; Figure S12: The low-VT 195Pt{1H} NMR spectra (107 MHz) of the mixture of [Bu4N]2[PtCl6] in d6-acetone after photolysis; Figure S13: The 1D 195Pt NMR spectra (107 MHz) of the photoreaction mixture of [Pe4N]2[PtCl6] in acetone recorded in d6-acetone at 298 K; Figure S14: The 2D-(195Pt-1H) NMR spectra (500 MHz for 1H) of the photoreaction mixture of [Pe4N]2[PtCl6] in acetone recorded in d6-acetone at 298 K; Figure S15: The 1D 195Pt{1H} NMR spectra (107 MHz) of the mixture of [Pr4N]2[PtCl6] photolysis in acetone recorded in d6-acetone at 298 K. (cross: unknown signal; asterix: olefin-Pt(II) signals); Figure S16: The 2D 195Pt-1H NMR spectra (500 MHz for 1H) of the mixture of photolysis of [Pr4N]2[PtCl6] in acetone recorded in d6-acetone at 298 K; Table S1: Crystallographic Data for [PPN]2[PtCl4] and [NPr4]2[PtCl4]; Figure S17: Theoretically possible conformers of pentadiene complexed by two trichloroplatinate(II) groups; Figure S18: The optimised structures of dinuclear bis{trichloroplatinate(II)} complexes of 1,3-pentadiene (S1 and S3) and 1,4-pentadiene (S5 and S6); Table S2: The Gibbs energies (298.15 K, 1 atm.) of conformers of pentadiene-platinum(II) complexes and relative values compared to structure 1; Table S3: Parameters of EPR experiments and observed g value in vary of samples.
Author Contributions
Conceptualisation, D.W.B.; methodology, D.W.B., M.Z.S., V.C. and C.E.H.D.; formal analysis; investigation, I.H.S., M.Z.S., A.S. and P.G.; data curation, I.H.S. and D.W.B.; writing—original draft preparation, I.H.S.; writing—review and editing, D.W.B., I.H.S., V.C. and C.E.H.D.; supervision, D.W.B., V.C. and C.E.H.D. A.C.W. collected the crystallographic data and solved and refined the structures. Experimental work was carried out by I.H.S., P.G. and A.S. Data were interpreted by I.H.S., P.G., A.S., C.E.H.D., V.C. and D.W.B. The manuscript was drafted by I.H.S. and then edited by D.W.B. with contributions from V.C. and C.E.H.D. Funding was obtained by I.H.S., V.C. and C.E.H.D. The project was conceived by D.W.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Indonesian Ministry of Education, Directorate General of Higher Education, the Department of Chemistry, Procter & Gamble, European Research Council (208589-BIOIONS).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are available in the Supporting Information and at https://doi.org/10.15124/fa0465d2-72a9-4af0-9dca-40d9e5184b96.
Acknowledgments
Support is gratefully acknowledged from the Indonesian Ministry of Education Directorate General of Higher Education (DIKTI) (scholarship to I.H.S.); Procter & Gamble and the Department of Chemistry, University of York (scholarship to P.G.); European Research Council (support to A.S. and C.E.H.D.).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Sethi, N.K.; Whitwood, A.C.; Bruce, D.W. Preparation of a Heterodimetallic Di-μ-Chlorido Complex of Palladium and Platinum. Eur. J. Inorg. Chem. 2013, 2013, 2078–2082. [Google Scholar] [CrossRef]
- Silalahi, I.H.; Sethi, N.K.; Shafikov, M.Z.; Chechik, V.; Whitwood, A.C.; Bruce, D.W. Unexpected, Photochemically Induced Activation of the Tetrabutylammonium Cation by Hexachloroplatinate(Iv). Chem. Commun. 2018, 54, 13682–13685. [Google Scholar] [CrossRef] [PubMed]
- Silalahi, I.H.; Heyam, A.P.; Goult, C.A.; Aguiar, P.; Karadakov, P.B.; Bruce, D.W. Nuclear Magnetic Resonance and Computational Study of Trans-(m2:h2,h2-1,3-butadiene)bis(trichloroplatinate(II)). Organometallics 2020, 39, 4723–4734. [Google Scholar] [CrossRef]
- Shulpin, G.B.; Nizova, G.V.; Shilov, A.E. Photoinduced reactions of PtCl62− with saturated hydrocarbons and other C–H containing compounds. J. Chem. Soc. Chem. Commun. 1983, 671–672. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nizova, G.V.; Lederer, P. Photoinduced reactions of organic compounds with transition metal complexes: IV. Thermal and photochemical reactions of PtCl62−- and PtCl42− with olefins and alcohols: Convenient synthesis of π-olefin complexes of platinum(II). J. Organomet. Chem. 1984, 275, 283–294. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nizova, G.V.; Kitaigorodskii, A.N.; Serdobov, M.V. Photoinduced reactions of organic compounds with transition metal complexes: III. Formation of a σ-methyl complex of platinum(IV) and a π-ethylene complex of platinum(II) in photochemical and thermal reactions of PtCl62− with methyl and ethyl derivatives of tin or germanium. thermal oxidative addition of Me4Sn to PtCl42− to afford the σ-methyl complex of platinum(IV). J. Organomet. Chem. 1984, 275, 273–282. [Google Scholar] [CrossRef]
- Kitaigorodskii, A.N.; Nekipelov, V.M.; Nikitaev, A.T.; Shul’pin, G.B. The reaction of PtCl62− with aromatic compounds to afford anionic σ-aryl complexes of platinum(IV). J. Organomet. Chem. 1984, 275, 295–301. [Google Scholar] [CrossRef]
- Nizova, G.V.; Serdobov, M.V.; Nikitaev, A.T.; Shul’pin, G.B. Photoinduced reactions of organic compounds with transition metal complexes: II. Reaction of PtCl62− with acetone to give a σ-acetonyl complex of platinum(IV). Detection of platinum(III) compounds by ESR. J. Organomet. Chem. 1984, 275, 139–144. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nizova, G.V. The reaction of PtCl62− with aromatic compounds to afford anionic σ-aryl complexes of platinum(IV): VII. Cleavage of aryl-element bonds (element = Hg, Sn, Pb or B). Arylation reactions of arenes and olefins with α-aryl complexes of platinum(IV) J. Organomet. Chem. 1984, 276, 109–114. [Google Scholar] [CrossRef]
- Serdobov, M.V.; Nizova, G.V.; Shulpin, G.B. Formation of organometallic complexes of platinum(II) and platinum(IV) in reactions of PtCl62− with alkanes, olefins and aromatics induced by γ-irradiation. J. Organomet. Chem. 1984, 265, C12–C14. [Google Scholar] [CrossRef]
- Nizova, G.V.; Shul’pin, G.B. The formation of s-arylplatinum(IV) complexes in the photochemical reaction of the hexachloroplatinate (PtCl62−) ion with arenes in methylene chloride (). Organomet. Chem. USSR 1990, 3, 231–232. [Google Scholar]
- Shul’pin, G.B. C–H functionalization: Thoroughly tuning ligands at a metal ion, a chemist can greatly enhance catalyst’s activity and selectivity. Dalton Trans. 2013, 42, 12794–12818. [Google Scholar] [CrossRef]
- Matthews, E.; Sen, A.; Yoshikawa, N.; Bergström, E.; Dessent, C.E.H. UV Laser Photoactivation of Hexachloroplatinate Bound to Individual Nucleobases in Vacuo as Molecular Level Probes of a Model Photopharmaceutical. Phys. Chem. Chem. Phys. 2016, 18, 15143–15152. [Google Scholar] [CrossRef]
- Kaufman, S.H.; Weber, J.M.; Pernpointner, M. Electronic Structure and UV Spectrum of Hexachloroplatinate Dianions in Vacuo. J. Chem. Phys. 2013, 139, 194310. [Google Scholar] [CrossRef]
- Cercola, R.; Wong, N.G.K.; Rhodes, C.; Olijnyk, L.; Mistry, N.S.; Hall, L.M.; Berenbeim, J.A.; Lynam, J.M.; Dessent, C.E.H. A ‘One Pot’ Mass Spectrometry Technique for Characterizing Solution- and Gas-Phase Photochemical Reactions by Electrospray Mass Spectrometry. RSC Adv. 2021, 11, 19500–19507. [Google Scholar] [CrossRef] [PubMed]
- Matni, A.; Boubekeur, L.; Mezailles, N.; Le Floch, P.; Geoffroy, M. Oxidation Products of Iminophosphorane and Bis-Iminophosphorane: An EPR Study. Chem. Phys. Lett. 2005, 411, 23–27. [Google Scholar] [CrossRef]
- Oh, S.; Morales-Collazo, O.; Keller, A.N.; Brennecke, J.F. Cation-Anion and Anion-CO2 Interactions in Triethyl(Octyl)Phosphonium Ionic Liquids with Aprotic Heterocyclic Anions (AHAs). J. Phys. Chem. B 2020, 124, 8877–8887. [Google Scholar] [CrossRef] [PubMed]
- Amir, H.; Kar, M.; O’Dell, L.A.; Forsyth, M.; Swadźba-Kwaśny, M.; Holbrey, J.D. New Tetrabutylphosphonium Organic Ionic Plastic Crystals Incorporating Borate Anions. J. Mater. Chem. A 2025, 13, 18842–18850. [Google Scholar] [CrossRef]
- Pregosin, P.S. Platinum-195 Nuclear Magnetic Resonance. Coord. Chem. Rev. 1982, 44, 247–291. [Google Scholar] [CrossRef]
- Goggin, P.L.; Goodfellow, R.J.; Haddock, S.R.; Taylor, B.F.; Marshall, I.R.H. The Platinum-195 Chemical Shift in Some Platinum(0) and Platinum(II) Complexes and Its Relationship to Their Structure. J. Chem. Soc. Dalton Trans. 1976, 459–467. [Google Scholar] [CrossRef]
- Clever, G.H.; Kawamura, W.; Tashiro, S.; Shiro, M.; Shionoya, M. Stacked Platinum Complexes of the Magnus’ Salt Type Inside a Coordination Cage. Angew. Chem. Int. Ed. 2012, 51, 2606–2609. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Tanaka, Y.; Tsubomura, T. [N,N’-Bis(2-Aminoethyl-kN)-N,N’-Bis(2-Ammonioethyl)Ethylenediamine-k2N,N’]Platinum(II) Bis(Tetrachloroplatinate) Trihydrate. Acta Cryst. C 1996, 52, 541–543. [Google Scholar] [CrossRef]
- Davies, M.S.; Fenton, R.R.; Huq, F.; Ling, E.C.H.; Hambley, T.W. Studies on the Nature and Strength of Pt...H(-N) Interactions. The Crystal Structures of Chloro[N-(2-Aminoethyl)-N-(2-Ammonioethyl)Ethane-1,2-Diamine]Platinum(II) Chloride and Dichloro [4,7-Diaza-1-Azoniacyclo. Aust. J. Chem. 2000, 53, 451–456. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Palatinus, L.; van der Lee, A. Symmetry Determination Following Structure Solution in P1. J. Appl. Crystallogr. 2008, 41, 975–984. [Google Scholar] [CrossRef]
- Palatinus, L.; Prathapa, S.J.; van Smaalen, S. EDMA: A Computer Program for Topological Analysis of Discrete Electron Densities. J. Appl. Crystallogr. 2012, 45, 575–580. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Hellweg, A.; Rappoport, D. Development of new auxiliary basis functions of the Karlsruhe segmented contracted basis sets including diffuse basis functions (def2-SVPD, def2-TZVPPD, and def2-QVPPD) for RI-MP2 and RI-CC calculations. Phys. Chem. Chem. Phys. 2014, 17, 1010–1017. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Cox, L.E.; Peters, D.G.; Wehry, E.L. Photoaquation of hexachloroplatinate(IV). J. Inorg. Nucl. Chem. Lett. 1972, 34, 297–305. [Google Scholar] [CrossRef]
- Wright, R.C.; Laurence, G.S. Production of platinum(III) by flash photolysis of PtCl62−. J. Chem. Soc. Chem. Commun. 1972, 132–133. [Google Scholar] [CrossRef]
- Pozdnyakov, I.P.; Glebov, E.M.; Matveeva, S.G.; Plyusnin, V.F.; Mel’nikov, A.A.; Chekalin, S.V. Primary photophysical and photochemical processes upon UV excitation of PtBr62− and PtCl62− complexes in water and methanol. Russ. Chem. Bull. 2015, 64, 1784–1795. [Google Scholar] [CrossRef]
- Glebov, E.M.; Plyusnon, V.F.; Grivin, V.P.; Venediktov, A.B.; Korenev, S.V. Photochemistry of PtBr62− in aqueous solution. Russ. Chem. Bull. 2007, 56, 2357–2363. [Google Scholar] [CrossRef]
- Glebov, E.M.; Kolomeets, A.V.; Pozdnyakov, I.P.; Grivin, V.P.; Plyusnon, V.F.; Tkachenko, N.V.; Lemmetyinen, H. Chain processes in the photochemistry of PtIV halide complexes in aqueous solutions. Russ. Chem. Bull. 2013, 62, 1540–1548. [Google Scholar] [CrossRef]
- Glebov, E.M.; Plyusnon, V.F. On the Cause of Solvent Effect in PtIVCl62− Photochemistry. High Energy Chem. 2021, 55, 203–211. [Google Scholar] [CrossRef]
- Matveeva, S.G.; Grivin, V.P.; Plyusnin, V.F.; Vasilchenko, D.B.; Glebov, E.M. Mechanism of chain photochemical reaction of (n-Bu4N)2[PtCl6] in chloroform. J. Photochem. Photobiol. A 2018, 359, 80–86. [Google Scholar] [CrossRef]
- Grivin, V.P.; Matveeva, S.G.; Fedunov, R.G.; Yanshole, V.V.; Vasilchenko, D.B.; Glebov, E.M. Photochemistry of (n-Bu4N)2[Pt(NO3)6] in acetonitrile. Photochem. Photobiol. Sci. 2024, 23, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Luinstra, G.A.; Wang, L.; Stahl, S.S.; Labinger, J.A.; Bercaw, J.E. C–H Activation by Aqueous Platinum Complexes: A Mechanistic Study. J. Organomet. Chem. 1995, 504, 75–91. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).