

Homogeneous Metal-Catalyzed Hydrogenation of CO2 Derivatives: Towards Indirect Conversion of CO2 to Methanol


Abstract
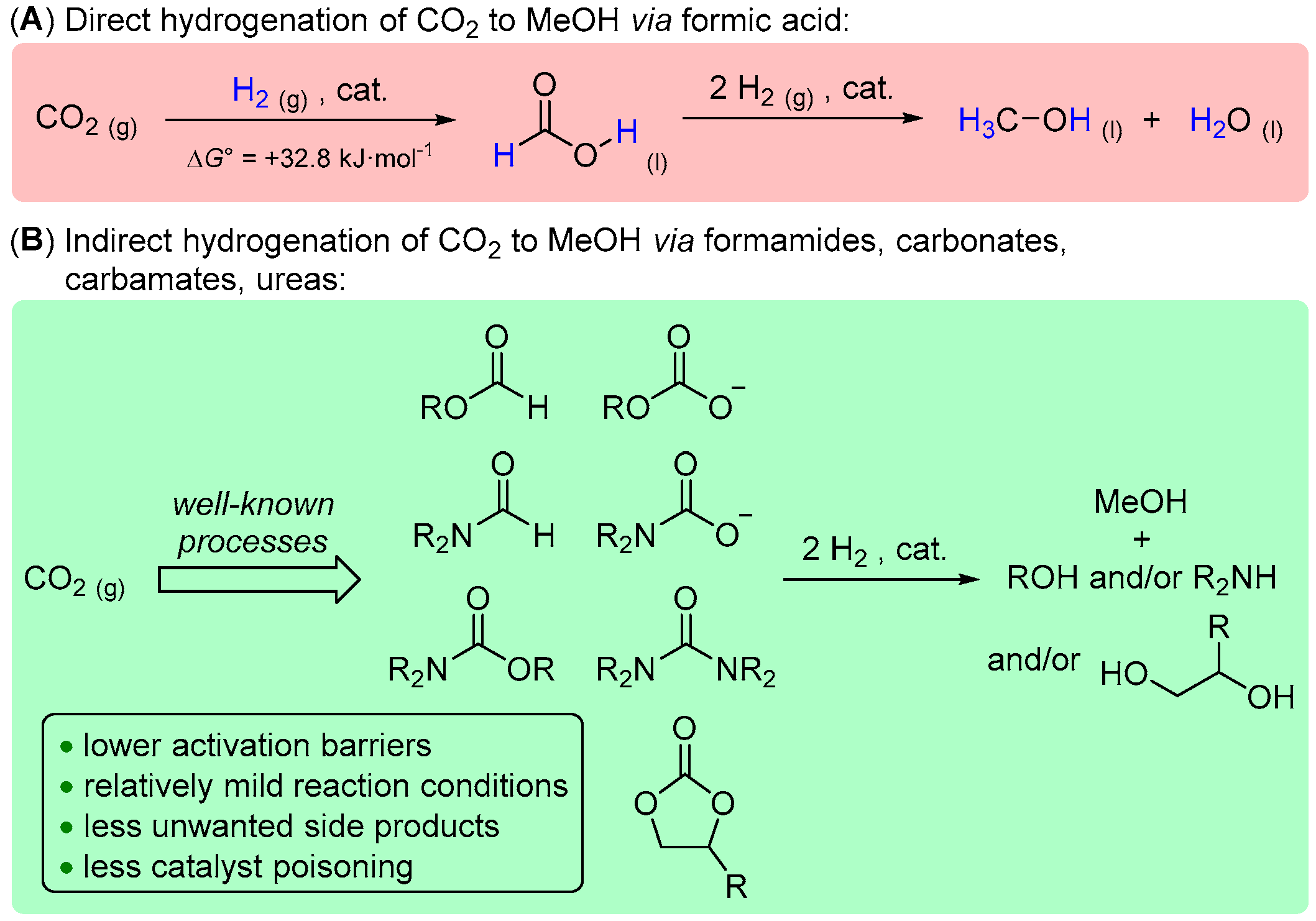
1. Introduction
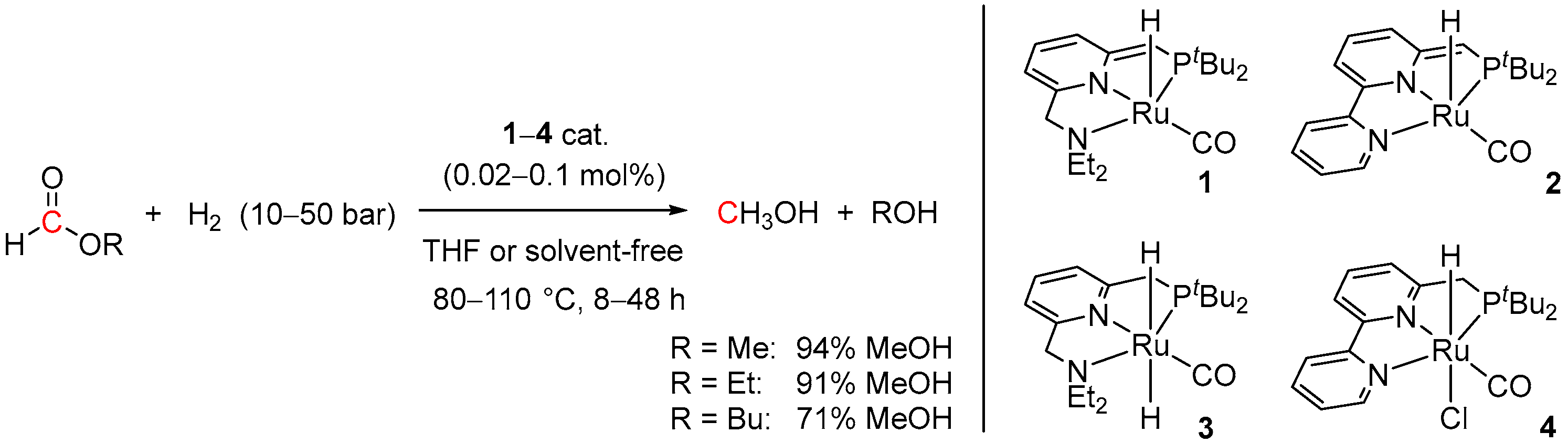
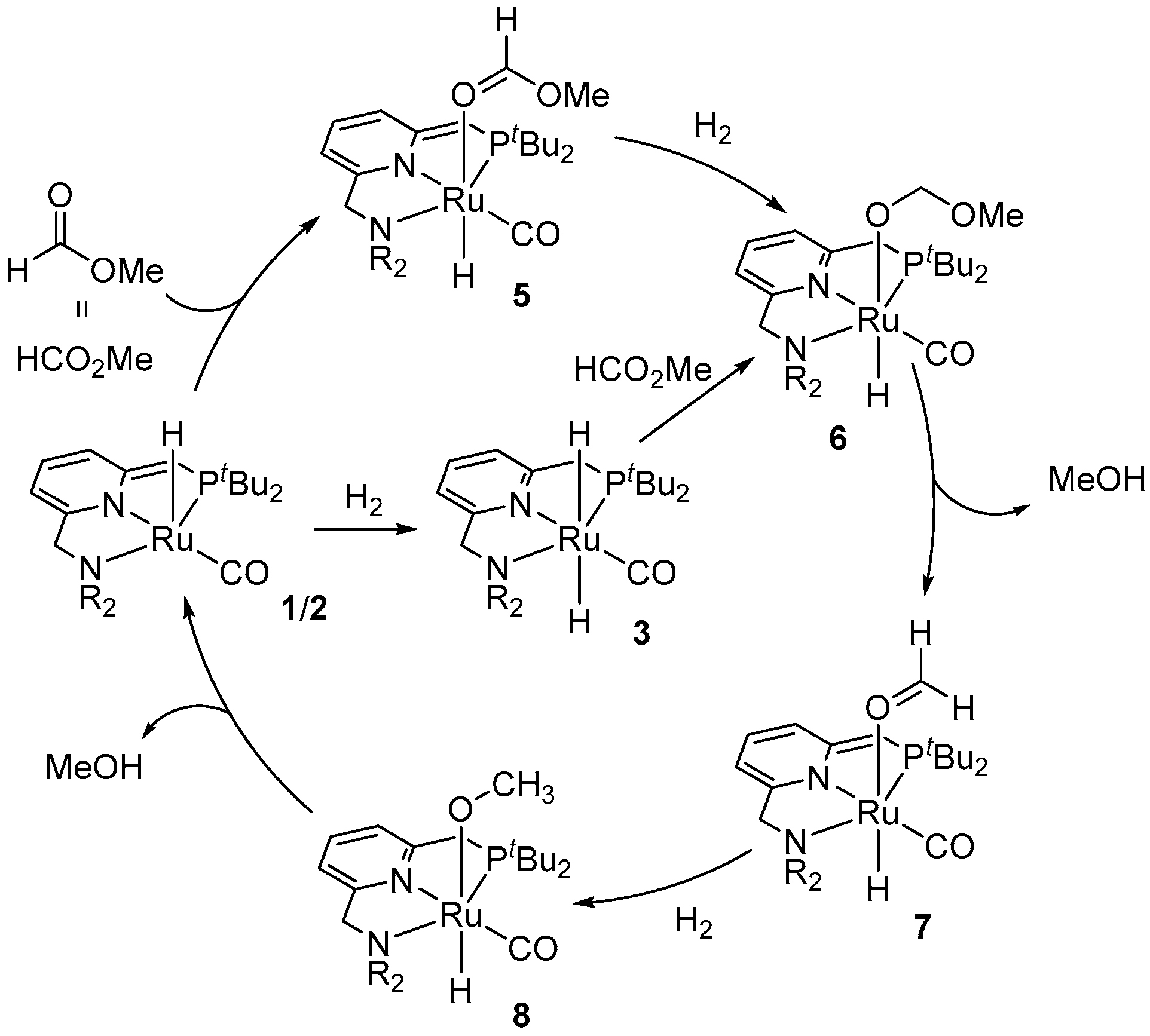
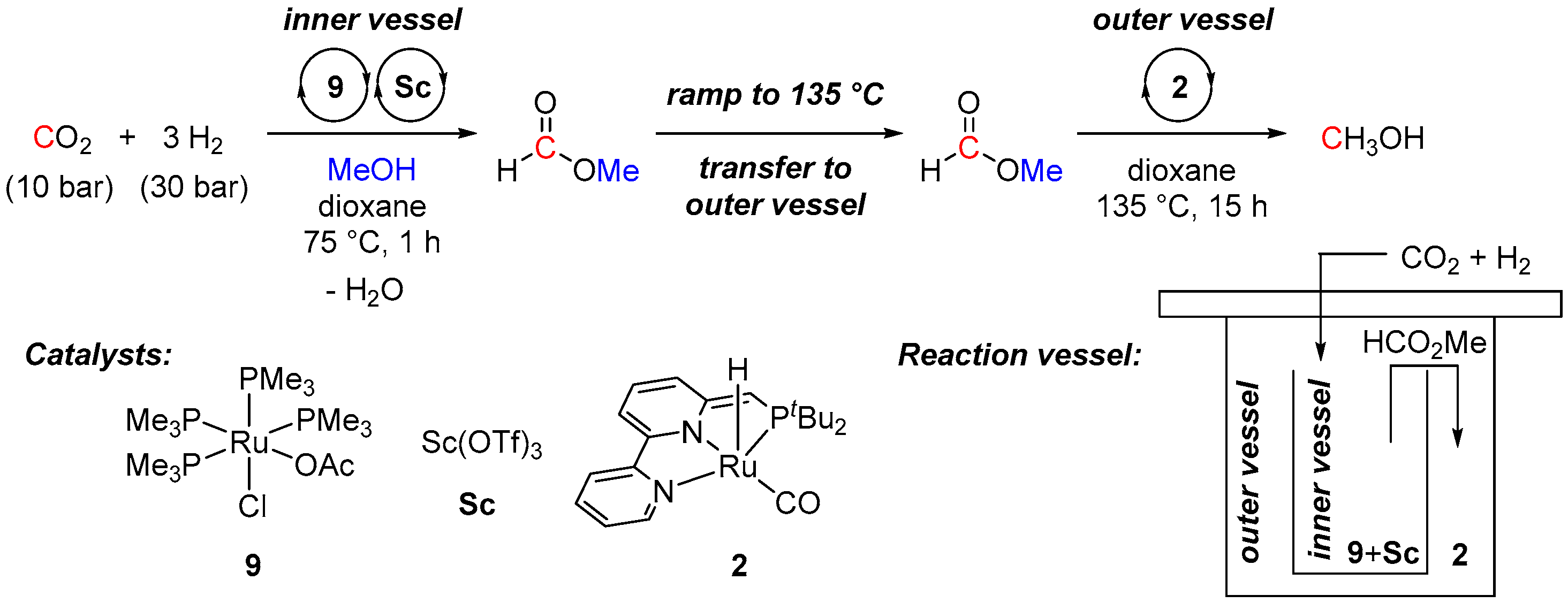
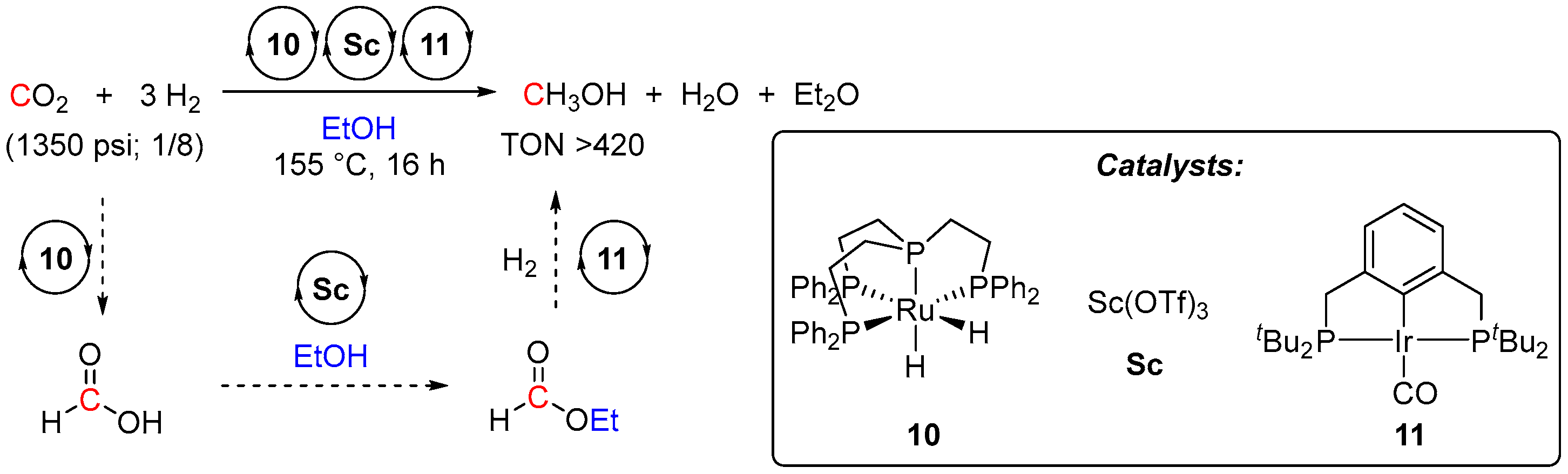
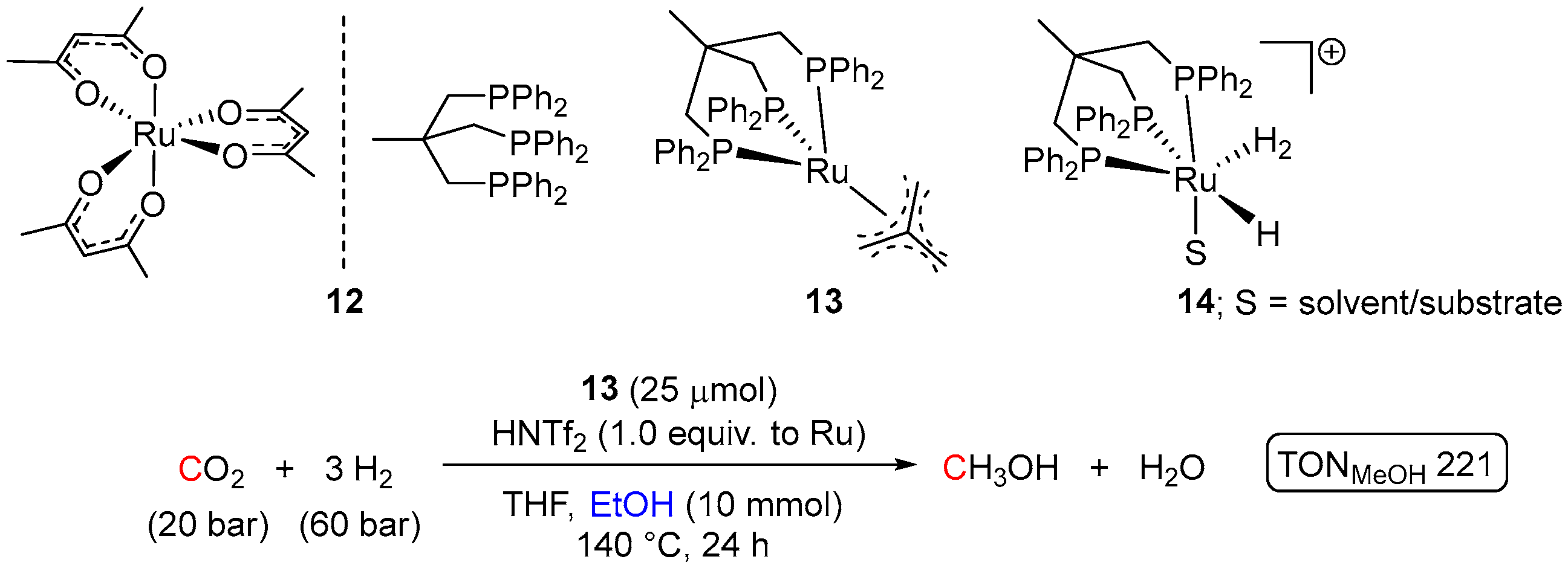
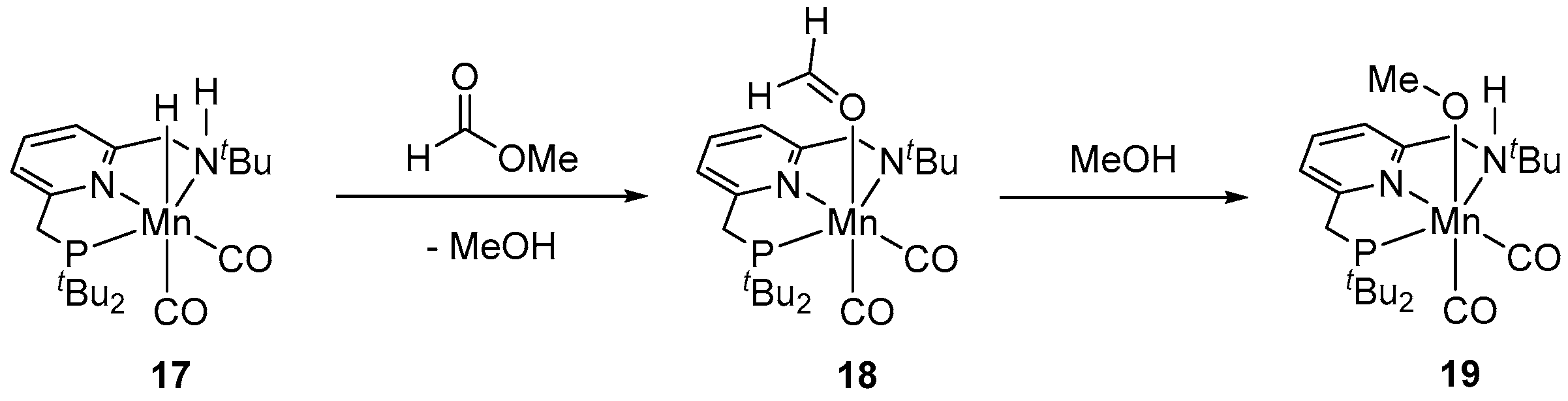
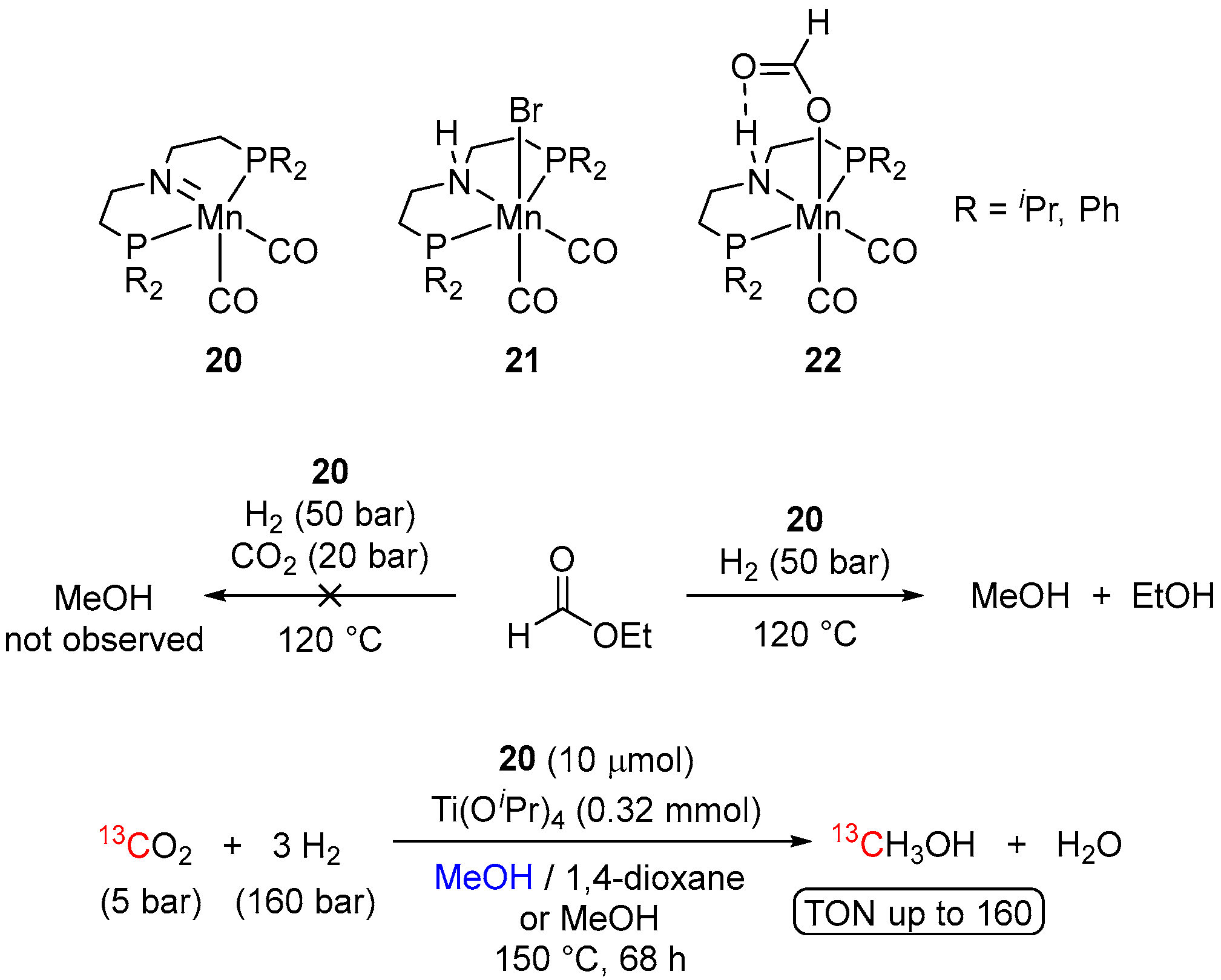
2. Catalytic Hydrogenation of Formates
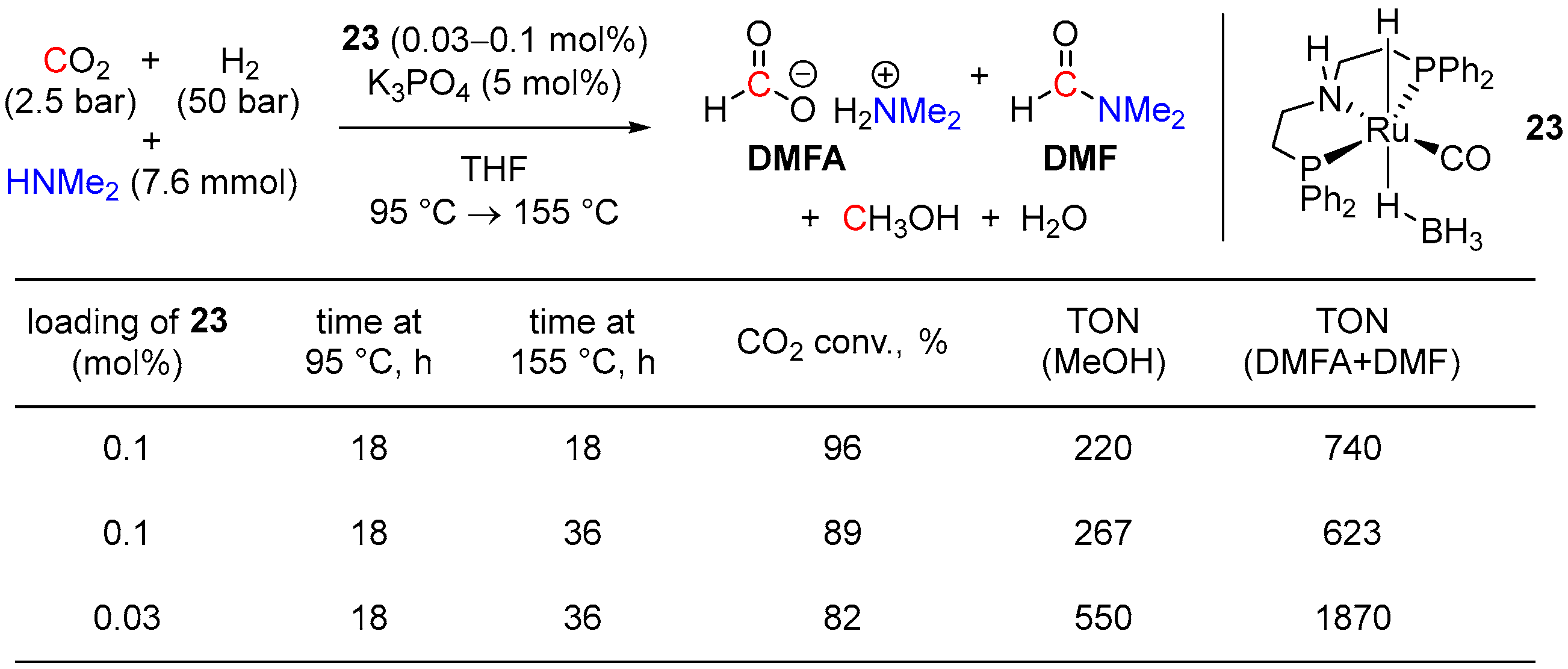
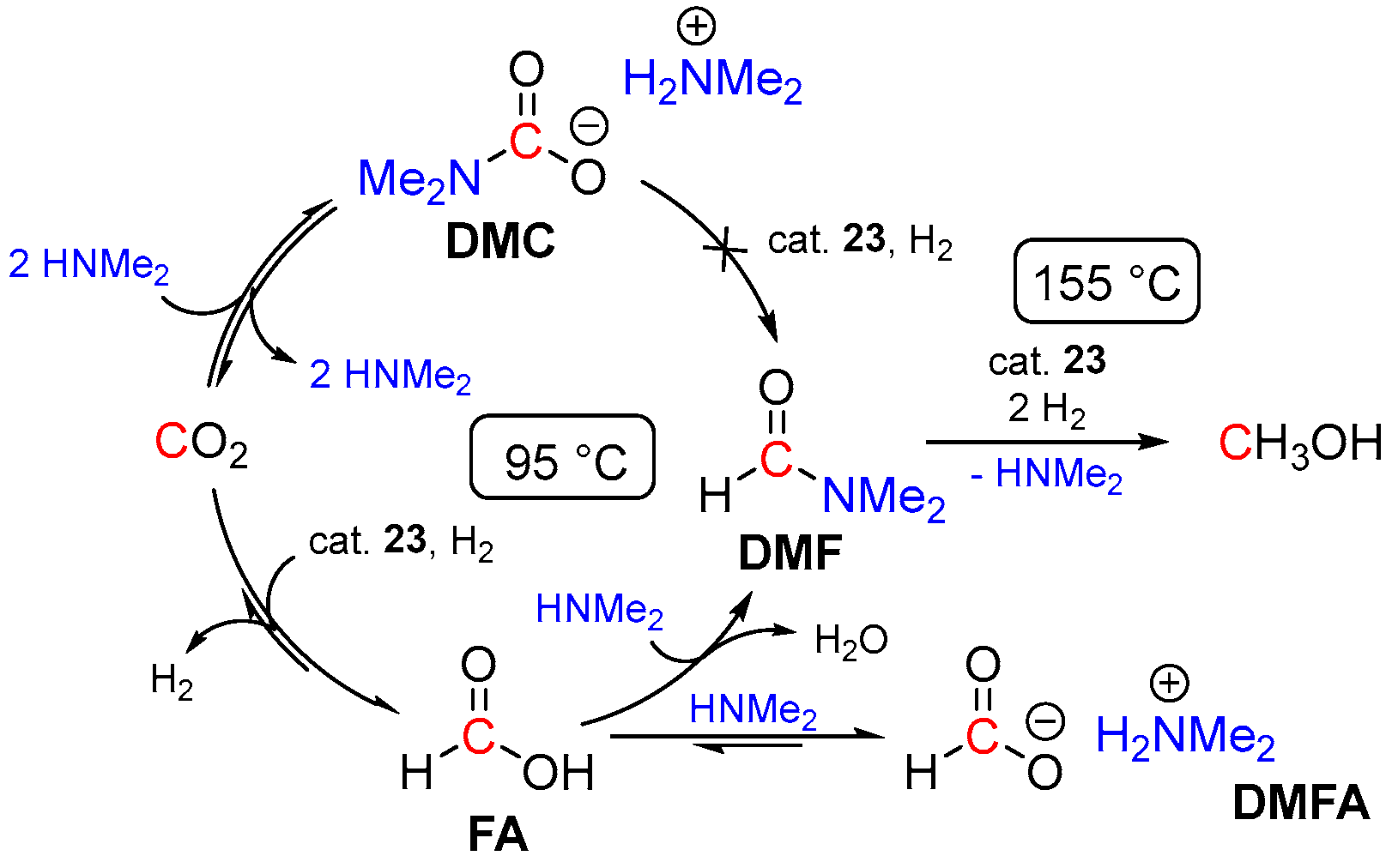
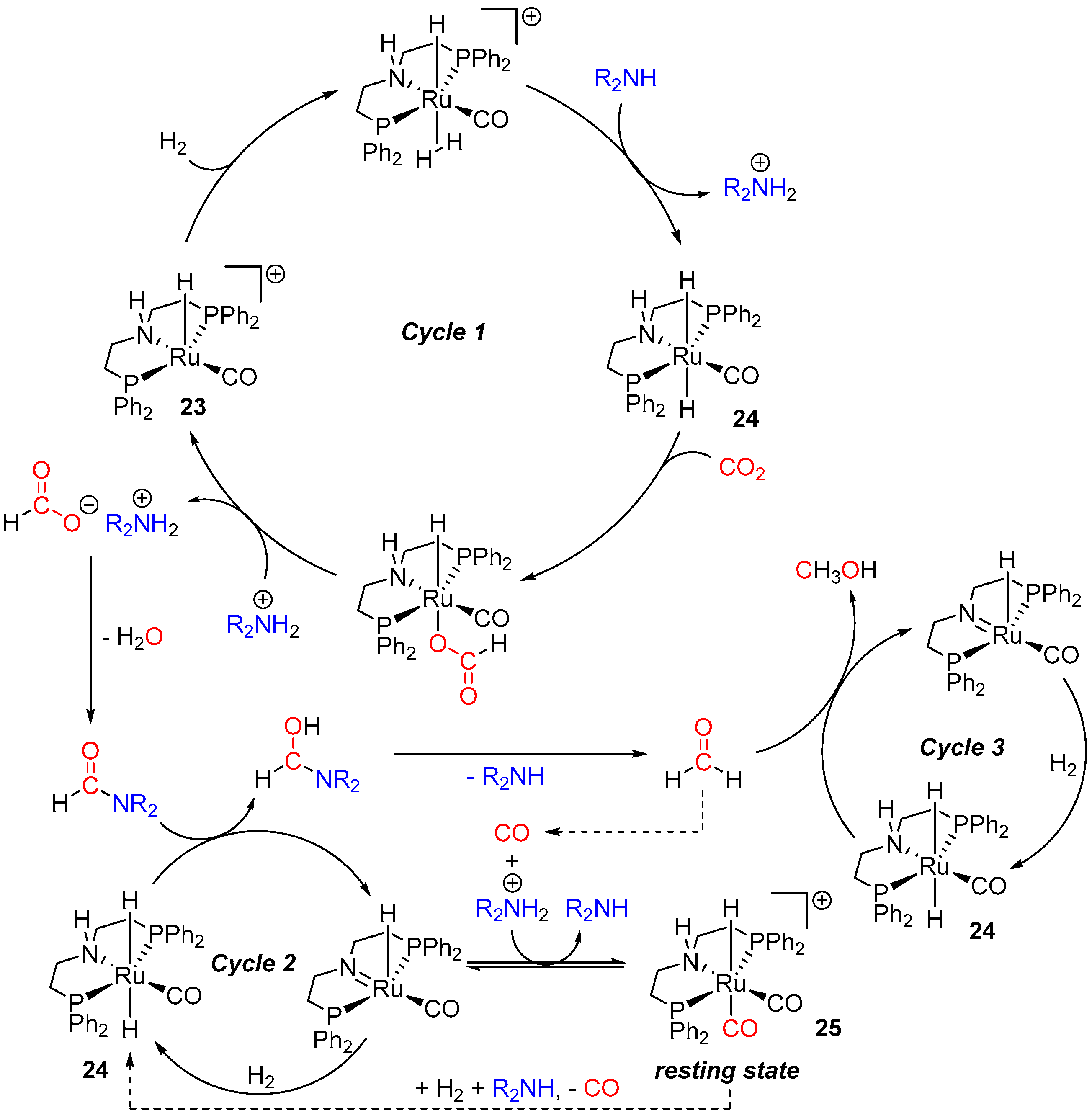
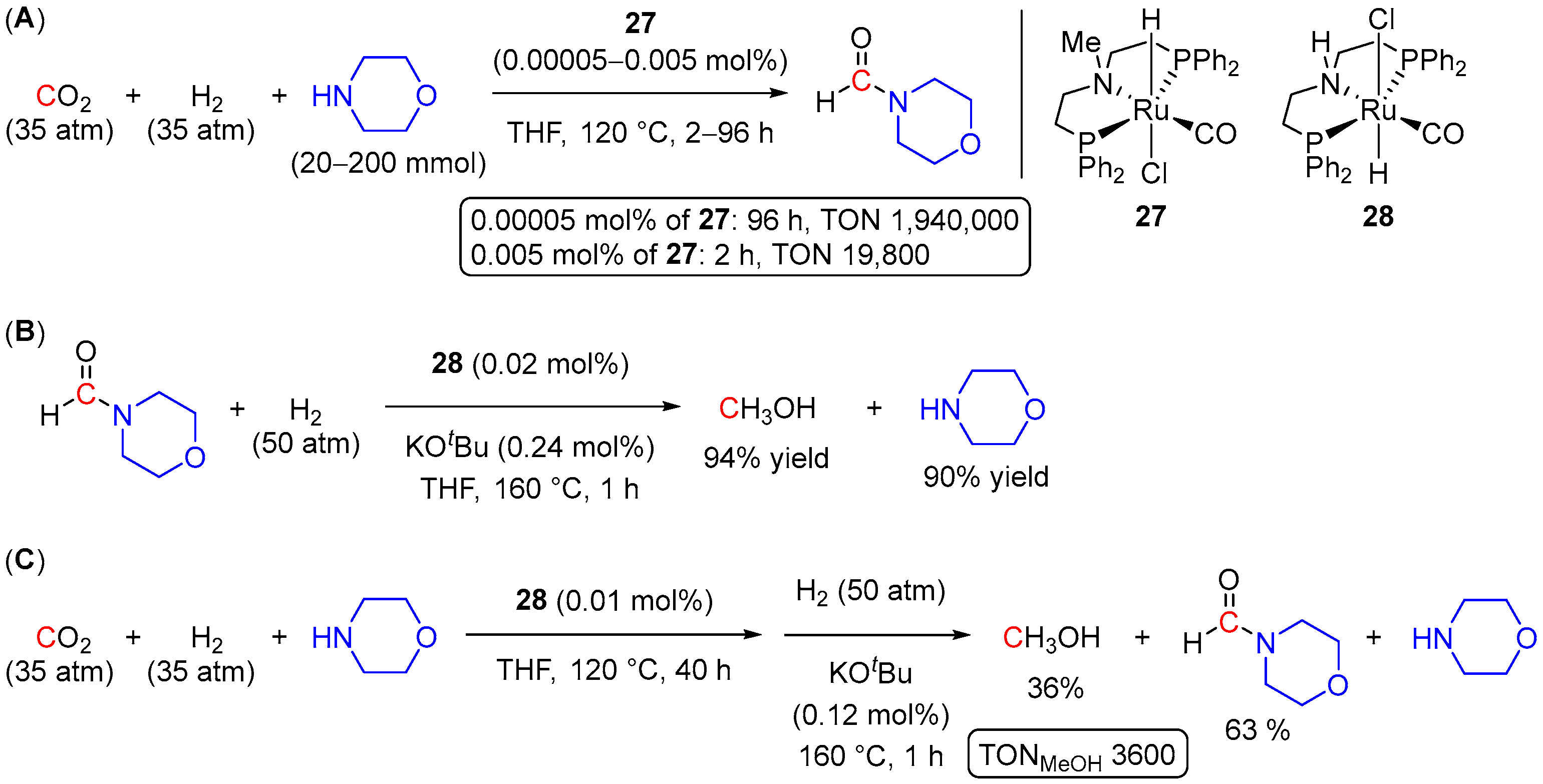
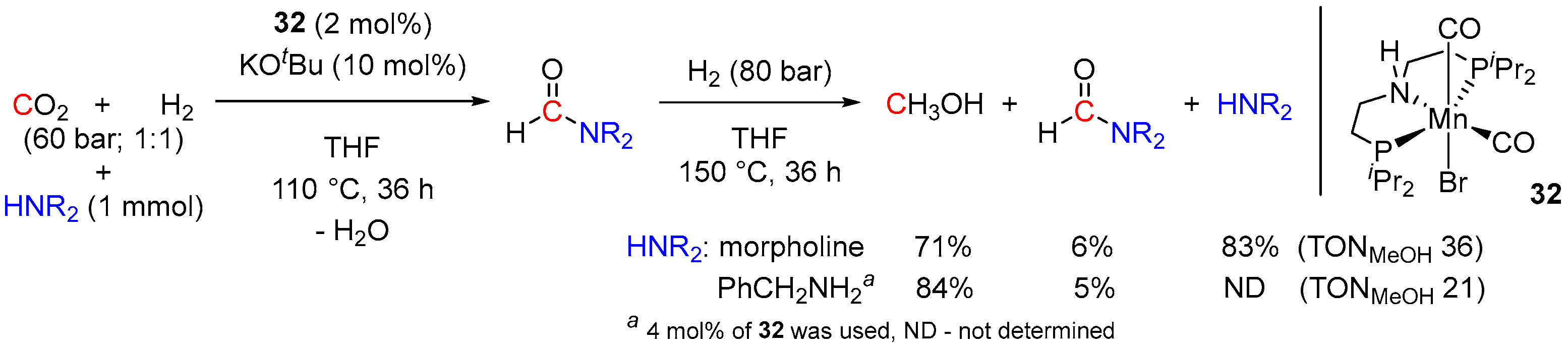
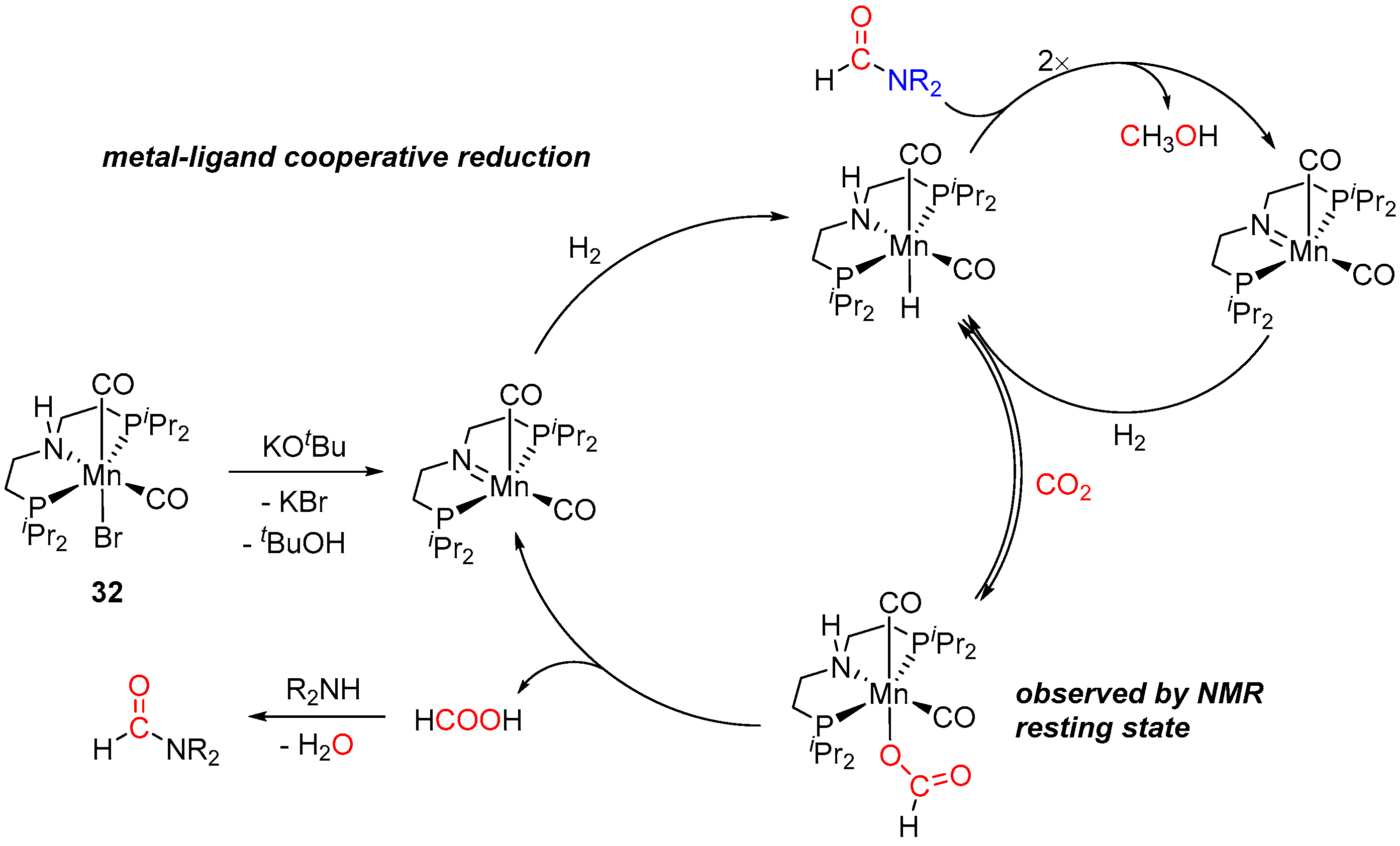
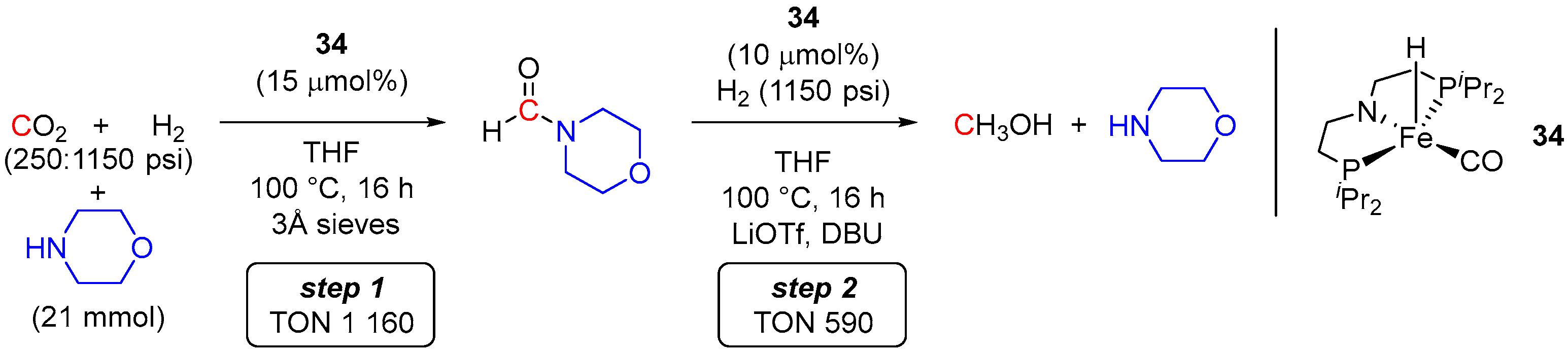
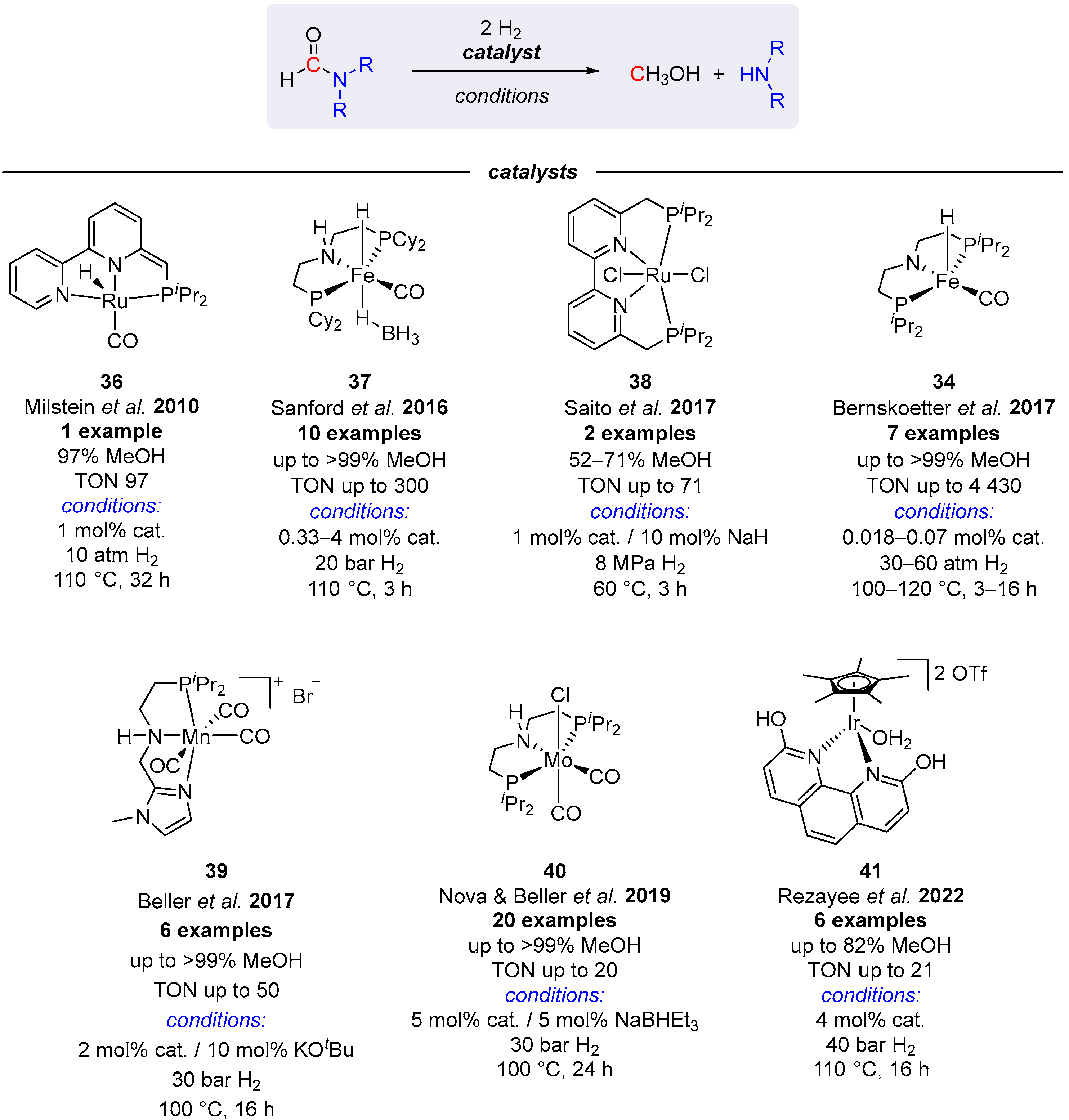
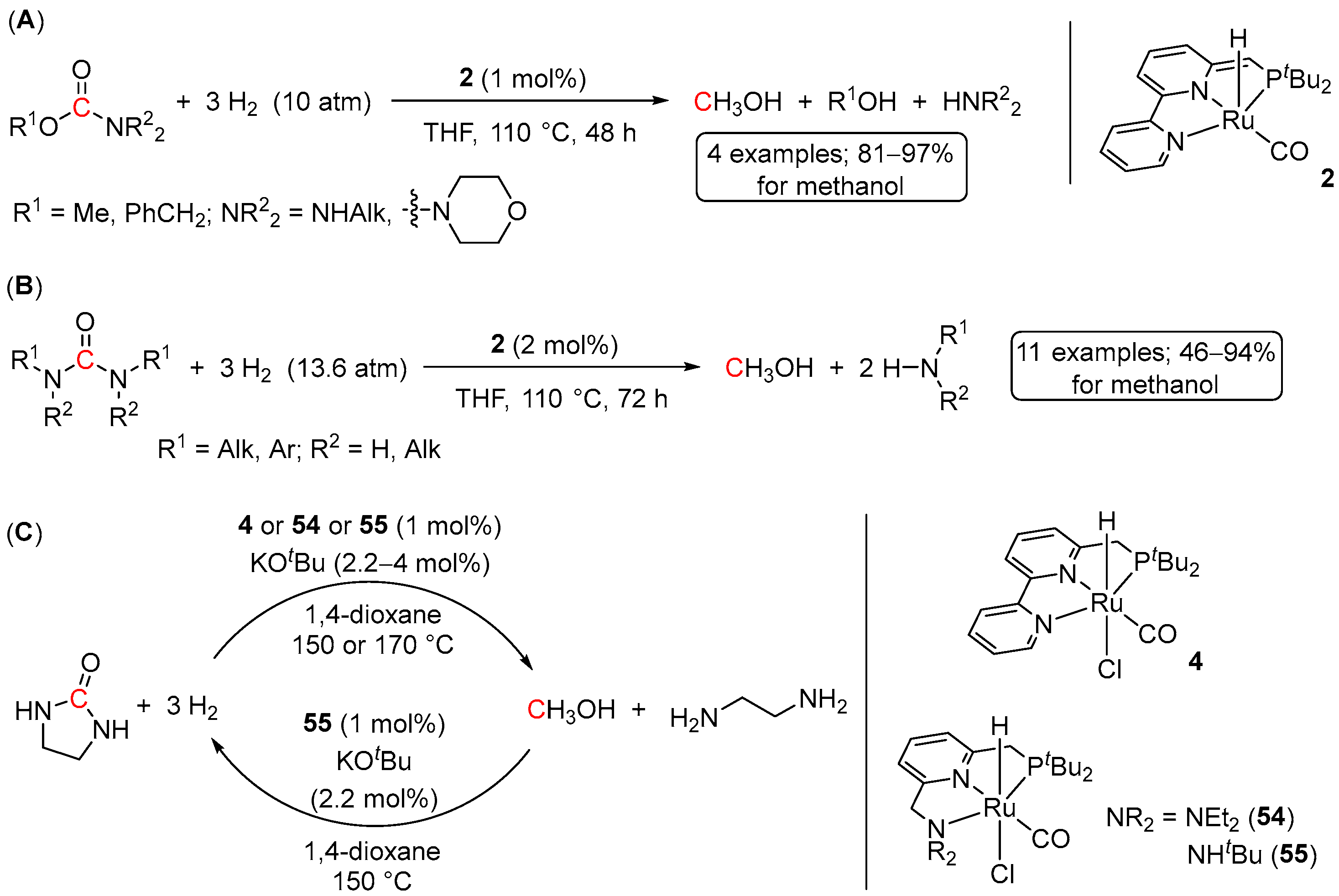
3. Catalytic Hydrogenation of Formamides
4. Catalytic Hydrogenation of Carbonates
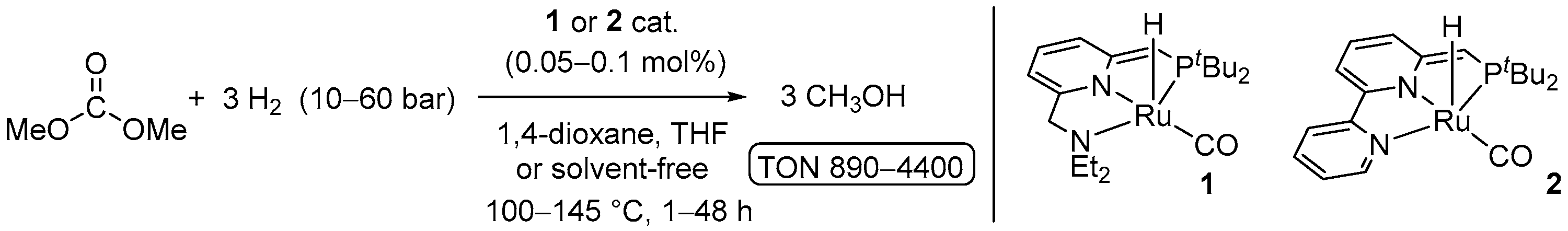
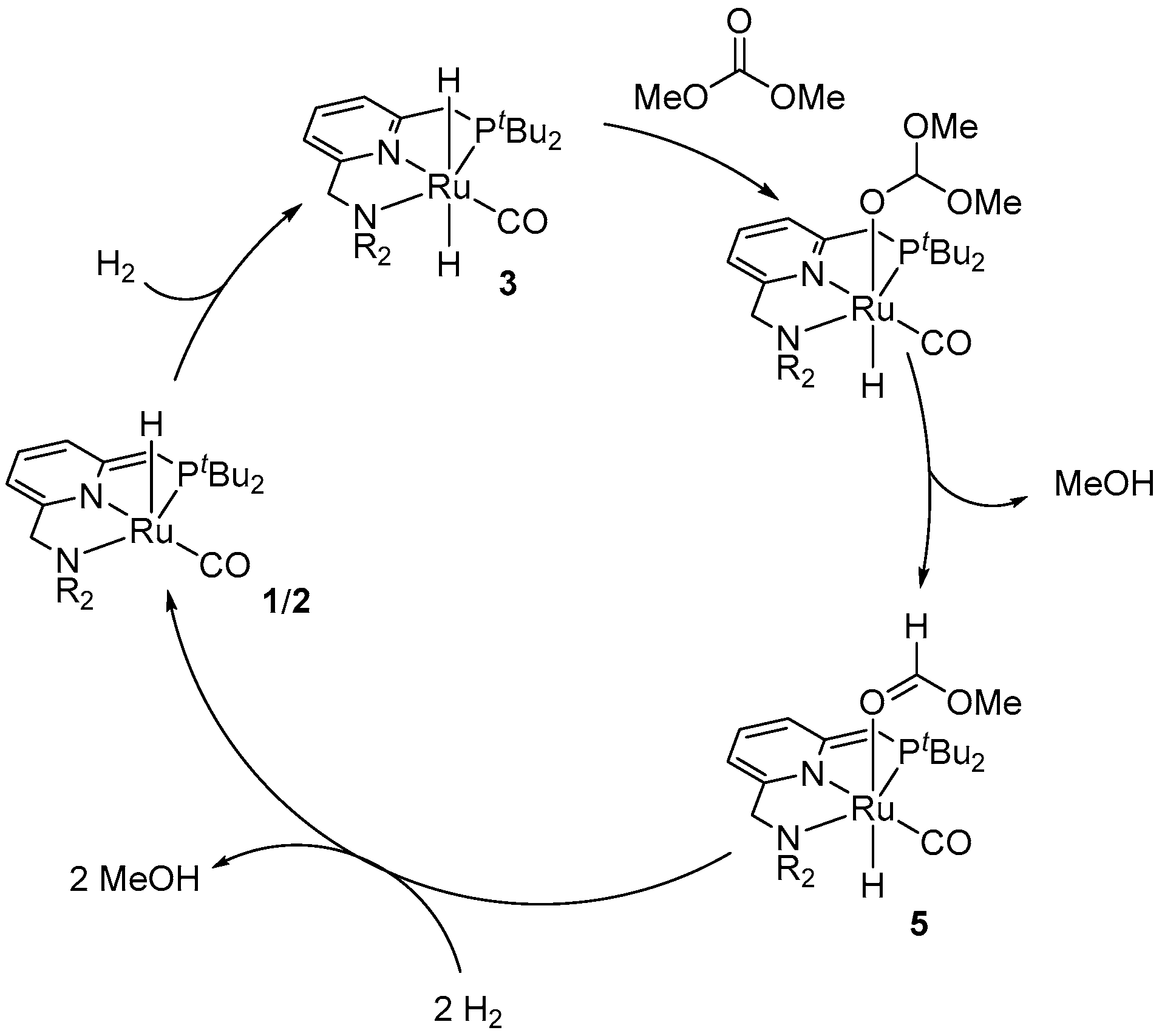
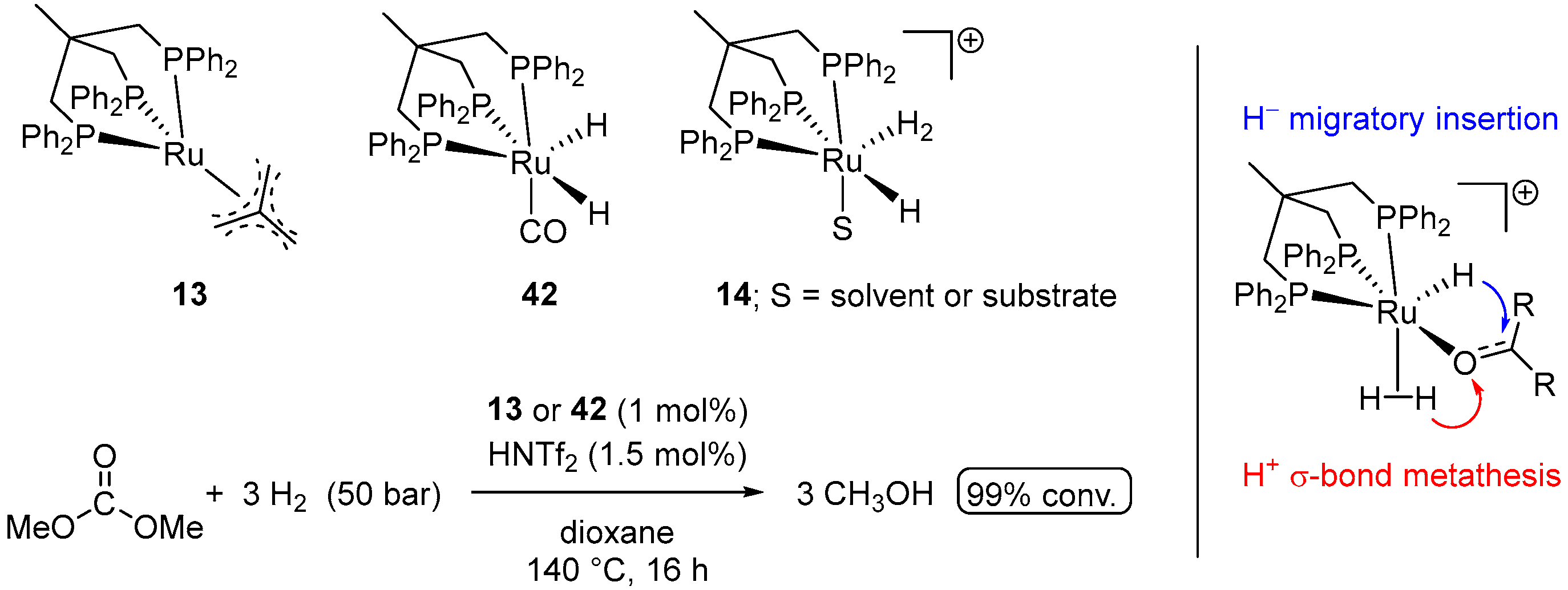
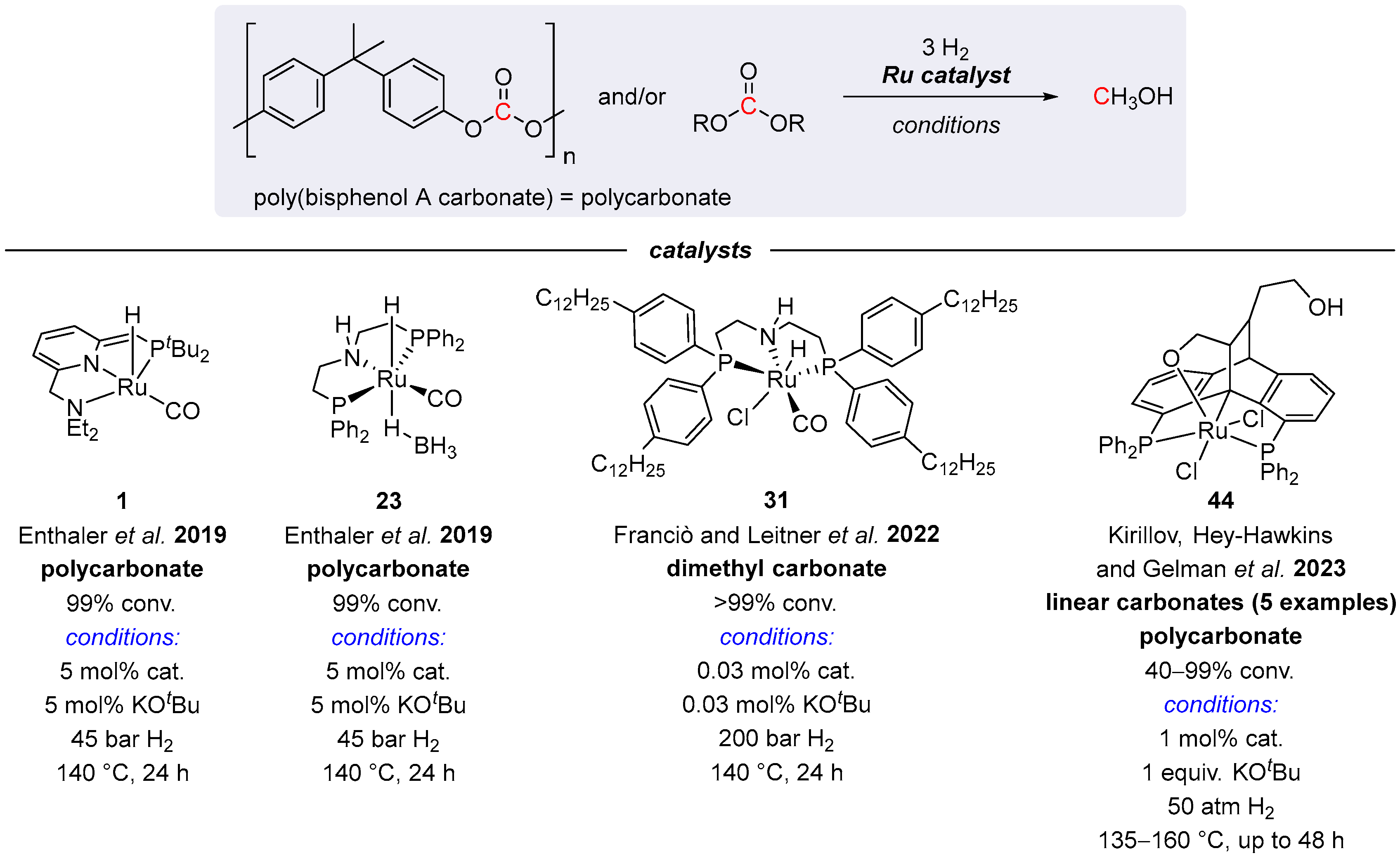
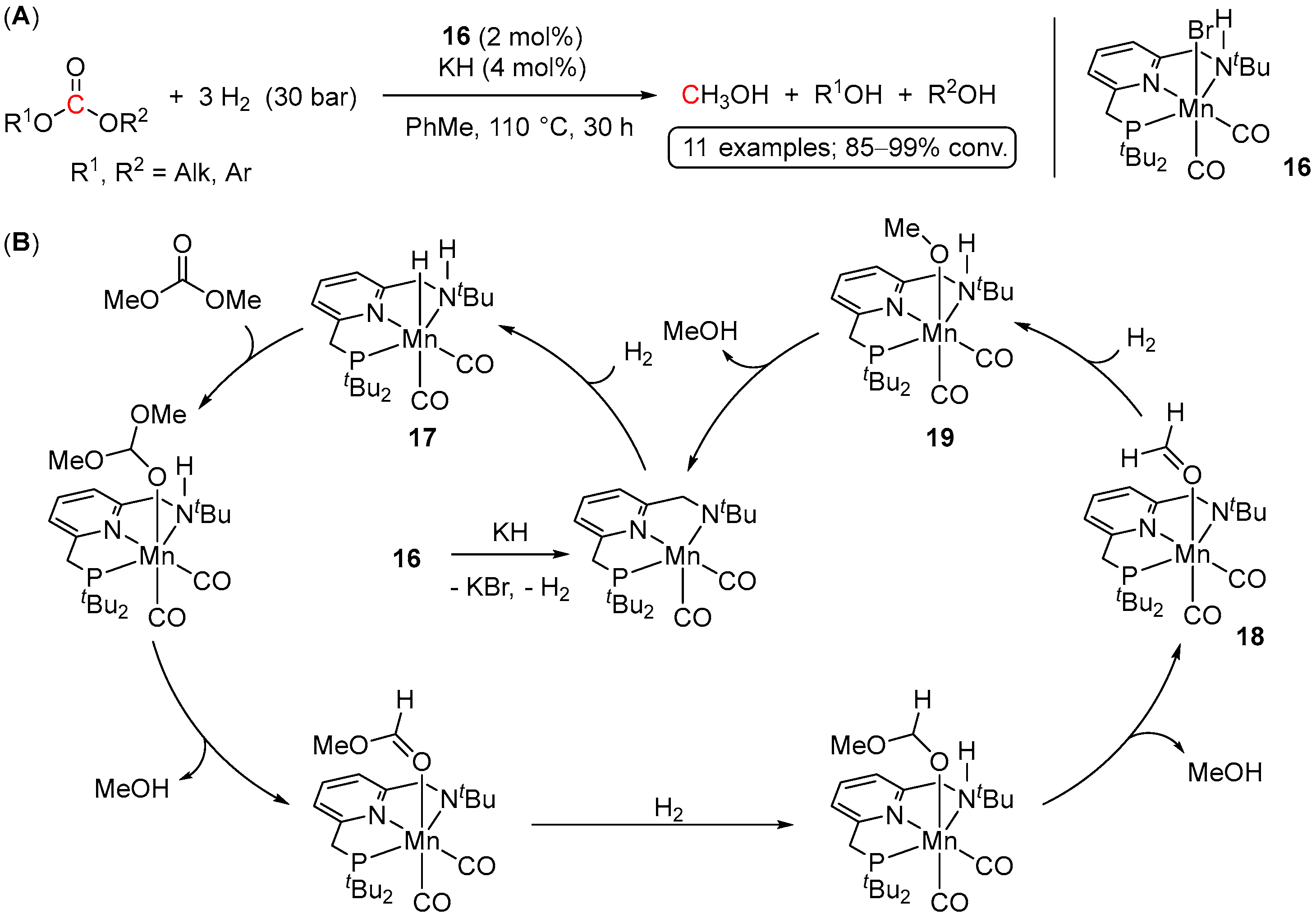
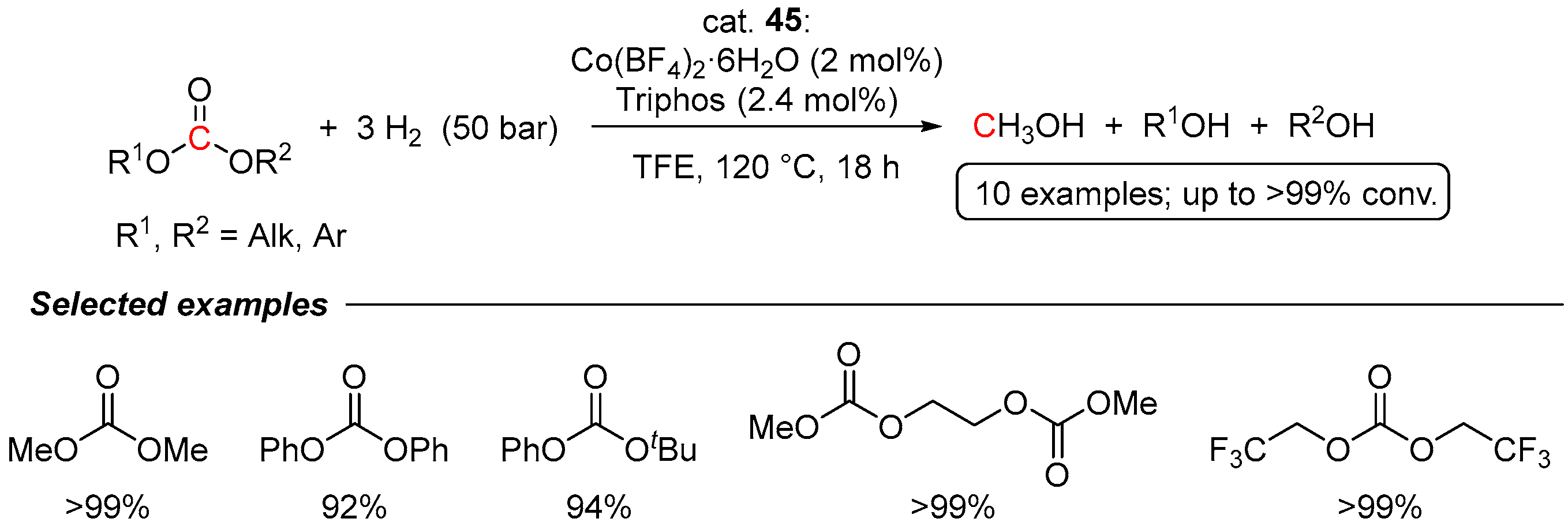
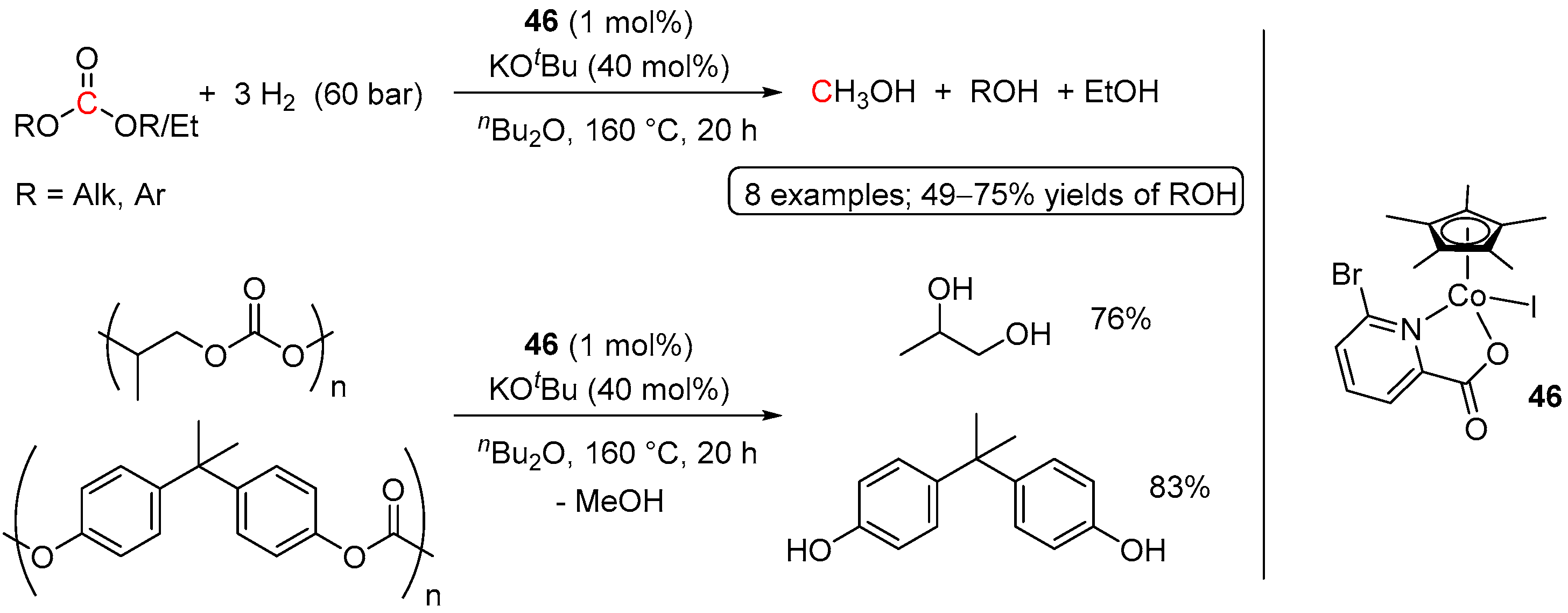
4.1. Hydrogenation of Acyclic Organic Carbonates
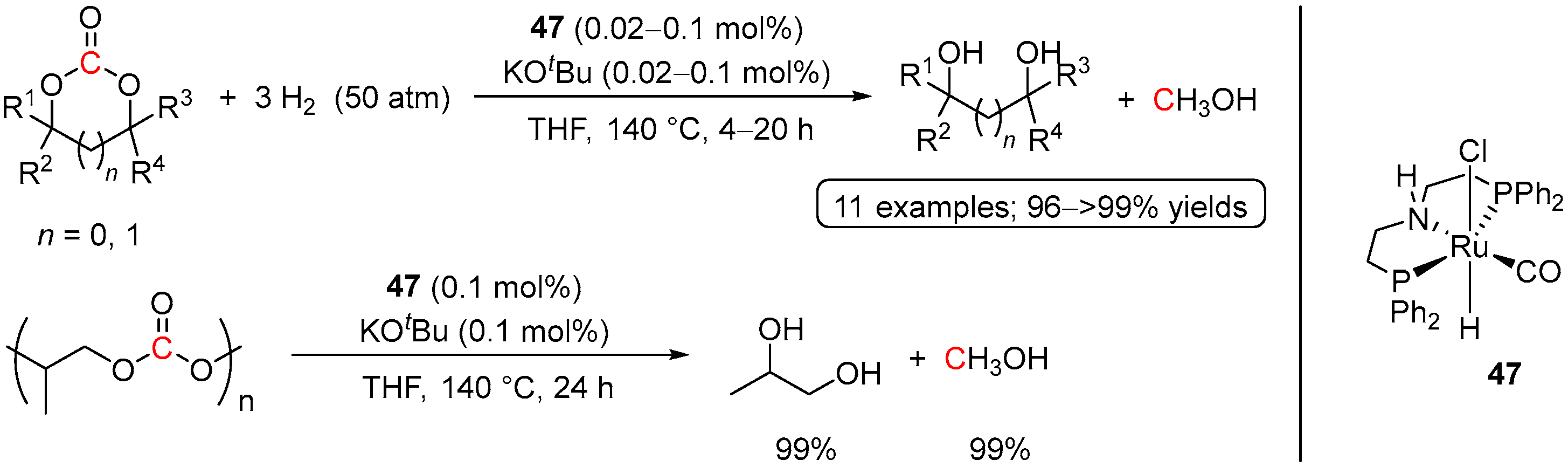
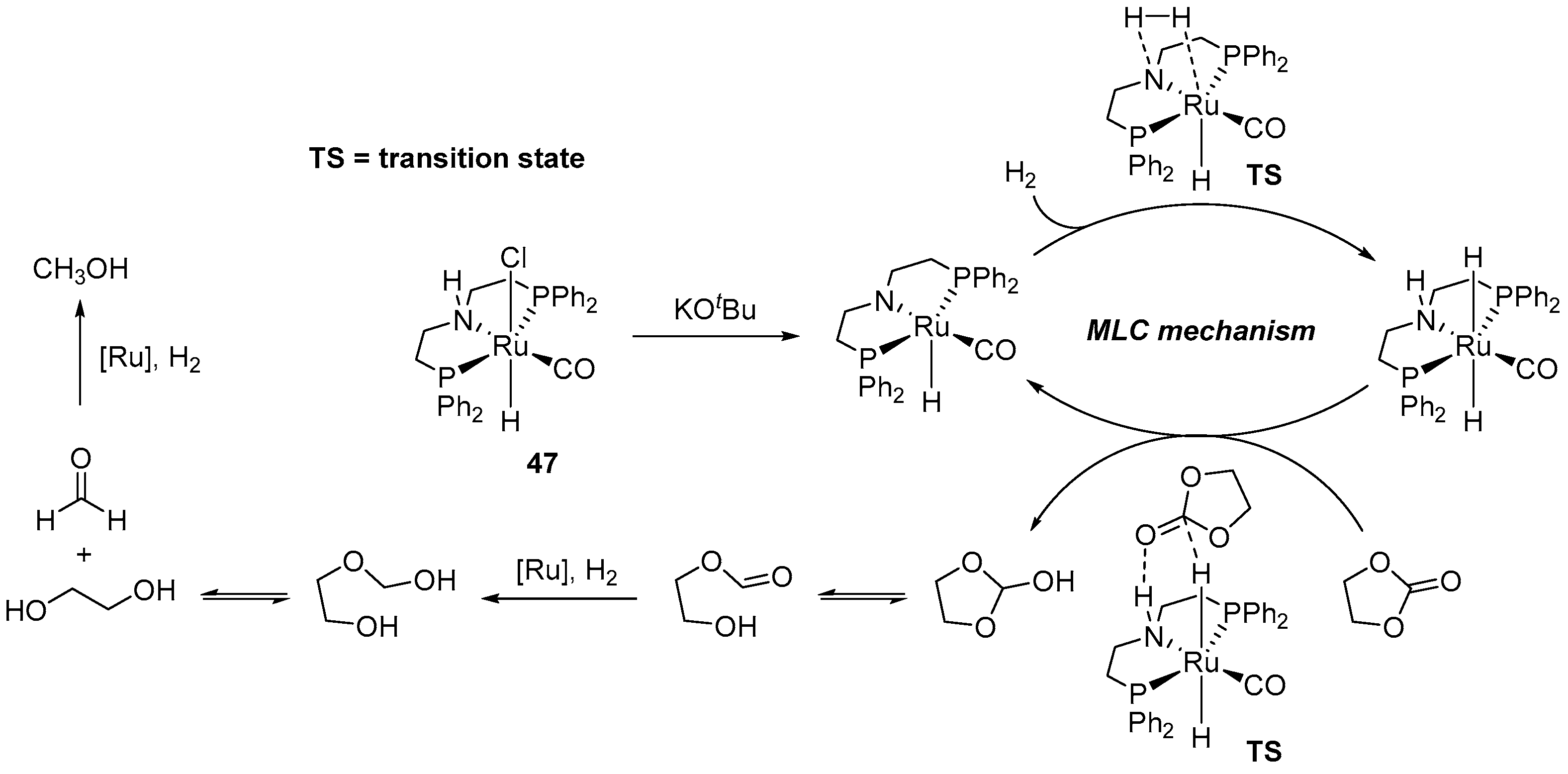
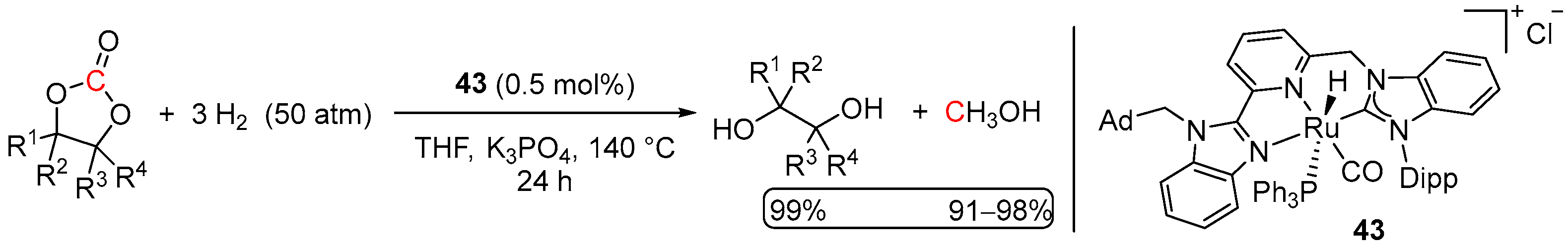
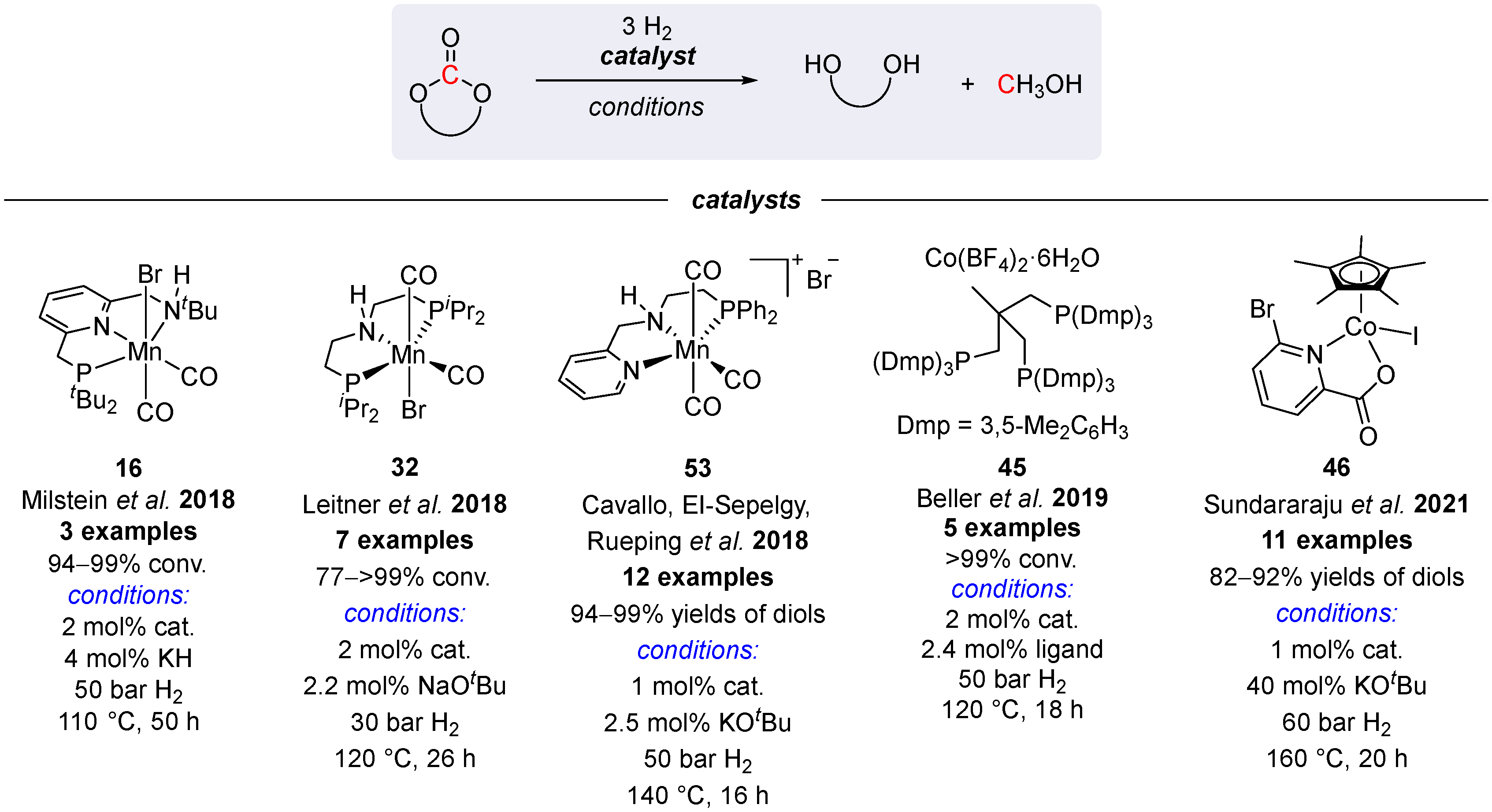
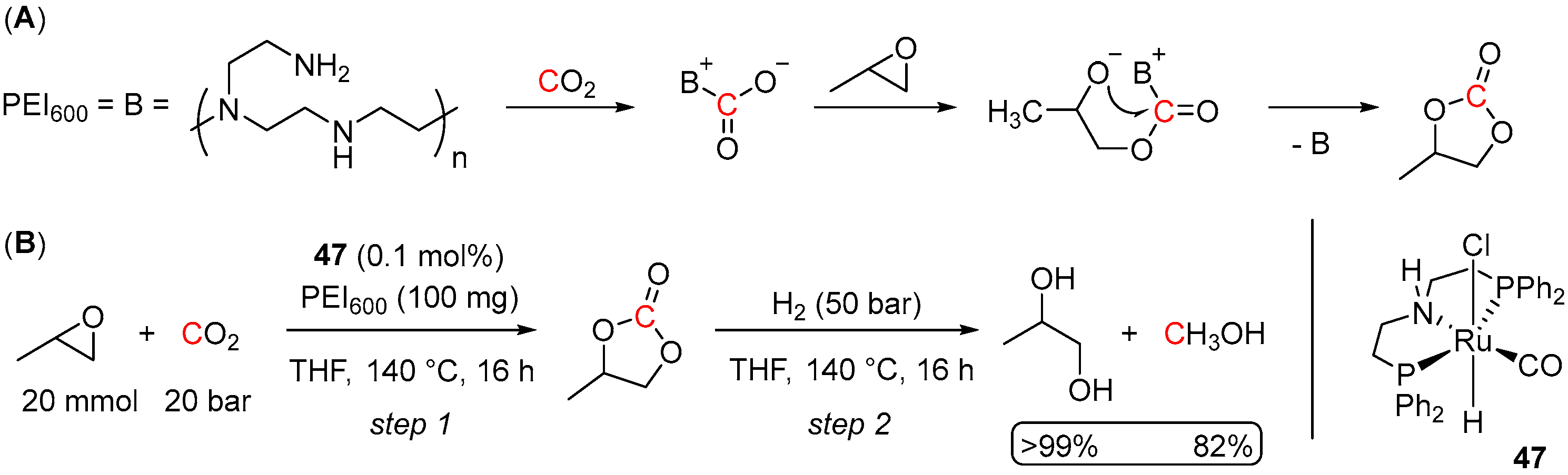
4.2. Hydrogenation of Cyclic Organic Carbonates
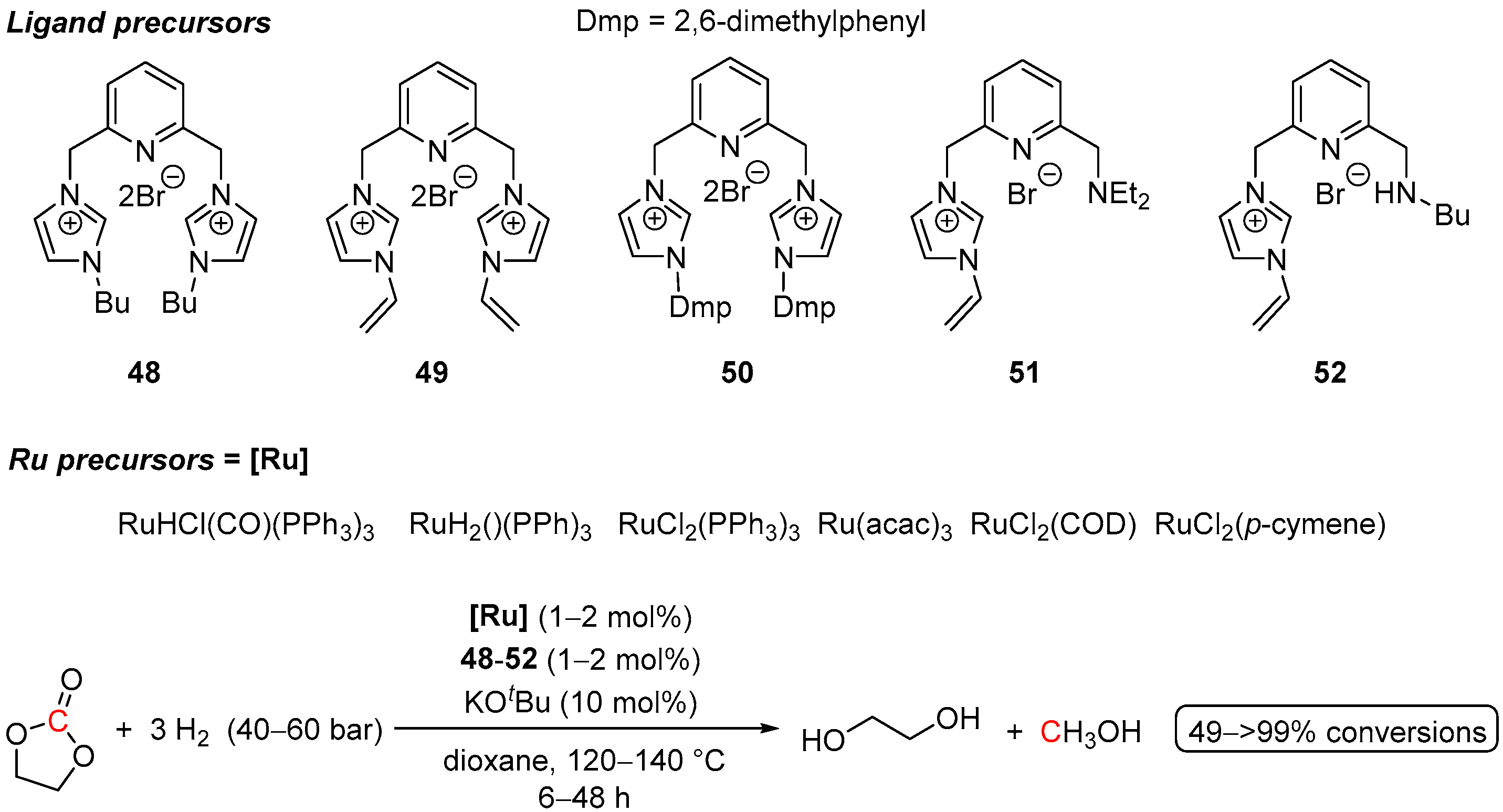
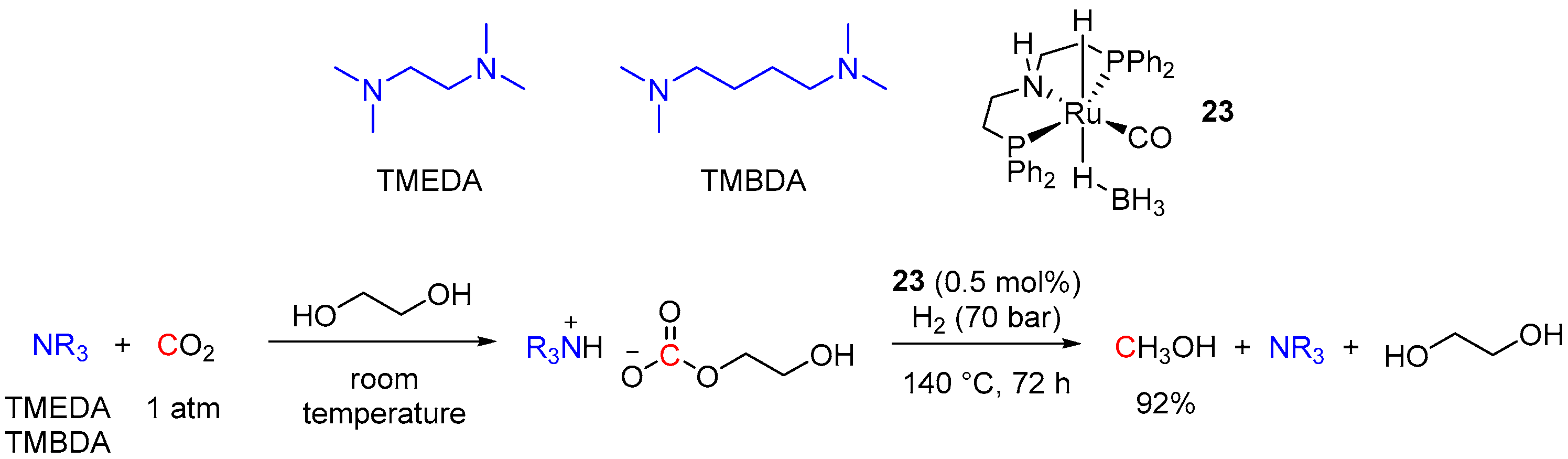
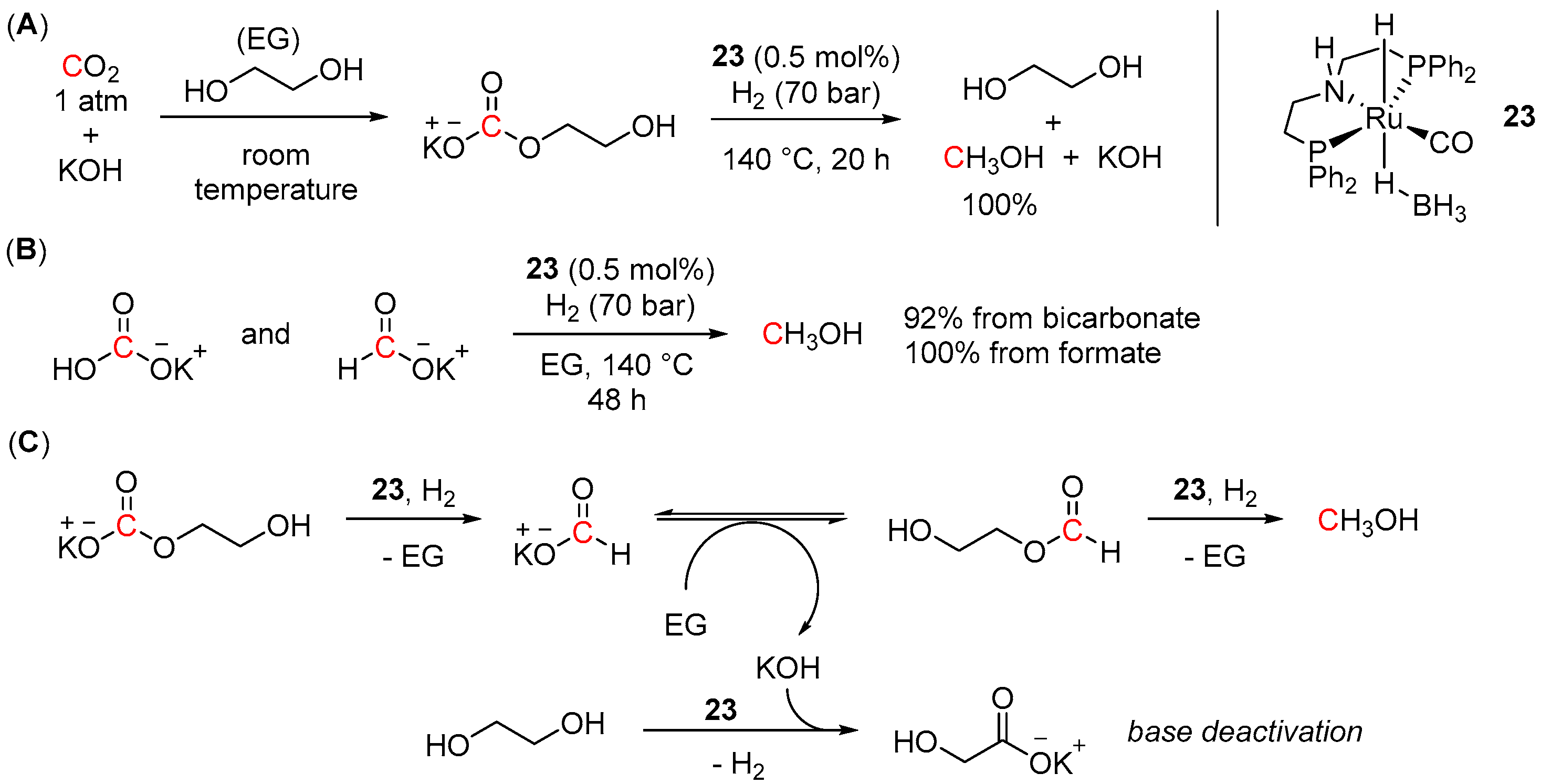
4.3. Hydrogenation of Carbonate Salts
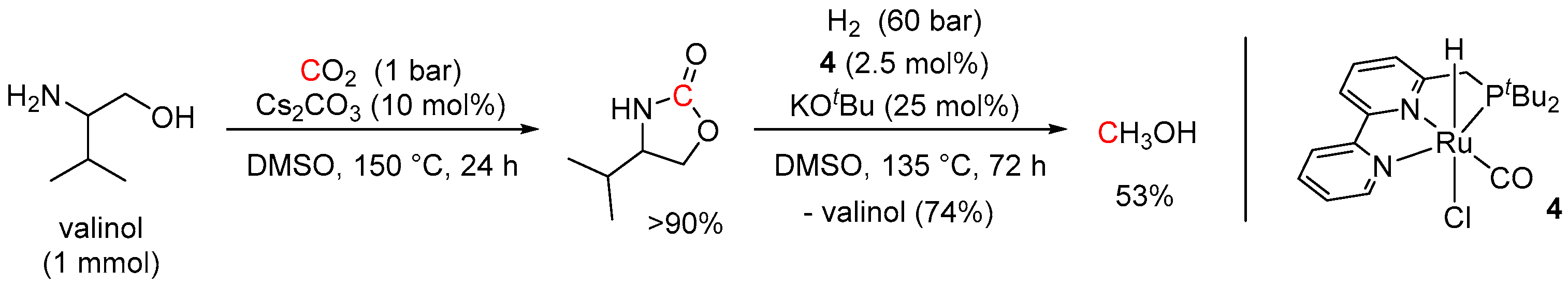
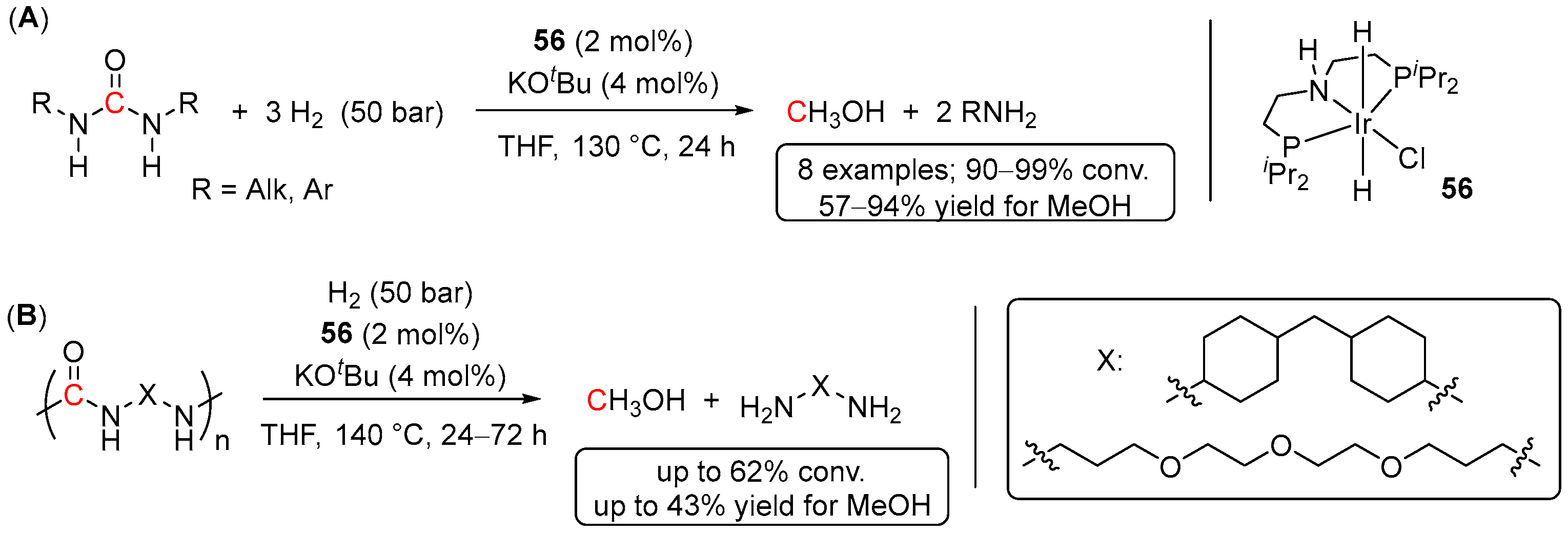
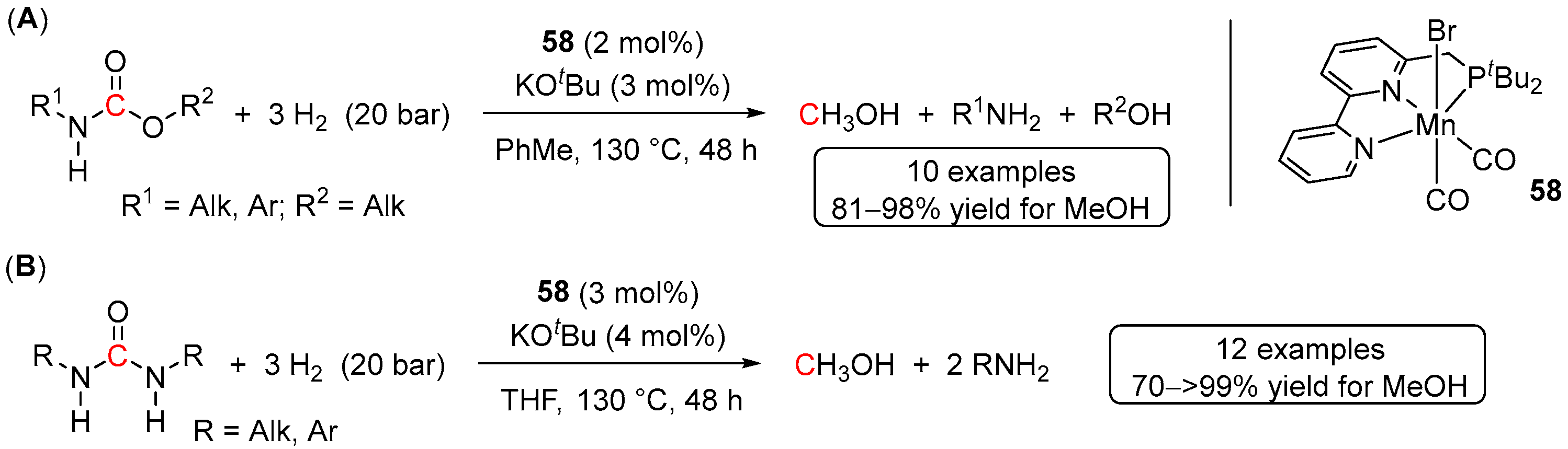
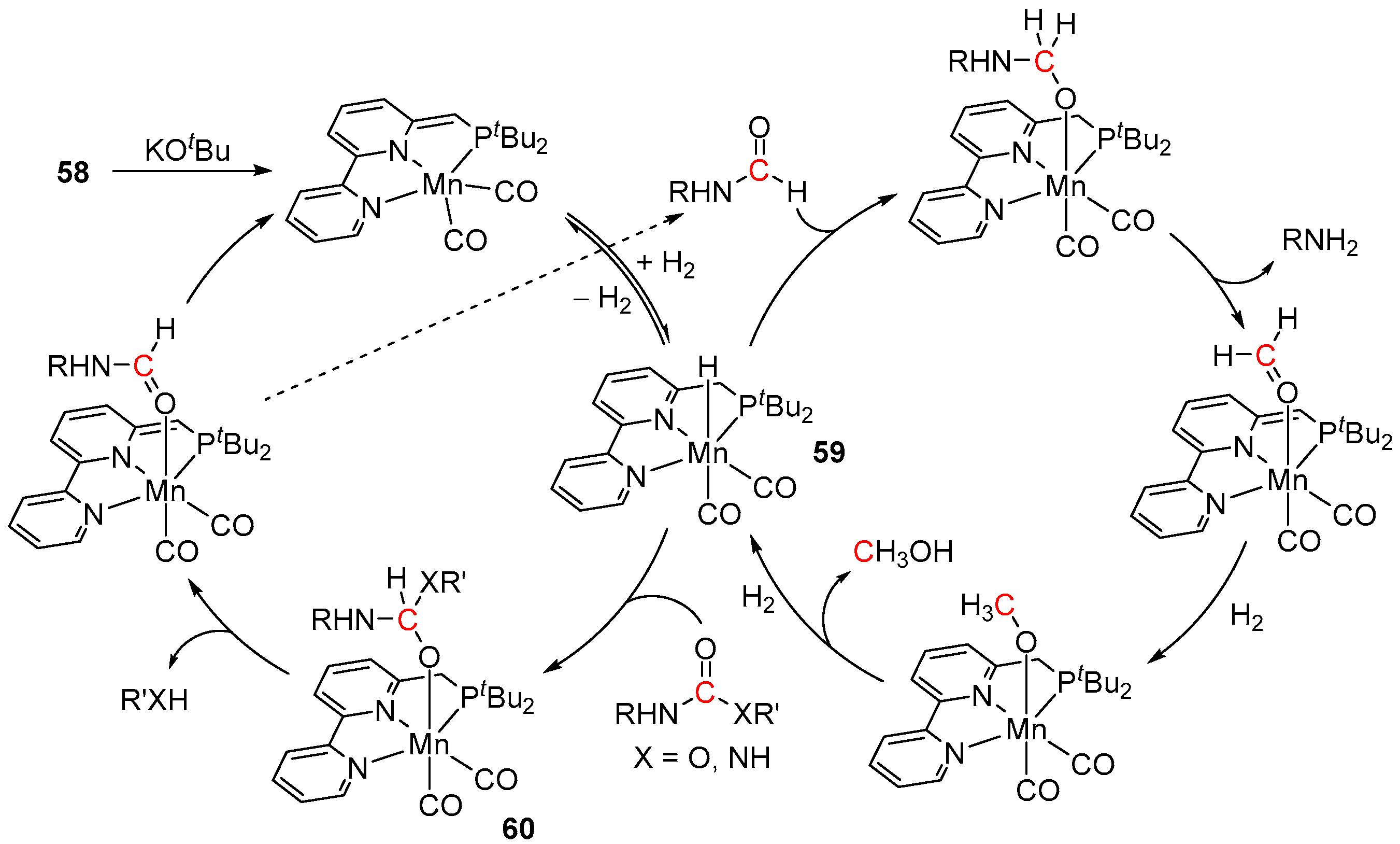
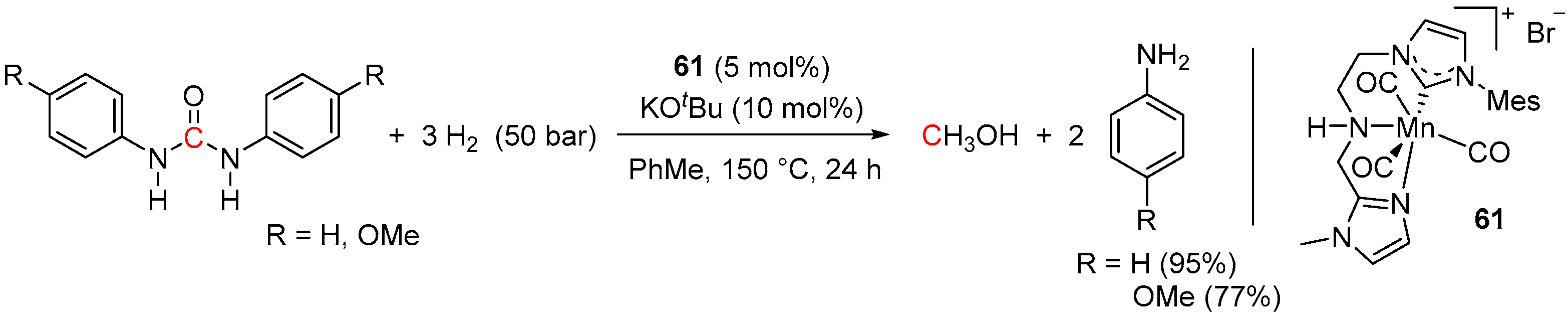
5. Catalytic Hydrogenation of Carbamates and Urea Derivatives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liu, G. (Ed.) Greenhouse Gases—Capturing, Utilization and Reduction; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Global Monitoring Laboratory. Trends in Atmospheric Carbon Dioxide. Available online: https://gml.noaa.gov/ccgg/trends/ (accessed on 14 June 2023).
- Key Renewables Trends; International Energy Agency: Paris, France, 2015.
- Shen, M.; Kong, F.; Tong, L.; Luo, Y.; Yin, S.; Liu, C.; Zhang, P.; Wang, L.; Chu, P.K.; Ding, Y. Carbon capture and storage (CCS): Development path based on carbon neutrality and economic policy. Carb. Neutrality 2022, 1, 37. [Google Scholar] [CrossRef]
- Dods, M.N.; Kim, E.L.; Long, J.R.; Weston, S.C. Deep CCS: Moving beyond 90% Carbon Dioxide Capture. Environ. Sci. Technol. 2021, 55, 8524–8534. [Google Scholar] [CrossRef]
- Osman, A.I.; Hefny, M.; Maksoud, M.I.A.A.; Elgarahy, A.M.; Rooney, D.W. Recent advances in carbon capture storage and utilization technologies: A review. Environ. Chem. Lett. 2021, 19, 797–849. [Google Scholar] [CrossRef]
- Gao, W.; Liang, S.; Wang, R.; Jiang, Q.; Zhang, Y.; Zheng, Q.; Xie, B.; Toe, C.Y.; Zhu, X.; Wang, J.; et al. Industrial carbon dioxide capture and utilization: State of the art and future challenges. Chem. Soc. Rev. 2020, 49, 8584–8686. [Google Scholar] [CrossRef] [PubMed]
- Knez, Ž.; Markočič, E.; Leitgeb, M.; Primožič, M.; Knez Hrnčič, M.; Škerget, M. Industrial applications of supercritical fluids: A review. Energy 2014, 77, 235–243. [Google Scholar] [CrossRef]
- Prasad, S.K.; Sangwai, J.S.; Byun, H.-S. A review of the supercritical CO2 fluid applications for improved oil and gas production and associated carbon storage. J. CO2 Util. 2023, 72, 102479. [Google Scholar] [CrossRef]
- Tan, Y.; Li, Q.; Xu, L.; Ghaffar, A.; Zhou, X.; Li, P. A critical review of carbon dioxide enhanced oil recovery in carbonate reservoirs. Fuel 2022, 328, 125256. [Google Scholar] [CrossRef]
- Ali, M.; Jha, N.K.; Pal, N.; Keshavarz, A.; Hoteit, H.; Sarmadivaleh, M. Recent advances in carbon dioxide geological storage, experimental procedures, influencing parameters, and future outlook. Earth Sci. Rev. 2022, 225, 103895. [Google Scholar] [CrossRef]
- Sahoo, P.K.; Zhang, Y.; Das, S. CO2-Promoted Reactions: An Emerging Concept for the Synthesis of Fine Chemicals and Pharmaceuticals. ACS Catal. 2021, 11, 3414–3442. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Das, S. Catalytic transformation of CO2 into C1 chemicals using hydrosilanes as a reducing agent. Green Chem. 2020, 22, 1800–1820. [Google Scholar] [CrossRef]
- Bai, S.-T.; De Smet, G.; Liao, Y.; Sun, R.; Zhou, C.; Beller, M.; Maes, B.U.W.; Sels, B.F. Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions. Chem. Soc. Rev. 2021, 50, 4259–4298. [Google Scholar] [CrossRef]
- Sun, R.; Liao, Y.; Bai, S.-T.; Zheng, M.; Zhou, C.; Zhang, T.; Sels, B.F. Heterogeneous catalysts for CO2 hydrogenation to formic acid/formate: From nanoscale to single atom. Energy Environ. Sci. 2021, 14, 1247–1285. [Google Scholar] [CrossRef]
- Dixneuf, P.H. A bridge from CO2 to methanol. Nat. Chem. 2011, 3, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Federsel, C.; Jackstell, R.; Beller, M. State-of-the-Art Catalysts for Hydrogenation of Carbon Dioxide. Angew. Chem. Int. Ed. 2010, 49, 6254–6257. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- ChemAnalyst. Decode the Future of Methanol. Available online: https://www.chemanalyst.com/industry-report/methanol-market-219 (accessed on 14 June 2023).
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef] [PubMed]
- For example: Sklyaruk, J.; Zubar, V.; Borghs, J.C.; Rueping, M. Methanol as the Hydrogen Source in the Selective Transfer Hydrogenation of Alkynes Enabled by a Manganese Pincer Complex. Org. Lett. 2020, 22, 6067–6071. [Google Scholar] [CrossRef]
- Lane, E.M.; Zhang, Y.; Hazari, N.; Bernskoetter, W.H. Sequential Hydrogenation of CO2 to Methanol Using a Pincer Iron Catalyst. Organometallics 2019, 38, 3084–3091. [Google Scholar] [CrossRef]
- Li, Y.-N.; Ma, R.; He, L.-N.; Diao, Z.-F. Homogeneous hydrogenation of carbon dioxide to methanol. Catal. Sci. Technol. 2014, 4, 1498–1512. [Google Scholar] [CrossRef]
- Sen, R.; Goeppert, A.; Prakash, G.K.S. Homogeneous Hydrogenation of CO2 and CO to Methanol: The Renaissance of Low-Temperature Catalysis in the Context of the Methanol Economy. Angew. Chem. Int. Ed. 2022, 61, e202207278. [Google Scholar] [CrossRef] [PubMed]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef] [PubMed]
- Buysch, H.J. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012; Volume 7, pp. 45–71. [Google Scholar]
- Schäffner, B.; Schäffner, F.; Verevkin, S.P.; Börner, A. Organic Carbonates as Solvents in Synthesis and Catalysis. Chem. Rev. 2010, 110, 4554–4581. [Google Scholar] [CrossRef]
- Dabral, S.; Schaub, T. The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef]
- ChemAnalyst. Decode the Future of Urea. Available online: https://www.chemanalyst.com/industry-report/urea-market-666 (accessed on 14 June 2023).
- Kumar, A.; Bhardwaj, R.; Mandal, S.K.; Choudhury, J. Transfer Hydrogenation of CO2 and CO2 Derivatives using Alcohols as Hydride Sources: Boosting and H2-Free Alternative Strategy. ACS Catal. 2022, 12, 8886–8903. [Google Scholar] [CrossRef]
- Gormley, R.J.; Rao, V.U.S.; Soong, Y. Methyl formate hydrogenolysis for low-temperature methanol synthesis. Appl. Catal. A 1992, 87, 81–101. [Google Scholar] [CrossRef]
- Iwasa, N.; Terashita, M.; Arai, M.; Takezawa, N. New catalytic functions of Pd and Pt catalysts for hydrogenolysis of methyl formate. React. Kinet. Catal. Lett. 2001, 74, 93–98. [Google Scholar] [CrossRef]
- Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L.J.W.; Milstein, D. Efficient hydrogenation of organic carbonates, carbamates and formates indicates alternative routes to methanol based on CO2 and CO. Nat. Chem. 2011, 3, 609–614. [Google Scholar] [CrossRef]
- Li, H.; Wen, M.; Wang, Z.-X. Computational Mechanistic Study of the Hydrogenation of Carbonate to Methanol Catalyzed by the RuIIPNN Complex. Inorg. Chem. 2012, 51, 5716–5727. [Google Scholar] [CrossRef]
- Huff, C.A.; Sanford, M.S. Cascade Catalysis for the Homogeneous Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2011, 133, 18122–18125. [Google Scholar] [CrossRef]
- Chu, W.-Y.; Culakova, Z.; Wang, B.T.; Goldberg, K.I. Acid-Assisted Hydrogenation of CO2to Methanol in a Homogeneous Catalytic Cascade System. ACS Catal. 2019, 9, 9317–9326. [Google Scholar] [CrossRef]
- Wesselbaum, S.; vom Stein, T.; Klankermayer, J.; Leitner, W. Hydrogenation of Carbon Dioxide to Methanol by Using a Homogeneous Ruthenium-Phosphine Catalyst. Angew. Chem. Int. Ed. 2012, 51, 7499–7502. [Google Scholar] [CrossRef]
- Leitner, W. Carbon Dioxide as a Raw Material: The Synthesis of Formic Acid and Its Derivatives from CO2. Angew. Chem. Int. Ed. 1995, 34, 2207–2221. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous Hydrogenation of Carbon Dioxide. Chem. Rev. 1995, 95, 259–272. [Google Scholar] [CrossRef]
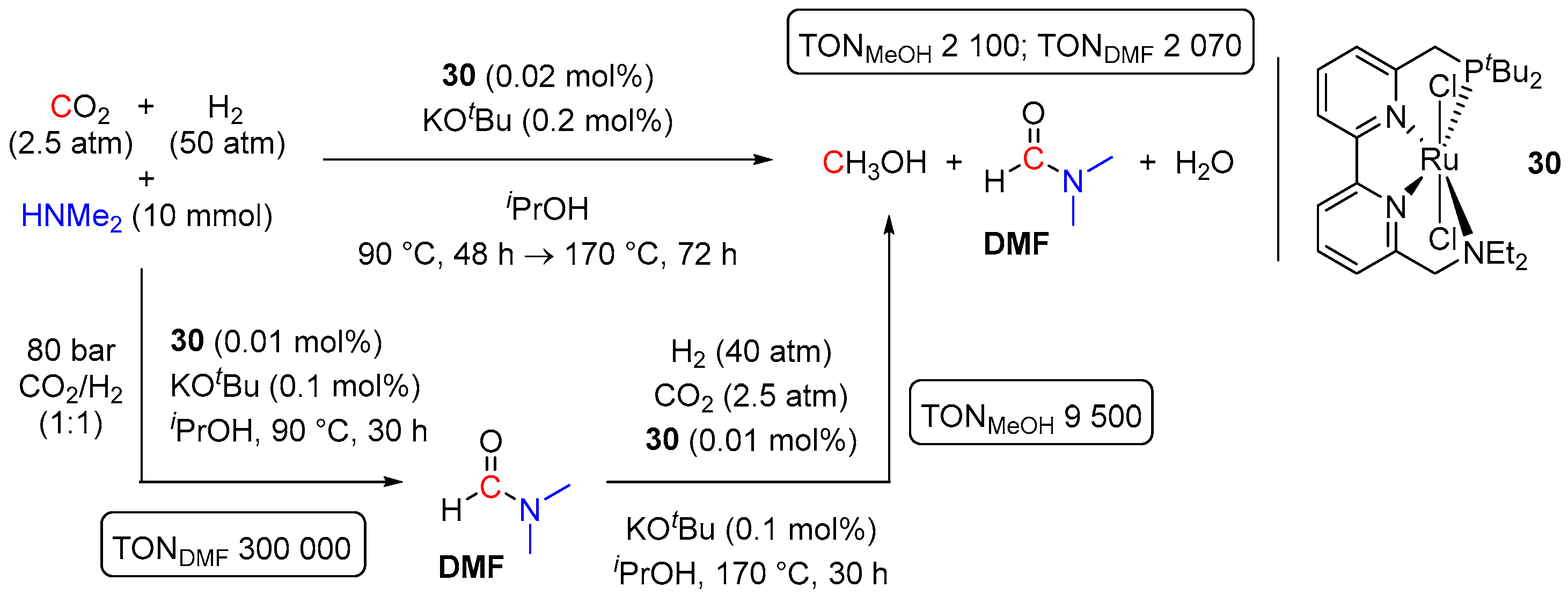
- Schneidewind, J.; Adam, R.; Baumann, W.; Jackstell, R.; Beller, M. Low-Temperature Hydrogenation of Carbon Dioxide to Methanol with a Homogeneous Cobalt Catalyst. Angew. Chem. Int. Ed. 2017, 56, 1890–1893. [Google Scholar] [CrossRef]
- Kumar, A.; Janes, T.; Espinosa-Jalapa, N.A.; Milstein, D. Manganese Catalyzed Hydrogenation of Organic Carbonates to Methanol and Alcohols. Angew. Chem. Int. Ed. 2018, 57, 12076–12080. [Google Scholar] [CrossRef] [PubMed]
- Kuβ, D.A.; Hölscher, M.; Leitner, M. Hydrogenation of CO2 to Methanol with Mn-PNP-Pincer Complexes in the Presence of Lewis Acids: The Formate Resting State Unleashed. ChemCatChem 2021, 13, 3319–3323. [Google Scholar]
- Kuβ, D.A.; Hölscher, M.; Leitner, W. Combined Computational and Experimental Investigation on the Mechanism of CO2 Hydrogenation to Methanol with Mn-PNP-Pincer Catalysts. ACS Catal. 2022, 12, 15310–15322. [Google Scholar]
- Khalimon, A.Y.; Gudun, K.A.; Hayrapetyan, D. Base Metal Catalysts for Deoxygenative Reduction of Amides to Amines. Catalysts 2019, 9, 490. [Google Scholar] [CrossRef]
- Khalimon, A.Y. Deoxygenative Hydroboration of Carboxamides: A Versatile and Selective Synthetic Approach to Amines. Dalton Trans. 2021, 50, 17455–17466. [Google Scholar] [CrossRef]
- Werkmeister, S.; Junge, K.; Beller, M. Catalytic Hydrogenation of Carboxylic Acid Esters, Amides, and Nitriles with Homogeneous Catalysts. Org. Process Res. Dev. 2014, 18, 289–302. [Google Scholar] [CrossRef]
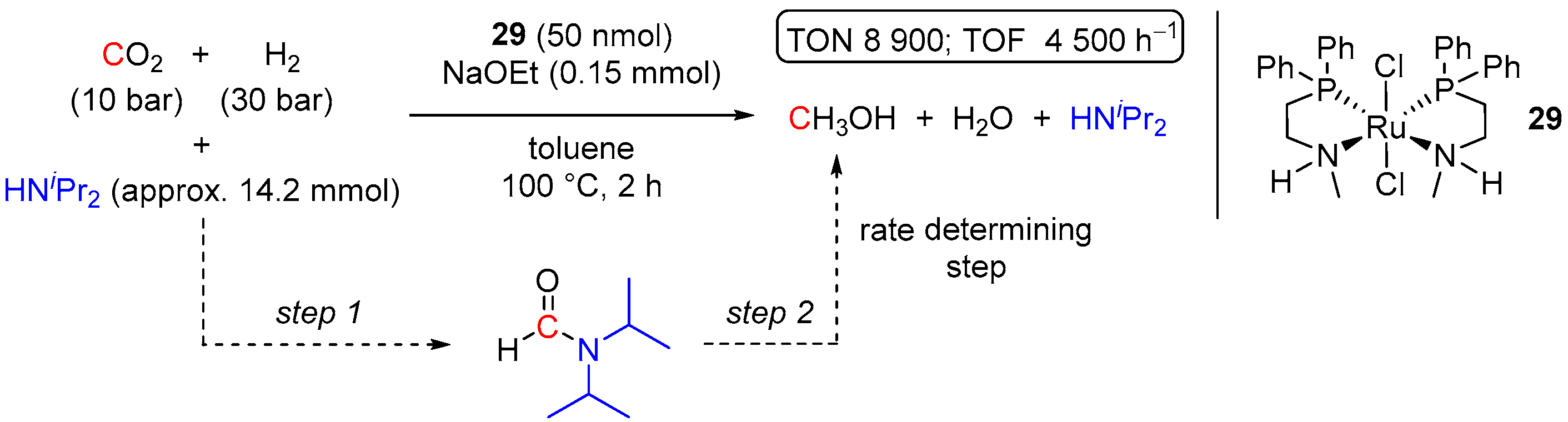
- Rezayee, N.M.; Huff, C.A.; Sanford, M.S. Tandem Amine and Ruthenium-Catalyzed Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Sen, R.; Kothandaraman, J.; Goeppert, A.; Chowdhury, R.; Munoz, S.B.; Haiges, R.; Prakash, G.K.S. Mechanistic Insights into Ruthenium-Pincer-Catalyzed Amine-Assisted Homogeneous Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2019, 141, 3160–3170. [Google Scholar] [CrossRef]
- Mandal, S.C.; Rawat, K.S.; Nandi, S.; Pathak, B. Theoretical insights into CO2 hydrogenation to methanol by a Mn-PNP complex. Catal. Sci. Technol. 2019, 9, 1867–1878. [Google Scholar] [CrossRef]
- Zhang, L.; Han, Z.; Zhao, X.; Wang, Z.; Ding, K. Highly Efficient Ruthenium-Catalyzed N-Formylation of Amines with H2 and CO2. Angew. Chem. Int. Ed. 2015, 54, 6186–6189. [Google Scholar] [CrossRef]
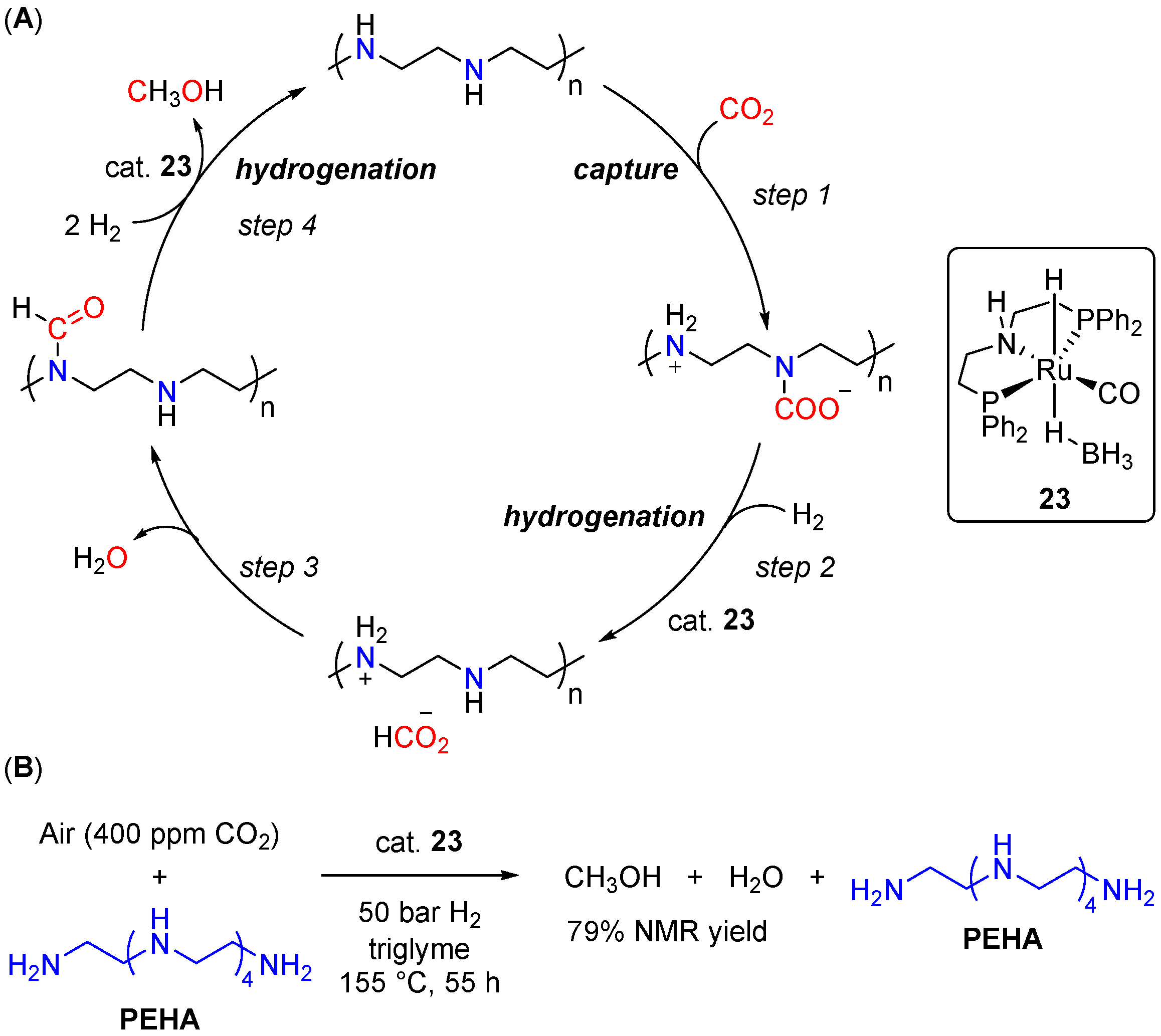
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.K.S. Conversion of CO2 from Air into Methanol Using a Polyamine and a Homogeneous Ruthenium Catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Sen, R.; Goeppert, A.; Prakash, G.K.S. Integrative CO2 Capture and Hydrogenation to Methanol with Reusable Catalyst and Amine: Toward a Carbon Neutral Methanol Economy. J. Am. Chem. Soc. 2018, 140, 1580–1583. [Google Scholar] [CrossRef]
- Everett, M.; Wass, D.F. Highly productive CO2 hydrogenation to methanol—A tandem catalytic approach via amide intermediates. Chem. Commun. 2017, 53, 9502–9504. [Google Scholar] [CrossRef]
- Zhang, F.-H.; Liu, C.; Li, W.; Tian, G.-L.; Xie, J.-H.; Zhou, Q.-L. An Efficient Ruthenium Catalyst Bearing Tetradentate Ligand for Hydrogenations of Carbon Dioxide. Chin. J. Chem. 2018, 36, 1000–1002. [Google Scholar] [CrossRef]
- Yoshimura, A.; Watari, R.; Kuwata, S.; Kayaki, Y. Poly(ethyleneimine)-Mediated Consecutive Hydrogenation of Carbon Dioxide to Methanol with Ru Catalysts. Eur. J. Inorg. Chem. 2019, 2019, 2375–2380. [Google Scholar] [CrossRef]
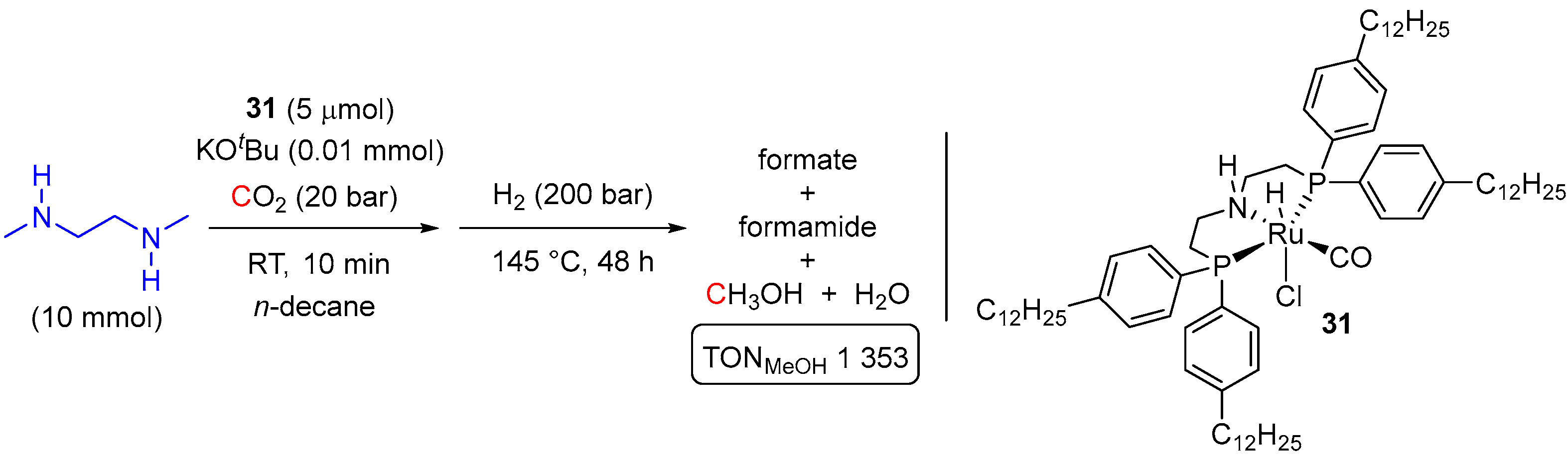
- Diehl, T.; Lanzerath, P.; Franciò, G.; Leitner, W. A Self-Separating Multiphasic System for Catalytic Hydrogenation of CO2 and CO2-Derivatives to Methanol. ChemSusChem 2022, 15, e202201250. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Goeppert, A.; Kothandaraman, J.; Prakash, G.K.S. Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide. ACS Catal. 2017, 7, 6347–6351. [Google Scholar] [CrossRef]
- Ribeiro, A.P.C.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Carbon dioxide-to-methanol single-pot conversion using a C-scorpionate iron(II) catalyst. Green Chem. 2017, 19, 4811–4815. [Google Scholar] [CrossRef]
- Bernskoetter, W.H.; Hazari, N. Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts. Acc. Chem. Res. 2017, 50, 1049–1058. [Google Scholar] [CrossRef]
- Wei, D.; Shi, X.; Junge, H.; Du, C.; Beller, M. Carbon neutral hydrogen storage and release cycles based on dual-functional roles of formamides. Nat. Commun. 2023, 14, 3726. [Google Scholar] [CrossRef]
- Balaraman, E.; Gnanaprakasam, B.; Shimon, L.J.W.; Milstein, D. Direct Hydrogenation of Amides to Alcohols and Amines under Mild Conditions. J. Am. Chem. Soc. 2010, 132, 16756–16758. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Samblanet, D.C.; Sanford, M.S. Iron-Catalyzed Hydrogenation of Amides to Alcohols and Amines. ACS Catal. 2016, 6, 6377–6383. [Google Scholar] [CrossRef]
- Miura, T.; Naruto, V.; Toda, K.; Shimomura, T.; Saito, S. Multifaceted catalytic hydrogenation of amides via diverse activation of a sterically confined bipyridine-ruthenium framework. Sci. Rep. 2017, 7, 1586. [Google Scholar] [CrossRef]
- Jayrathne, U.; Zhang, Y.; Hazari, N.; Bernskoetter, W.H. Selective Iron-Catalyzed Deaminative Hydrogenation of Amides. Organometallics 2017, 36, 409–416. [Google Scholar] [CrossRef]
- Papa, V.; Cabrero-Antonino, J.R.; Alberico, E.; Spanneberg, A.; Junge, K.; Junge, H.; Beller, M. Efficient and selective hydrogenation of amides to alcohols and amines using a well-defined manganese-PNN pincer complex. Chem. Sci. 2017, 8, 3576–3585. [Google Scholar] [CrossRef]
- Leischner, T.; Suarez, L.A.; Spennenberg, A.; Junge, K.; Nova, A.; Beller, M. Highly selective hydrogenation of amides catalysed by a molybdenum pincer complex: Scope and mechanism. Chem. Sci. 2019, 10, 10566–10576. [Google Scholar] [CrossRef] [PubMed]
- Ravn, A.K.; Rezayee, N.M. The Investigation of a Switchable Iridium Catalyst for the Hydrogenation of Amides: A Case Study of C–O Versus C–N Bond Scission. ACS Catal. 2022, 12, 11927–11933. [Google Scholar] [CrossRef]
- Dibenedetto, A.; Angelini, A. Synthesis of Organic Carbonates. In Advances in Inorganic Chemistry; Aresta, M., van Eldik, R., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 66, pp. 25–81. [Google Scholar]
- Parker, H.L.; Sherwood, J.; Hunt, A.J.; Clark, J.H. Cyclic Carbonates as Green Alternative Solvents for the Heck Reaction. ACS Sustain. Chem. Eng. 2014, 2, 1739–1742. [Google Scholar]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kühn, F.E. Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef] [PubMed]
- vom Stein, T.; Meuresch, M.; Limper, D.; Schmitz, M.; Hölscher, M.; Coetzee, J.; Cole-Hamilton, D.L.; Klamkermayer, J.; Leitner, W. Highly Versatile Catalytic Hydrogenation of Carboxylic and carbonic Acid Derivatives using a Ru-Triphos Complex: Molecular Control over Selectivity and Substrate Scope. J. Am. Chem. Soc. 2014, 136, 13217–13225. [Google Scholar] [CrossRef]
- Geilen, F.M.A.; Engendahl, B.; Hölscher, M.; Klamkermayer, J.; Leitner, W. Selective Homogeneous Hydrogenation of Biogenic carboxylic Acids with [Ru(TriPhos)H]+: A Mechanistic Study. J. Am. Chem. Soc. 2011, 133, 14349–14358. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, H.; Chen, J.; Le, Z.-G.; Tu, T. Synthesis, Characterization and Catalytic Application of Pyridine-bridged N-Heterocyclic carbine-Ruthenium Complexes in the Hydrogenation of Carbonates. Chem. Asian J. 2017, 12, 2809–2812. [Google Scholar] [CrossRef]
- Alberti, C.; Eckelt, S.; Enthaler, S. Ruthenium-Catalyzed Hydrogenative Depolymerization of End-of-Life Poly(bisphenol A carbonate). ChemistrySelect 2019, 4, 12268–12271. [Google Scholar] [CrossRef]
- Kindler, T.-O.; Alberti, C.; Sundermeier, J.; Enthaler, S. Hydrogenative Depolymerization of End-of-Life Poly(Bisphenol A Carbonate) catalyzed by a Ruthenium MACHO-Complex. ChemistryOpen 2019, 8, 1410–1412. [Google Scholar] [CrossRef]
- Mujahed, S.; Richter, M.S.; Kirillov, E.; Hey-Hawkins, E.; Gelman, D. A High-valent Ru-PCP Pincer Catalyst for the Hydrogenation of Organic Carbonates. Isr. J. Chem. 2023, e202300037. [Google Scholar] [CrossRef]
- Ferretti, F.; Scharnagl, F.K.; Dall’Anese, A.; Jackstell, R.; Dastgir, S.; Beller, M. Additive-free cobalt-catalyzed hydrogenation of carbonates to methanol and alcohols. Catal. Sci. Technol. 2019, 9, 3548–3553. [Google Scholar] [CrossRef]
- Dahiya, P.; Gangwar, M.K.; Sundararaju, B. Well-defined Cp*Co(III)-catalyzed Hydrogenation of Carbonates and Polycarbonates. ChemCatChem 2021, 13, 934–939. [Google Scholar] [CrossRef]
- Han, Z.; Rong, L.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. Catalytic Hydrogenation of Cyclic Carbonates: A Practical Approach from CO2 and Epoxides to Methanol and Diols. Angew. Chem. Int. Ed. 2012, 51, 13041–13045. [Google Scholar] [CrossRef]
- Wu, X.; Ji, L.; Ji, Y.; Elageed, E.H.M.; Gao, G. Hydrogenation of ethylene carbonate catalyzed by lutidine-bridged N-heterocyclic carbine ligands and ruthenium precursors. Catal. Commun. 2016, 85, 57–60. [Google Scholar] [CrossRef]
- Zubar, V.; Lebedev, Y.; Azofra, L.M.; Cavallo, L.; El-Sepegly, O.; Rueping, M. Hydrogenation of CO2-Derived Carbonates and Polycarbonates to Methanol and Diols by Metal-Ligand Cooperative Manganese Catalysis. Angew. Chem. Int. Ed. 2018, 57, 13439–13443. [Google Scholar] [CrossRef] [PubMed]
- Kaithal, A.; Hölscher, M.; Leitner, W. Catalytic Hydrogenation of Cyclic Carbonates using Manganese Complexes. Angew. Chem. Int. Ed. 2018, 57, 13449–13453. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Heldebrant, D.J. Catalytic coproduction of methanol and glycol in one pot from epoxide, CO2, and H2. RSC Adv. 2020, 10, 42557–42563. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Dagle, R.A.; Dagle, V.L.; Davidson, S.D.; Walter, E.D.; Burton, S.D.; Hoyt, D.W.; Heldebrant, D.J. Condensed-phase low temperature heterogeneous hydrogenation of CO2 to methanol. Catal. Sci. Technol. 2018, 8, 5098–5103. [Google Scholar] [CrossRef]
- Lao, D.B.; Galan, B.R.; Linehan, J.C.; Heldebrant, D.J. The steps of activating a prospective CO2 hydrogenation catalyst with combined CO2 capture and reduction. Green Chem. 2016, 18, 4871–4874. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Heldebrant, D.J. Towards environmentally benign capture and conversion: Heterogeneous metal catalyzed CO2 hydrogenation in CO2 capture solvents. Green Chem. 2020, 22, 828–834. [Google Scholar] [CrossRef]
- Rainbolt, J.E.; Koech, P.K.; Yonker, C.R.; Zheng, F.; Main, D.; Weaver, M.L.; Linehan, J.C.; Helderant, D.J. Anhydrous tertiary alkanolamines as hybrid chemical and physical CO2 capture reagents with pressure-swing regeneration. Energy Environ. Sci. 2011, 4, 480–484. [Google Scholar] [CrossRef]
- Sen, R.; Koch, C.J.; Goeppert, A.; Prakash, G.K.S. Tertiary Amine-Ethylene Glycol Based Tandem CO2 Capture and Hydrogenation to Methanol: Direct Utilization of Post-Combustion CO2. ChemSusChem 2020, 13, 6318–6322. [Google Scholar]
- Sen, R.; Goeppert, A.; Kar, S.; Prakash, G.K.S. Hydroxide Based Integrated CO2 Capture from Air and Conversion to Methanol. J. Am. Chem. Soc. 2020, 142, 4544–4549. [Google Scholar] [CrossRef]
- Pritchard, J.; Filonenko, G.A.; van Putten, R.; Hensen, E.J.M.; Pidko, E.A. Heterogeneous and homogeneous catalysts for the hydrogenation of carboxylic acid derivatives: History, advances and future directions. Chem. Rev. 2015, 44, 3808–3833. [Google Scholar]
- Ohgomori, Y.; Mori, S.; Yoshida, S.-I.; Watanabe, Y. The Role of Amide Solvents in the Formation of ethylene Glycol from Synthesis Gas. J. Mol. Catal. 1987, 40, 223–228. [Google Scholar] [CrossRef]
- Imperato, G.; Höger, S.; Lenoir, D.; König, B. Low melting sugar-urea-salt mixtures as solvents for organic reactions—Estimation of polarity and use in catalysis. Green Chem. 2006, 8, 1051–1055. [Google Scholar] [CrossRef]
- Kocienski, P.J. Protecting Groups, 3rd ed.; Thieme: New York, NY, USA, 2005. [Google Scholar]
- Balaraman, E.; Ben-David, Y.; Milstein, D. Unprecedented Catalytic Hydrogenation of Urea Derivatives to Amines and Methanol. Angew. Chem. Int. Ed. 2011, 50, 11702–11705. [Google Scholar] [CrossRef]
- Xie, Y.; Hu, P.; Ben-David, Y.; Milstein, D. A Reversible Liquid Organic Carrier System Based on Methanol-Ethylenediamine and Ethylene Urea. Angew. Chem. Int. Ed. 2019, 58, 5105–5109. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Garg, J.A.; Milstein, D. Combining Low-Pressure CO2 Capture and Hydrogenation to Form Methanol. ACS Catal. 2015, 5, 2416–2422. [Google Scholar] [CrossRef]
- Kumar, A.; Luk, J. Catalytic Hydrogenation of Urea Derivatives and Polyureas. Eur. J. Org. Chem. 2021, 2021, 4546–4550. [Google Scholar] [CrossRef]
- Das, U.K.; Kumar, A.; Ben-David, Y.; Iron, M.A.; Milstein, D. Manganese Catalyzed Hydrogenation of Carbamates and Urea Derivatives. J. Am. Chem. Soc. 2019, 141, 12962–12966. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Li, H.; Wang, Y.; Liu, Q. A Tailored Versatile and Efficient NHC-Based NNC-Pincer Manganese Catalyst for Hydrogenation of Polar Unsaturated Compounds. Angew. Chem. Int. Ed. 2023, 62, e202301042. [Google Scholar] [CrossRef] [PubMed]













































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
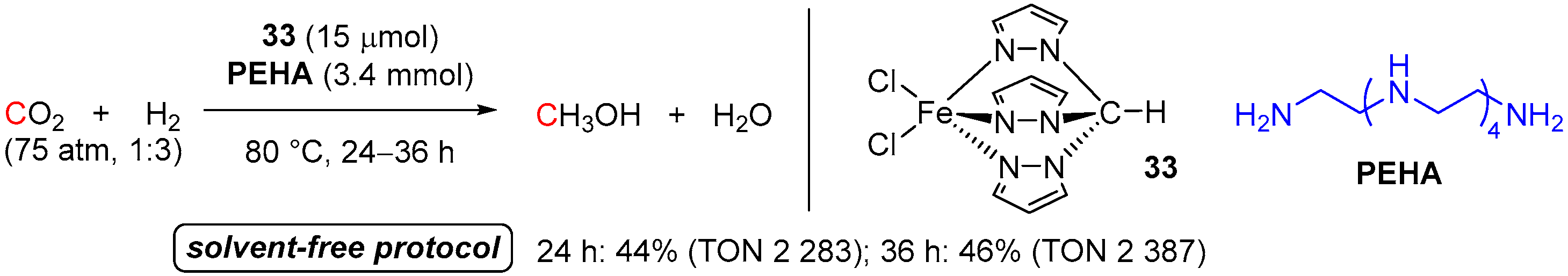{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
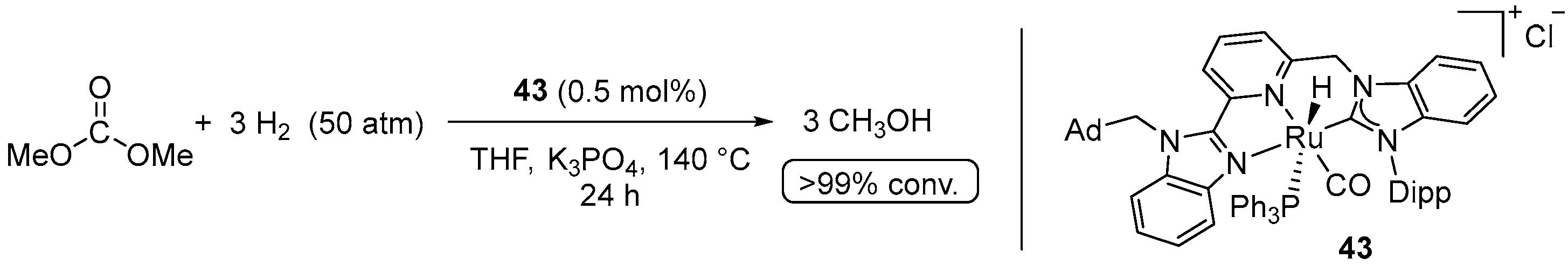{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
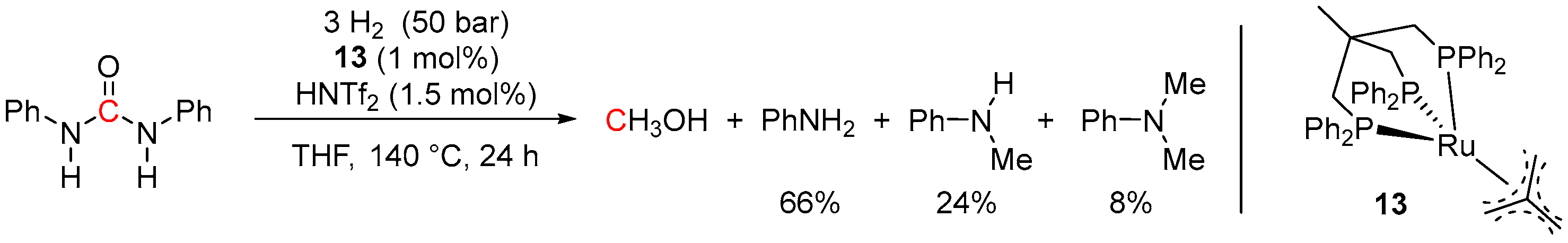{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Catalyst | T, °C/t, h/P(H2), bar b | TONmax for MeOH | By-Products c | Ref. |
|---|---|---|---|---|---|---|
| Formates | ||||||
| 1 | 1 (Ru) | 110/48/10 | 1155 | - d | [34] | |
| 2 | 2 (Ru) | 110/14/50 | 4700 | - d | [34] | |
| 3 | 3 (Ru) | 110/48/10 | 1215 | - d | [34] | |
| 4 | 4 (Ru) | 110/48/10 | 1365 | - d | [34] | |
| 5 | 11 (Ir) | 155/16/~83 | - d | HCOOMe e | [37] | |
| 6 | 13 (Ru) | 140/24/30 | 77 | - d | [38] | |
| 7 | 15 (Co) | 100/24/70 | 12 | - d | [41] | |
| 8 | 16 (Mn) | 110/30/30 | 43 | - d | [42] | |
| 9 | 20 (Mn) | 120/45/50 | - d | HCOOMe e | [43] | |
| Formamides | ||||||
| 10 | 23 (Ru) | 155/18/50 | 99 | - d | [48] | |
| 11 | 28 (Ru) | 160/1/~51 | 4700 | - d | [51] | |
| 12 | 30 (Ru) | 170/30/~41 | 9500 | - d | [55] | |
| 13 | 32 (Mn) | 150/24/70 | 128 | - d | [58] | |
| 14 | 34 (Fe) | 120/16/62 | 4950 | - d | [23] | |
| 15 | 36 (Ru) | 110/32/~10 | 97 | - d | [62] | |
| 16 | 37 (Fe) | 110/3/20 | 300 | - d | [63] | |
| 17 | 38 (Ru) | 60/3/80 | 71 | - d | [64] | |
| 18 | 39 (Mn) | 100/16/30 | 50 | - d | [66] | |
| 19 | 40 (Mo) | 100/24/30 | 20 | - d | [67] | |
| 20 | 41 (Ir) | 110/16/40 | 21 | - d | [68] | |
| 21 | 57 (Ru) | 180/20/30 | 210 | - d | [54] | |
| Acyclic carbonates | ||||||
| 22 | 1 (Ru) | 145/1/~61 | 2500 | - d | [34] | |
| 145/3.5/~41 | 2500 | - d | [34] | |||
| 23 | 2 (Ru) | 110/14/~51 | 4400 | - d | [34] | |
| 24 | 13 (Ru) | 140/16/50 | 94 (6219 in neat Me2CO3) | - d | [73] | |
| 25 | 16 (Mn) | 110/30/30 | 50 | - d | [42] | |
| 26 | 31 (Ru) | 140/24/200 | 3333 | - d | [57] | |
| 27 | 43 (Ru) | 140/24/~51 | 200 | - d | [75] | |
| 28 | 44 (Ru) | 135/24/~51 | 99 | - d | [78] | |
| 29 | 45 (Co) | 120/18/50 | 50 | - d | [79] | |
| 30 | 46 (Co) | 160/20/60 | 75 | - d | [80] | |
| Cyclic carbonates | ||||||
| 31 | 16 (Mn) | 110/50/50 | 50 | - d | [42] | |
| 32 | 32 (Mn) | 120/26/30 | 50 | - d | [84] | |
| 33 | 43 (Ru) | 140/24/~51 | 196 | - d | [75] | |
| 34 | 44 (Ru) | 130/20/~51 | 98 | - d | [78] | |
| 35 | 45 (Co) | 120/18/50 | 50 | - d | [79] | |
| 36 | 46 (Co) | 160/20/60 | 92 | - d | [80] | |
| 37 | 47 (Ru) | 140/4/~51 | 4950 | - d | [81] | |
| 38 | RuHCl(CO)(PPh3)3/48 | 130/12/50 | 44 | - d | [82] | |
| Carbonate salts | ||||||
| 39 | ammonium carbonate | 23 (Ru) | 140/72/70 | 184 | - d | [90] |
| 40 | potassium carbonate | 23 (Ru) | 140/20/70 | 200 | - d | [91] |
| 41 | potassium bicarbonate | 23 (Ru) | 140/48/70 | 184 | - d | [91] |
| 42 | potassium formate | 23 (Ru) | 140/48/70 | 200 | - d | [91] |
| Carbamates | ||||||
| 43 | 2 (Ru) | 110/48/~10 | 97 | - d | [34] | |
| 44 | 4 (Ru) | 135/72/60 | ~24 | - d | [98] | |
| 45 | 58 (Mn) | 130/24/50 | 49 | - d | [100] | |
| Urea derivatives | ||||||
| 46 | 2 (Ru) | 110/72/~14 | 47 | - d | [96] | |
| 47 | 4 (Ru) | 170/96/60 | 87 | - d | [97] | |
| 48 | 13 (Ru) | 140/24/50 | - d | Methylamines (32%) f | [73] | |
| 49 | 54 (Ru) | 170/168/60 | 79 | - d | [97] | |
| 50 | 55 (Ru) | 170/168/60 | 81 | - d | [97] | |
| 51 | 56 (Ir) | 130/24/50 | 47 | - d | [99] | |
| 52 | 58 (Mn) | 130/48/20 | 33 | - d | [100] | |
| 53 | 61 (Mn) | 150/24/50 | 19 | - d | [101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andizhanova, T.; Adilkhanova, A.; Khalimon, A.Y. Homogeneous Metal-Catalyzed Hydrogenation of CO2 Derivatives: Towards Indirect Conversion of CO2 to Methanol. Inorganics 2023, 11, 302. https://doi.org/10.3390/inorganics11070302
Andizhanova T, Adilkhanova A, Khalimon AY. Homogeneous Metal-Catalyzed Hydrogenation of CO2 Derivatives: Towards Indirect Conversion of CO2 to Methanol. Inorganics. 2023; 11(7):302. https://doi.org/10.3390/inorganics11070302
Chicago/Turabian StyleAndizhanova, Tolganay, Aziza Adilkhanova, and Andrey Y. Khalimon. 2023. "Homogeneous Metal-Catalyzed Hydrogenation of CO2 Derivatives: Towards Indirect Conversion of CO2 to Methanol" Inorganics 11, no. 7: 302. https://doi.org/10.3390/inorganics11070302
APA StyleAndizhanova, T., Adilkhanova, A., & Khalimon, A. Y. (2023). Homogeneous Metal-Catalyzed Hydrogenation of CO2 Derivatives: Towards Indirect Conversion of CO2 to Methanol. Inorganics, 11(7), 302. https://doi.org/10.3390/inorganics11070302