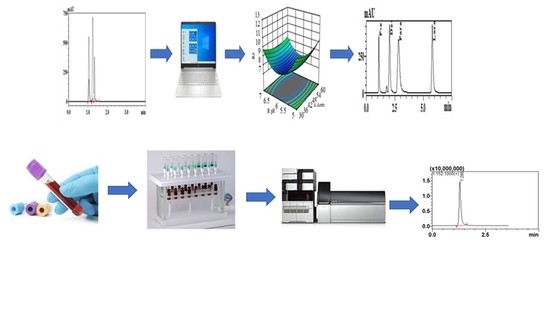
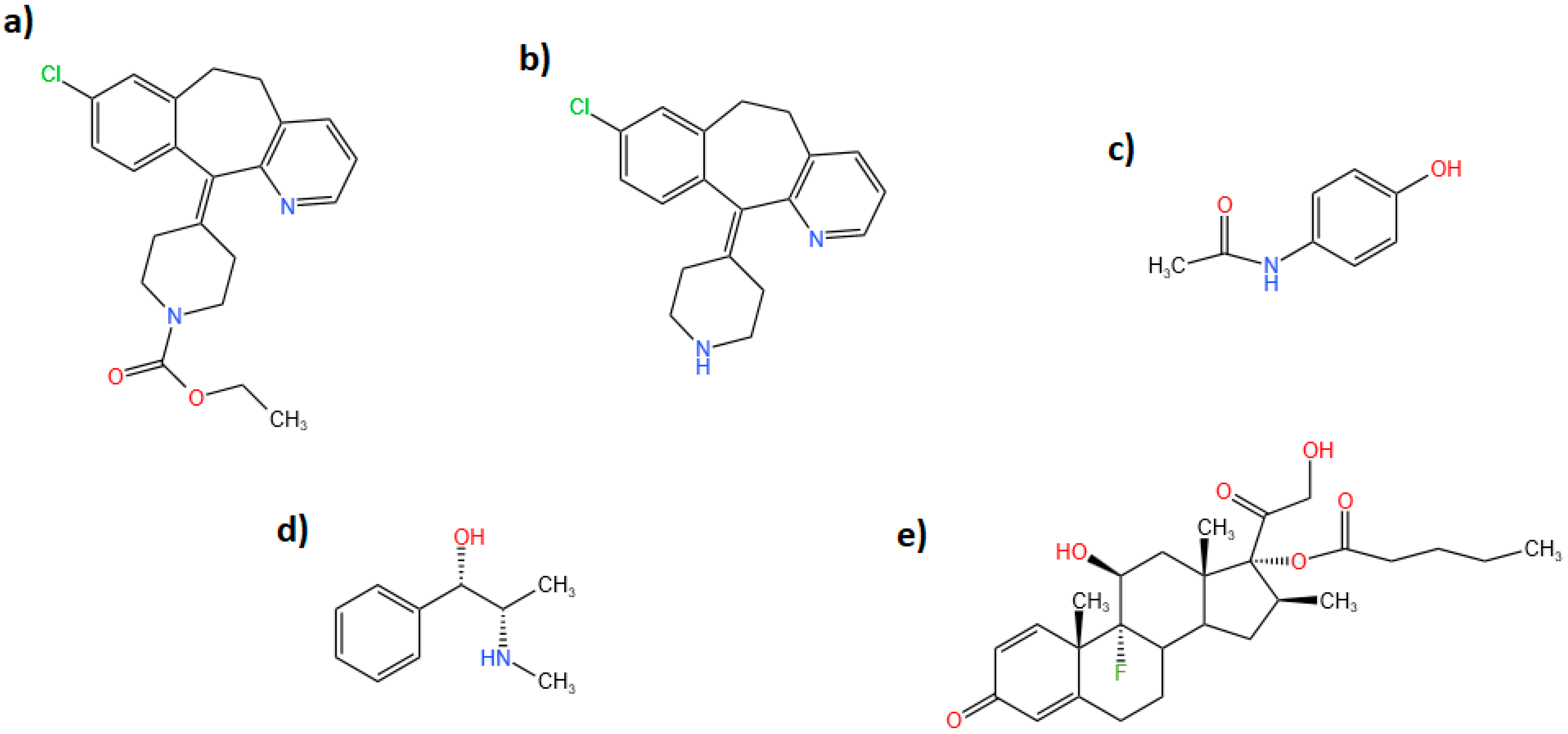
During the literature survey, a very limited number of reported methods were found for the determination of loratadine in its ternary mixture [
5,
6,
7,
8,
9,
10,
11]. However, to our knowledge, there are no reported chromatographic methods for the separation and quantification of the quaternary mixture loratadine, its active metabolite desloratadine, paracetamol, and pseudoephedrine. This is due to the very similar interaction of the mentioned analytes with the different stationary phases. In the present study, determination of loratadine, paracetamol, and pseudoephedrine in a tablet dosage form was attempted on C18 column using HPLC-PDA. The separation of the analytes was challenging due to the overlapping of paracetamol and pseudoephedrine peaks.
Additionally, the determination of the mixture with the pharmacologically active desloratadine in human plasma and urine was studied using SPE-HPLC-MS. Betamethasone valerate was used as an internal standard for the mass spectrometry method.
To achieve our goal, a QbD approach was used to separate the analytes over a minimum number of trials. Most of the published chromatographic methods use OFAT to develop a chromatographic method that can separate the tested mixture with an acceptable resolution. However, OFAT requires a large number of experiments, and it does not enable the full understanding of the main and interaction effects of the studied factors. Response surface study with a central composite design was applied to optimize the studied factors. The produced random 12 runs for the screening step and the other 20 runs for the optimization step are presented in the electronic
supplementary material, Table S1.
3.1. Screening Step
Irregular fractional two-level factorial design was implemented for the screening of all the factors and how they could affect the studied responses: resolution between each analyte and the next one and run time (R
t). Two levels for each factor were used: % acetonitrile at 30% and 70%, flow rate at 0.5 mL/min and 2 mL/min, pH 3 and 7, and oven temperature at 25 and 40 °C. The total number of runs for the screening step was 12, as presented in the electronic
supplementary material, Table S1. The factors were studied through a factorial study type using a two-level factorial design type. The process order of the developed screening model was main effects only and the type was a factorial model. The significance level to assess the effect of the model terms was alpha = 0.05. The ANOVA for the selected factorial model showed that only three factors were significant. The three factors were % acetonitrile (factor A), flow rate (factor B), and pH (factor C). The remaining factor (oven temperature) was a statistically insignificant factor, as presented in
Table 1. Pareto charts which indicate the significant factors for each response are presented in the electronic
supplementary material, Figure S1.
3.2. Optimization Step
Only the significant factors from the screening step were further studied for optimization. The study type was a response surface, and the design type was a central composite. The process order of the developed optimization model was quadratic, and the model type was polynomial. For optimization, 20 runs were carried out, as presented in the electronic
supplementary material, Table S1. The factors were studied at five different levels. ANOVA results are shown in
Table 1. Results show that the quadratic model was suggested for all the responses. The relationship between the different responses (y) and the significant factors (x) is shown in the following equation:
where x1, x2, and x3 are the studied factors acetonitrile %, flow rate, and pH, respectively.
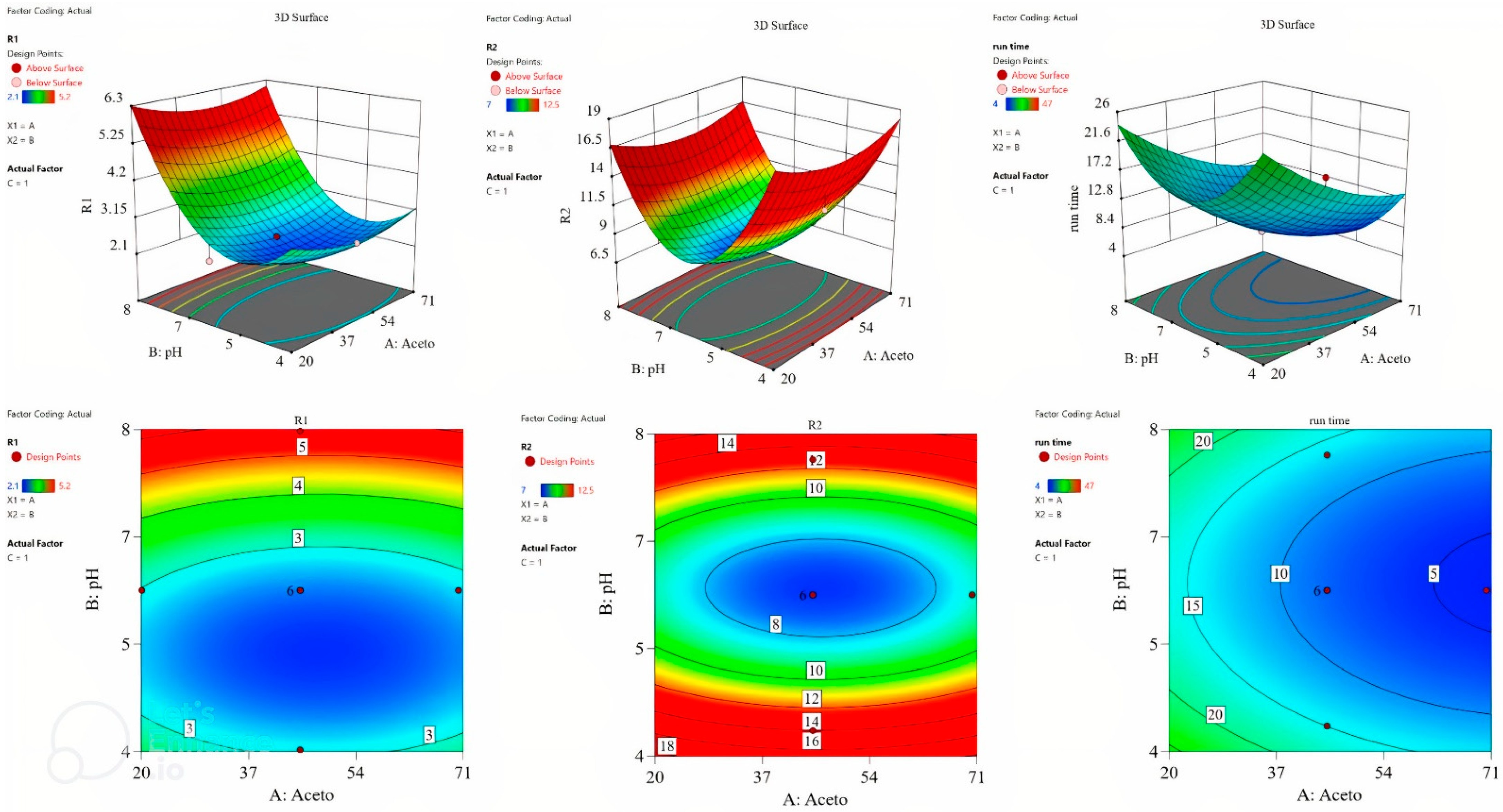
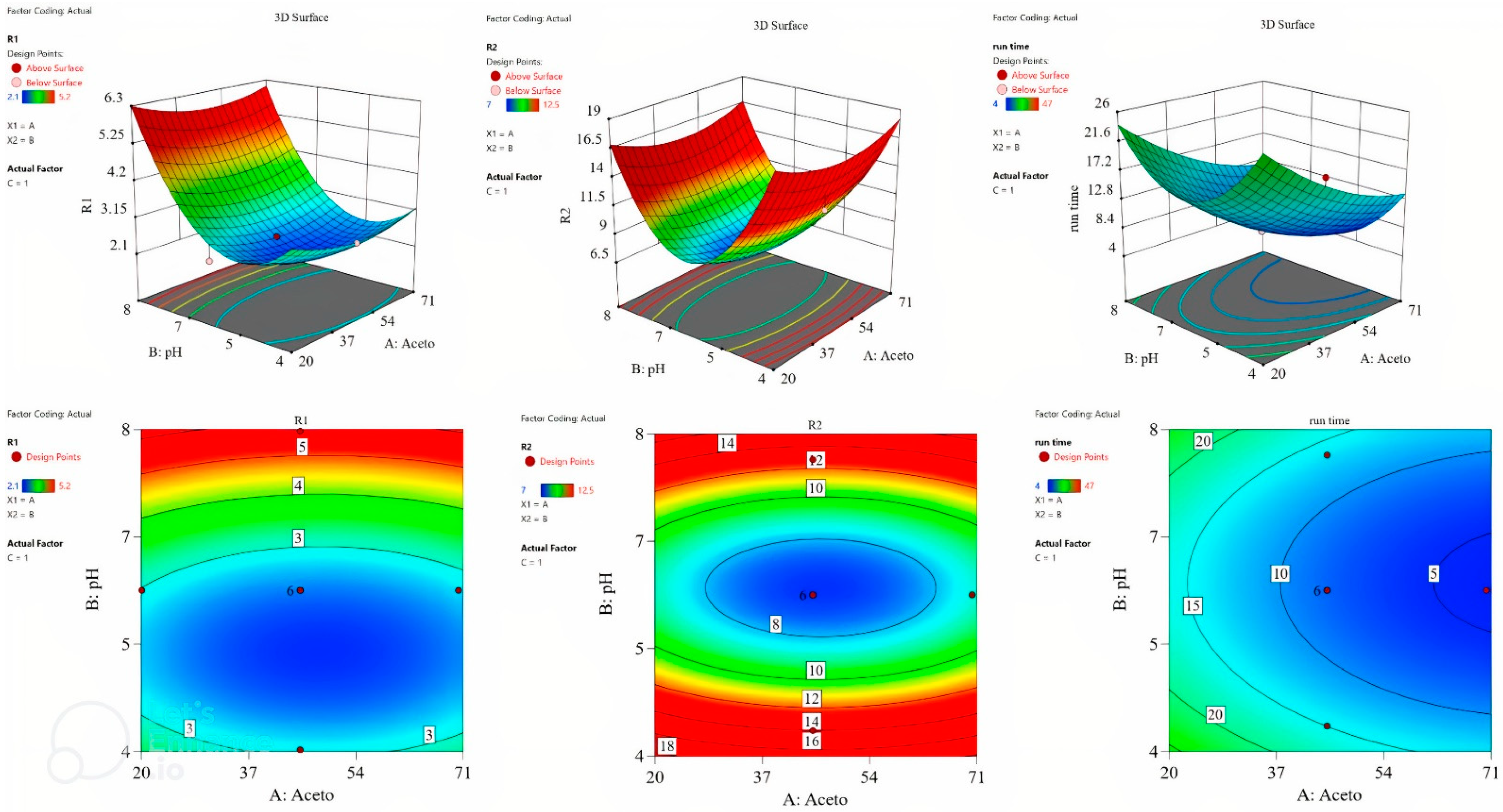
The surface plot and the contour plot are shown in
Figure 2, revealing the relationship between the factors and the studied responses.
The numerical optimization tool of the software was utilized to optimize the responses. The desirability criteria for R1 (resolution between pseudoephedrine and paracetamol) and R2 (resolution between paracetamol and loratadine) was set to the maximum, whereas it was set to the minimum for run time. The optimization tool suggested that 50% acetonitrile, pH 5.5, and 1.1 mL/min flow rate were the optimum conditions, reaching a desirability of 0.849.
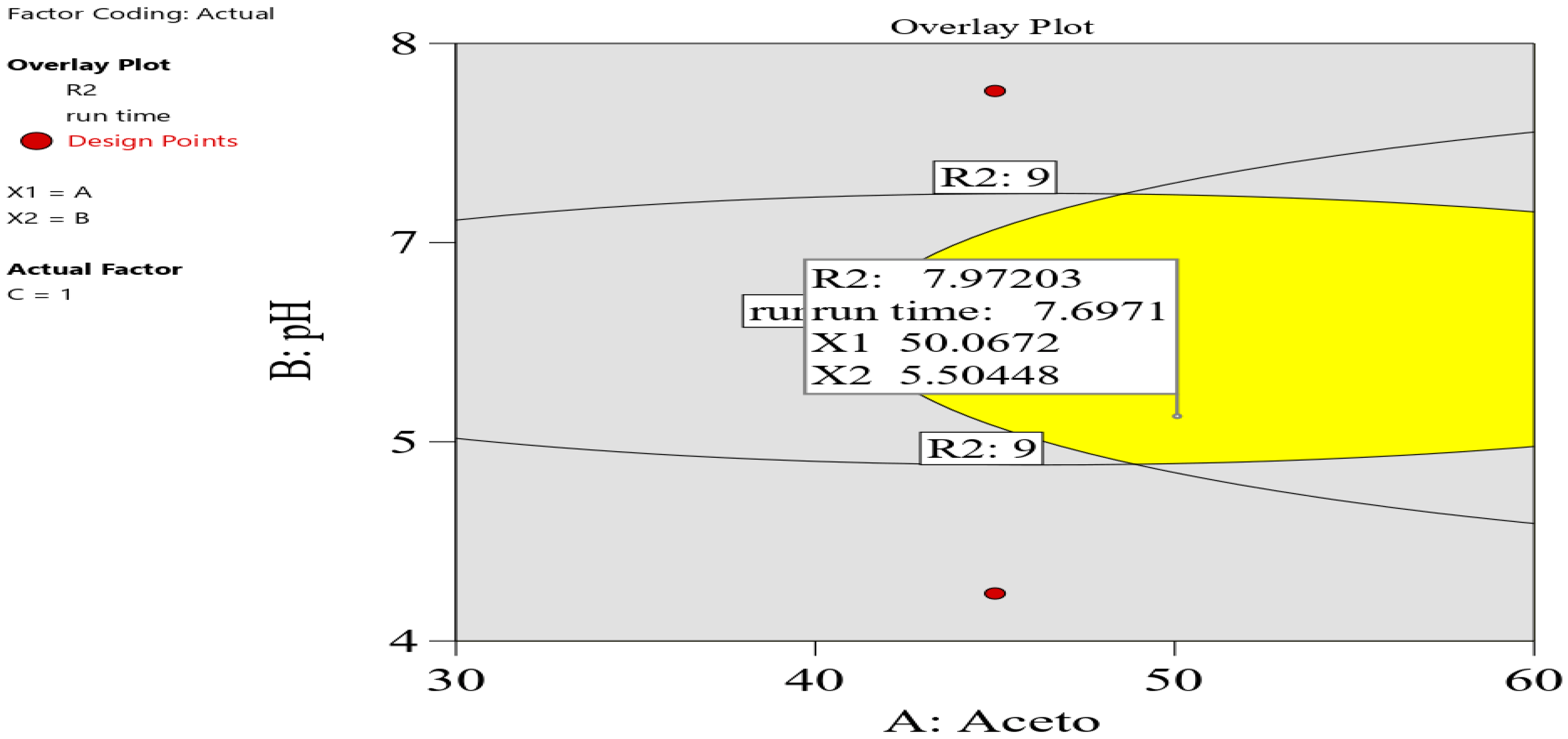
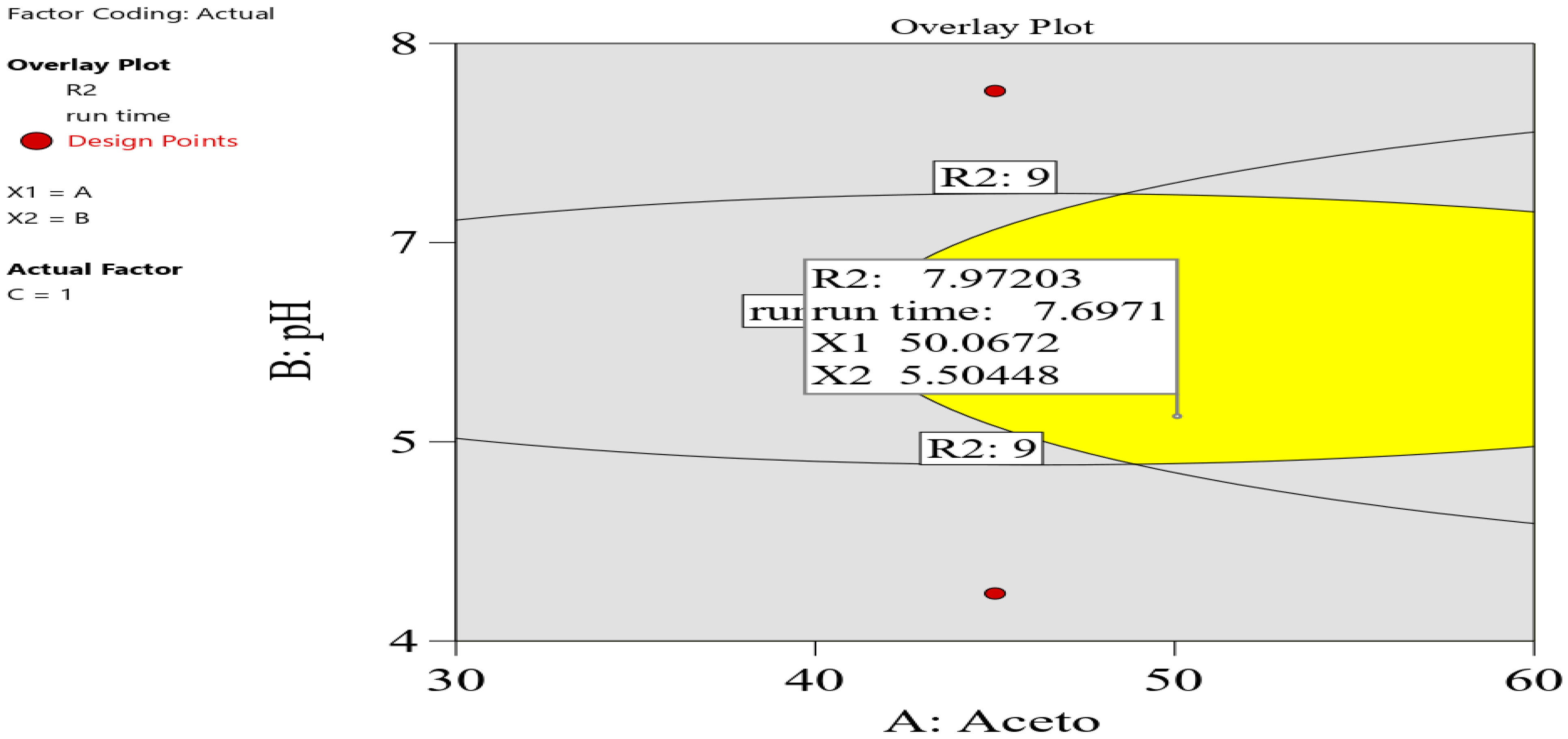
In the graphical optimization, the setting was as follows: R1 and R2 were set at 2 as the lower acceptable limit, and 10 min was set as the maximum run time. The overlay plot shows the optimum conditions, as shown in
Figure 3. Yellow indicates sweet-spot areas, whereas gray was assigned to areas that do not fit in the model.
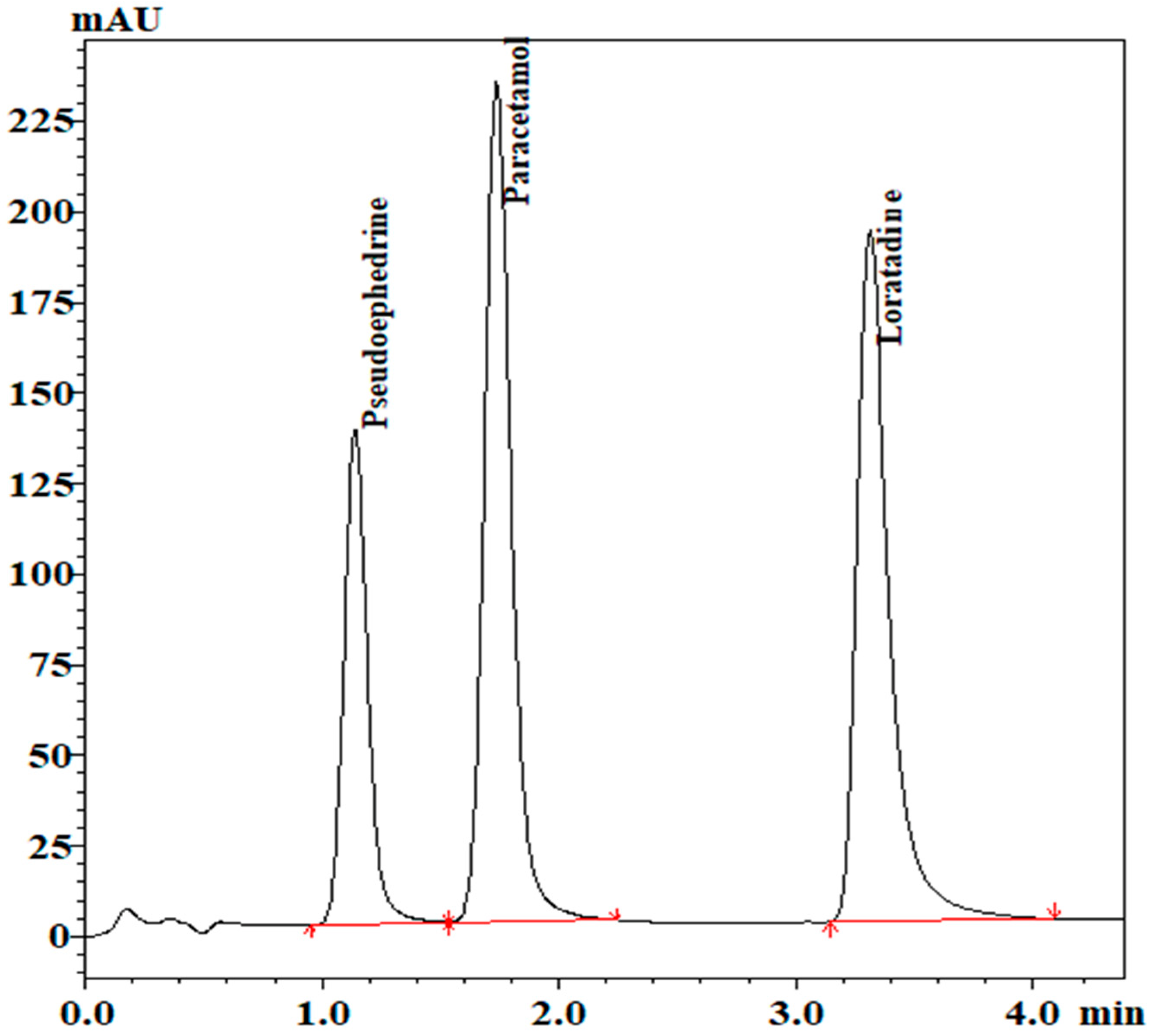
Loratadine was the reason for the long run time. So, gradient elution was used to shorten the run time. The gradient mobile phase was as follows:
The employed mobile phases were phase A: acetonitrile; phase B: potassium phosphate buffer, pH 5.5. The gradient was as follows: 50% phase B was used with a flow rate of 1.1 mL/min for 2 min, which was then decreased linearly to 30% B from 2 min to 3 min. Then, the gradient went back to the original conditions from 3 min to 4 min.
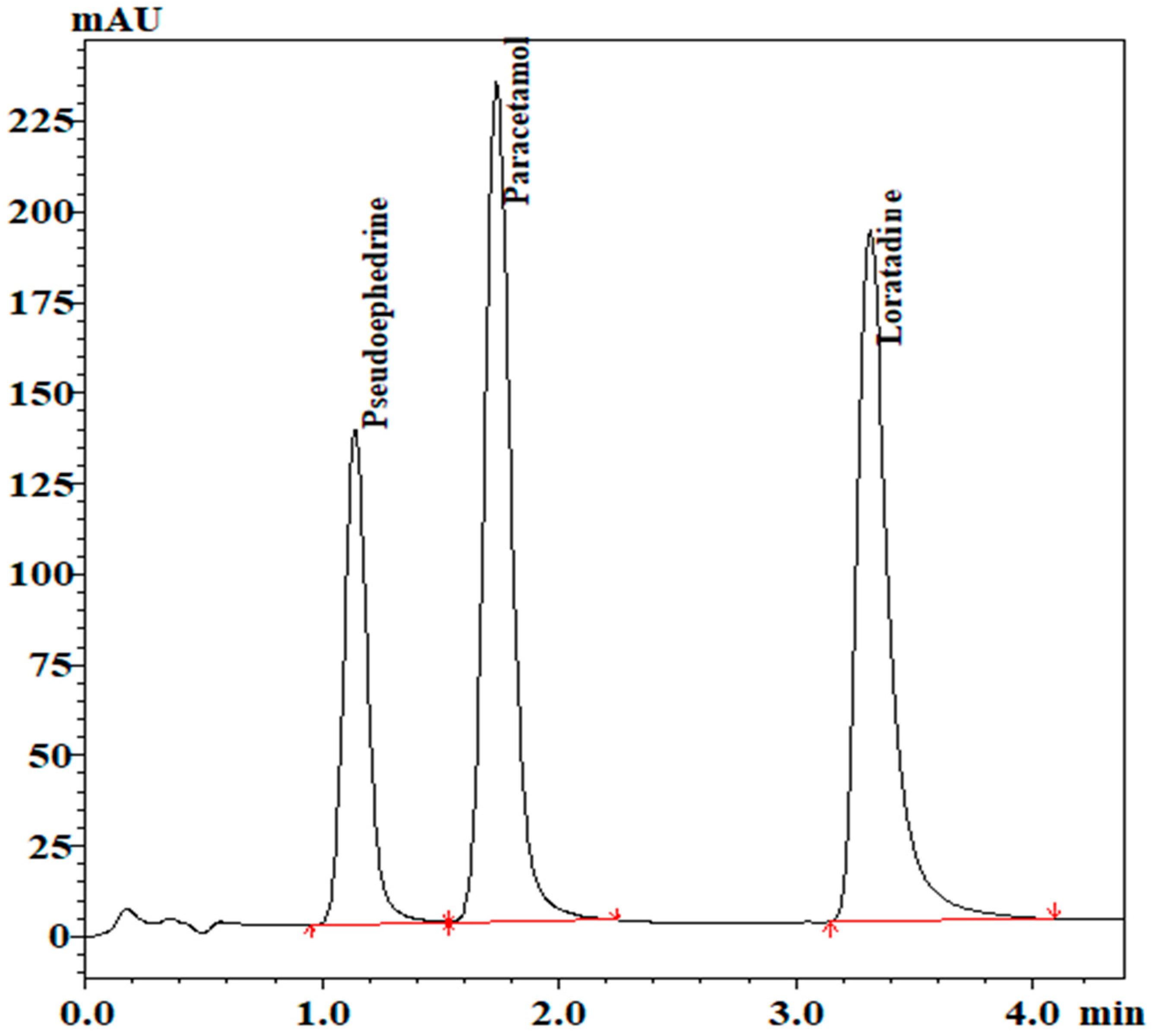
The chromatogram for separation of the analytes is shown in
Figure 4. System suitability parameters are shown in
Table 2.
Validation of the proposed method was carried out as per ICH guidelines [
21] regarding linearity, accuracy, precision, LOD and LOQ, and robustness. All the parameters were within the acceptable limits. Calibration curves are represented in
Figure S3 in the electronic
supplementary material. Validation parameters and results of the marketed dosage form are shown in
Table 3, whereas robustness results are represented in
Table S2 in the electronic
supplementary material.
3.3. SPE-HPLC-MS
To our knowledge, there are no reported HPLC-MS methods for determination of loratadine, its active metabolite desloratadine, paracetamol, and pseudoephedrine in human plasma and urine. So, the present study represents the first method for determination of the mixture in biological fluids by LC-MS.
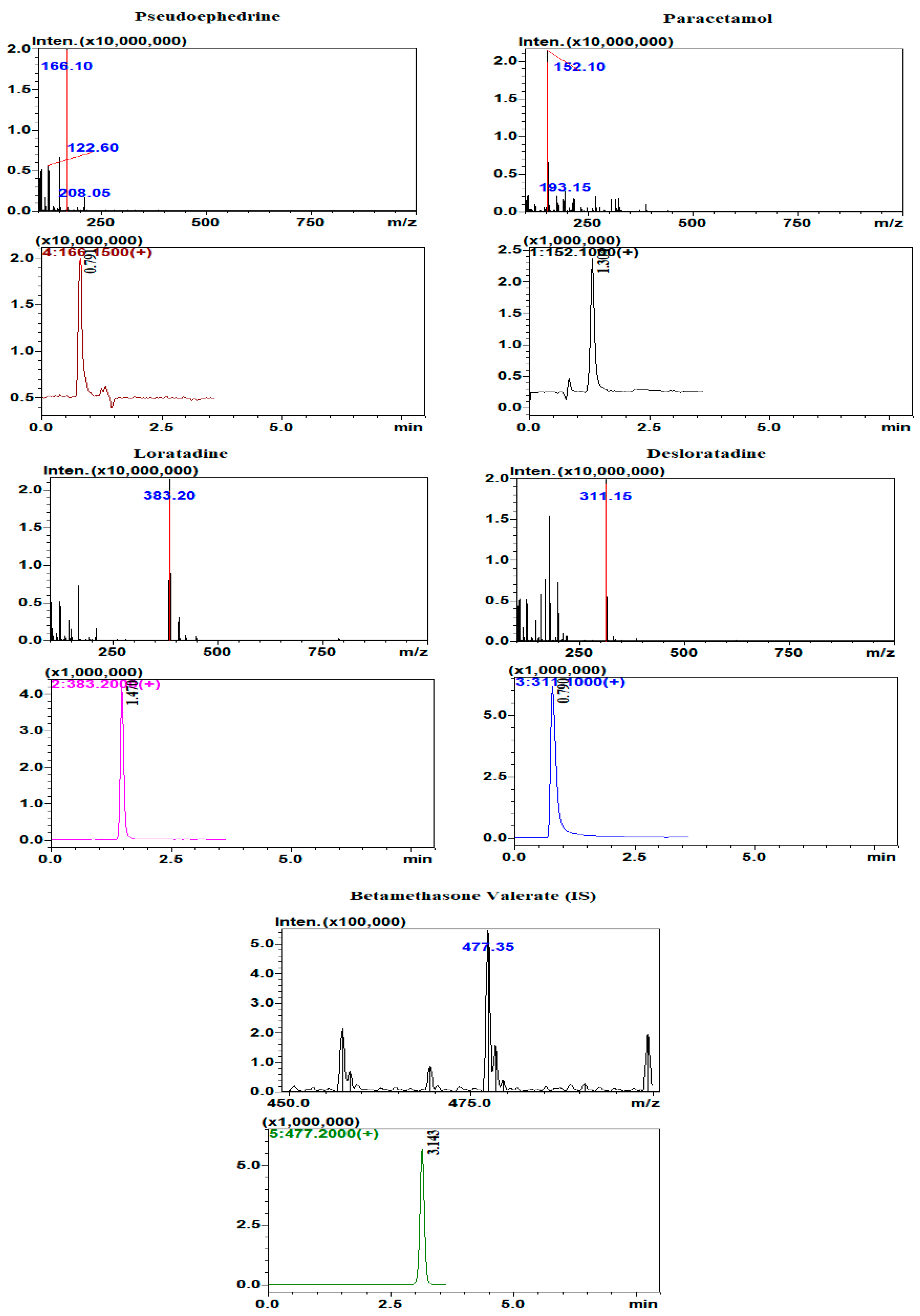
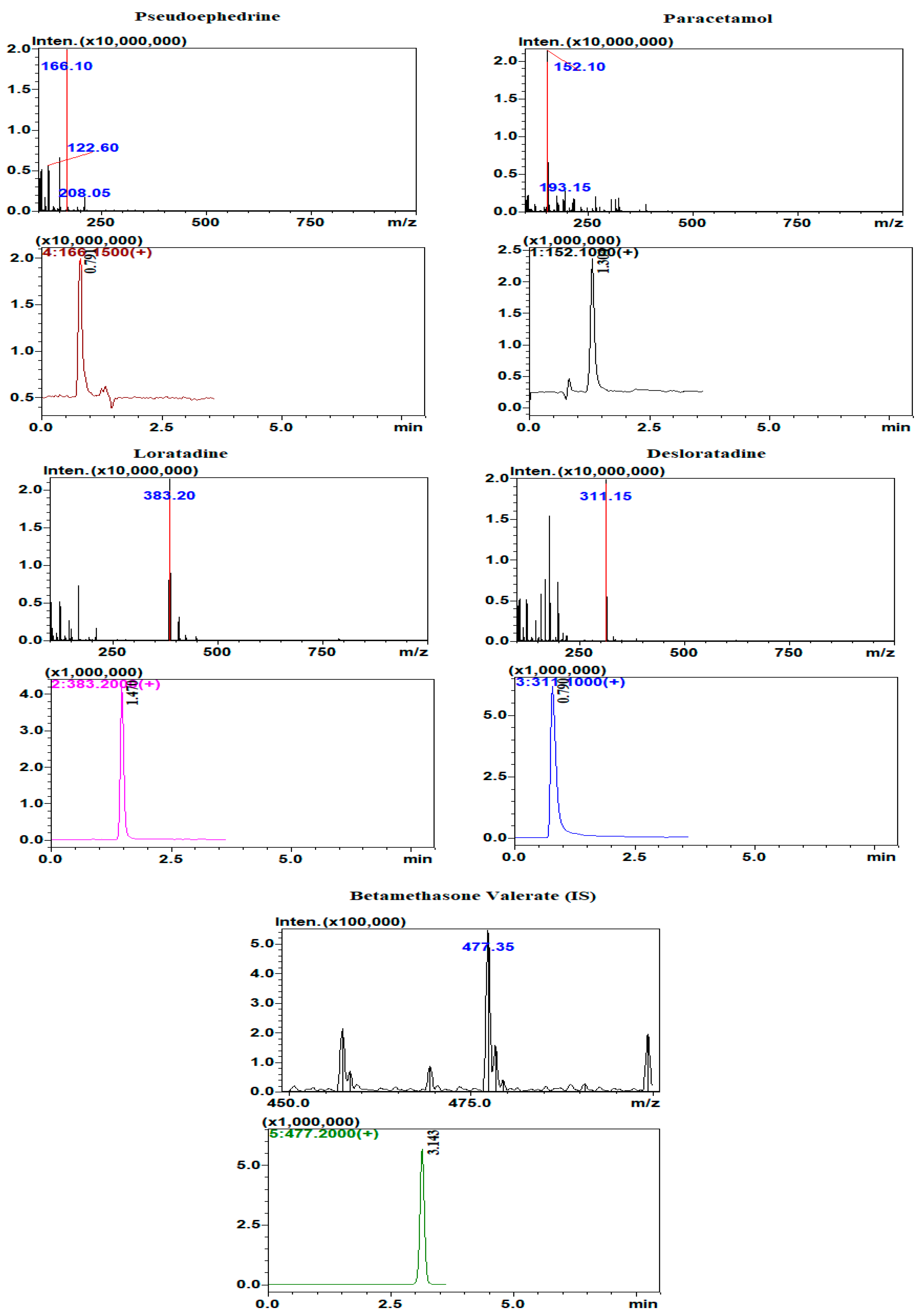
The very high sensitivity and selectivity of HPLC-MS makes it the technique of choice for determination of drugs and metabolites in biological body fluids. The separation of the four analytes and the IS was attempted using several mobile phase compositions and ratios. The optimum separation was obtained using a mobile phase composed of: phase A: 0.2% formic acid in acetonitrile; and phase B: 0.2% formic acid. The mobile phase ratio was 70% phase A: 30% phase B with a flow rate of 1.2 mL/min. The run time was about 3.5 min. Determination of the analytes and the internal standard was carried out under positive ionization mode. Selected ion monitoring (SIM) was used for the detection and quantification of the analytes where the protonated parent ions [M + H]+ of loratadine, desloratadine, paracetamol, pseudoephedrine, and betamethasone valerate (IS) were detected and determined at m/z 383.2, 311.2, 152.1, 166.15, and 477.2, respectively. The full MS scan and SIM chromatograms showing the retention times of the analyzed drugs are shown in
Figure 5.
Validation of the HPLC-MS method was carried out as per FDA guidelines for bioanalytical method validation regarding linearity, accuracy, precision, stability, recovery, and matrix effects [
22]. Calibration curves are represented in
Figure S4 in the electronic
supplementary material.
Extraction of the analytes from human plasma and urine was performed using solid phase extraction, where the recovery % for all the analytes was above 96 %. Selectivity of the method was tested by extracting and analyzing three different batches of blank human plasma and urine, where no interference from endogenous substances was detected at the m/z and retention times of the analytes.
The sensitivity of the method was tested via calculating LOQ and LOD. The lowest concentration that could be quantified for all the analytes was 0.5 ng/mL for all analytes where RSD did not exceed 20%.
Validation parameters of the developed method are shown in
Table 4 and
Table 5 and stability results are shown in
Table 6.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}