Abstract
A chiral analytical methodology was developed by nano-liquid chromatography (nano-LC) enabling the enantiomeric separation of two chiral drugs, lacosamide (novel antiepileptic drug) and colchicine (antiuremic drug), commercialized as pure enantiomers. A capillary column lab-packed with an amylose tris(3,5-dimethylphenylcarbamate) chiral stationary phase was used in a lab-assembled nano-LC system. Lacosamide and colchicine enantiomers were separated in less than 8.0 and 9.0 min, respectively, with resolution values of 1.6 and 2.3, using 20 nL of sample and 1.8 µL of mobile phase per analysis. The analytical characteristics of the proposed methodology were evaluated according to the International Council for Harmonisation (ICH) guidelines, showing good analytical performance with good recoveries (97–98% and 100–103%) and precision values (relative standard deviation (RSD) <10.5 and <3.0%) for lacosamide and colchicine enantiomers, respectively. LODs were 1.7 and 2.0 µg/mL for (S)- and (R)-lacosamide, respectively, and 1.0 µg/mL for both colchicine enantiomers. Additionally, the developed methodology enabled to detect a 0.1% of the enantiomeric impurities, fulfilling the ICH regulation requirements. The method was applied to the determination of lacosamide and colchicine enantiomers in different pharmaceutical formulations to ensure their quality control. The content of the enantiomeric impurities was below a 0.1% and the amount of (R)-lacosamide and (S)-colchicine agreed with their labeled contents.
1. Introduction
Nowadays, many of the drugs commercialized possess stereogenic centers, so they can exist as one or more pairs of enantiomers, which can react differently in biological processes. For this reason, the separation and determination of enantiomers of chiral drugs is a hot topic within the pharmaceutical field due to the impact that chirality may have on human health. Despite that enantiomers have the same atoms, bonds, and physicochemical properties, in presence of a chiral environment (e.g., the human body) they can exhibit different biological, pharmacokinetic, and pharmacodynamic activities [1]. In this sense, the desired biological or pharmacological activity of the drug is usually related mainly to a single enantiomer, whereas the other is frequently inactive, less active, has different activity, or can even be toxic [2]. As a result, the current trend is to commercialize drugs formulated in their enantiopure form, which means containing only the enantiomer responsible for the pharmaceutical activity. Indeed, it is better to employ single enantiomer formulations, as they enable using lower doses, provide lower interindividual variability and higher safety margin, as well as leading to less drug interactions and less side effects [3]. Nevertheless, it is necessary to develop sensitive and potent analytical methodologies to ensure the quality control of the pharmaceutical formulations marketed as pure enantiomers and guarantee the absence of their enantiomeric impurities. Accordingly, the International Council for Harmonisation (ICH), the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have set different guidelines for the correct validation of analytical methods and the marketing of chiral compounds [4,5,6]. These guidelines establish that the enantiomer responsible for the pharmacological activity must be identified and quantified and, in addition, a rigorous control of the presence of the enantiomeric impurities must be performed, which must not exceed of 0.1% in the pure enantiomeric drug formulation, referred to as the majority enantiomer.
In order to develop chiral analytical methodologies for the enantiomeric determination of drugs, separation techniques based on chromatographic and electrophoretic principles, such as TLC, HPLC, GC, CE, and SFC have been extensively employed [2,3,7,8]. Among them, HPLC is by far the technique of choice due to its relative simplicity in which the direct chiral separation is performed by using a chiral stationary phase (CSP). There are many types of CSPs with different chiral entities, such as cyclodextrins, polysaccharides, glycopeptides, antibiotics, pirkle-type selectors, proteins, crown-ether derivatives, ligand exchangers, ion exchangers, etc. [3,9]. Among them, polysaccharide-type CSPs with amylose- and cellulose-derived selectors are the most popular due to their powerful chiral recognition ability towards different compounds and their compatibility with aqueous and organic mobile phases [8]. On the other hand, miniaturization is a current trend with high impact in analytical chemistry which results in the decrease in the consumption of samples and organic solvents and the reduction of waste, saving time without losing efficiency and sensitivity [10]. In this sense, nano-LC emerged as a microseparation technique based on the miniaturization of HPLC, which uses capillary chromatographic columns with a small internal diameter (between 10 and 100 μm) [11]. The capillary contains the CSP and the mobile phase is delivered at low flow rates (10–700 nL/min), enabling minimum consumption of samples, mobile phase, and time without losing separation efficiency. Due to its advantages, it is presented as an alternative to conventional HPLC, enabling to develop sustainable analytical methods as it can drastically reduce the consumption of mobile phase and sample volumes [12].
Lacosamide and colchicine are two chiral drugs, which are commercialized as pure enantiomers. Therefore, they have to be subjected to a rigorous quality control to ensure the absence of their enantiomeric impurities. Lacosamide (Figure 1A) is one of the latest antiepileptic drugs approved in USA and Europe for adjunctive treatment of partial-onset seizures and diabetic neuropathic pain [2]. The (R)-configuration of this drug is the active enantiomer, whereas the (S)-enantiomer is inactive [1]. Nevertheless, the majority of the methods published for the analysis of lacosamide are achiral methodologies [13]. To date, only two works have described the enantiomeric separation of lacosamide by HPLC using polysaccharide coated chiral columns [14,15]. Nevertheless, to the best of our knowledge, its separation by nano-LC has never been reported before. On the other hand, colchicine (Figure 1B) is a chiral antiuremic drug in which both enantiomers show activity, but the (S)-enantiomer is more powerful and less toxic than the (R)-configuration [16]. Until now, few works have described the enantiomeric separation of colchicine [17,18,19]. The separation of colchicine enantiomers was first reported by TLC in 1993, but the analysis time took around 3.5 h [17]. More recently, our research team developed the first CE methodology enabling the enantiomeric separation of colchicine and its application to the analysis of pharmaceutical formulations [18]. Colchicine was also used as model compound to evaluate a vancomycin functionalized polymer-based monolith chiral capillary column in nano-LC [19] but the enantiomeric separation was not optimized nor applied to the analysis of pharmaceutical formulations.
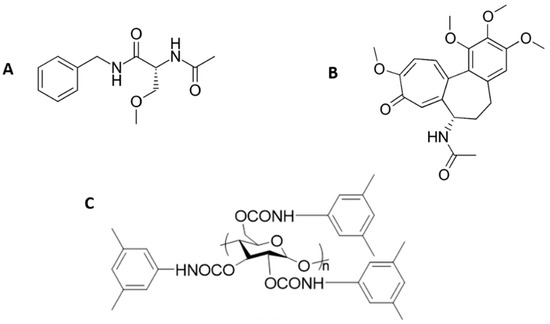
Figure 1.
Chemical structure of (A) lacosamide, (B) colchicine, and (C) the amylose tris (3,5-dimethylphenylcarbamate) chiral stationary phase employed.
Therefore, the aim of this work was to develop a chiral methodology by nano-LC, enabling for the first time the quality control of both lacosamide and colchicine pharmaceutical formulations by this microseparation technique. For this purpose, a capillary column was lab-packed with a commercial amylose tris(3,5-dimethylphenylcarbamate) CSP (Figure 1C) and was coupled to a lab-assembled nano-LC system. Different parameters were optimized to achieve the best enantiomeric separation conditions. In addition, the analytical parameters of the proposed method were evaluated following the ICH guidelines, and to demonstrate its feasibility it was applied to the determination of lacosamide and colchicine enantiomers in different single-enantiomer pharmaceutical formulations to ensure their quality control.
2. Materials and Methods
2.1. Chemicals, Reagents, and Standard Solutions
All reagents used were of analytical grade. Methanol (MeOH) and acetonitrile (ACN), 2-propanol and n-hexane were obtained from Scharlab S.L. (Barcelona, Spain), diethylamine (DEA) was from Merck (Darmstadt, Germany) and ethanol was acquired from Panreac (Barcelona, Spain). The water employed was purified in a Milli-Q system from Millipore (Bedford, MA, USA). Fused-silica capillaries (375 µm O.D. × 100 µm I.D.) were purchased from Ruifeng Chromatography Ltd. (Hebei, China).
(R,S)-colchicine was acquired from Santa Cruz Biotechnology (Dallas, TX, USA), (S)-colchicine was from Sigma-Aldrich (St. Louis, MO, USA), (R)-lacosamide was obtained from European Directorate for the Quality of Medicines (Strasbourg, France) and (S)-lacosamide from Clearsynth Labs LTD (Mumbai, India). The commercial pharmaceutical formulation of colchicine was obtained from a drug store in Alcalá de Henares (Madrid, Spain). As indicated on its label, the colchicine pharmaceutical formulation contained 0.5 mg colchicine and 5.0 mg of dicycloverine (an antispasmodic and gastric antisecretory drug) per capsule. The pharmaceutical formulation of lacosamide was acquired in its injectable form from a laboratory authorized to commercialize this drug. As indicated on its label, it had a concentration of 10,000 mg/L of lacosamide.
Stock standard solutions of colchicine (2000 mg/L) and lacosamide (1000 mg/L) enantiomers were prepared in ethanol and kept at −20 °C. These stock standard solutions were then properly diluted with ethanol until the desired concentration to achieve working standard solutions with the analytes at different concentration levels. The sample solution of the commercial pharmaceutical formulation of colchicine was obtained by grinding six capsules and homogenizing the resulting powder of the formulation. Then, 3 mL of ethanol were added to the grinded powder and the mixture was centrifuged at 4000 rpm for 20 min at 25 °C. The supernatant was collected and brought to a known volume. In the case of the pharmaceutical solution of lacosamide, an appropriate volume of the injectable drug was measured and diluted with ethanol to achieve the desired concentration. The resultant pharmaceutical formulation solutions were passed through a 0.45 µm nylon filter and kept at 4 °C until their analysis.
2.2. Preparation of the Capillary Column
LUX-Amylose-1 commercial stationary phase (amylose tris(3,5-dimethylphenylcarbamate), 5 µm particle size) was obtained from Phenomenex (Torrance, CA, USA). The chemical structure of the stationary phase is shown in Figure 1C. A capillary column (150 mm × 100 µm I.D.) was lab-packed with this stationary phase by the slurry packing method by Column Scientific Ltd. (Xiamen, China) following a previous reported procedure [20].
2.3. Nano-LC Analysis
The chromatographic separation was carried out on a laboratory-assembled nano-LC instrument consisting of a manual Valco four-port injection valve equipped with a 20 nL internal loop (Houston, TX, USA), a Shimadzu SPD-15C UV detector (Kyoto, Japan) with a lab-made on-column detection cell and a DiNA nano gradient pump (Tokyo, Japan). The amylose-based capillary stationary phase was coupled to the nano-LC instrument. Separation was performed at room temperature in isocratic mode, using as mobile phase the mixture n-hexane/2-propanol/DEA (80/20/0.2, v/v/v). To reduce the flow rate at nL/min levels, a stainless-steel T piece (Cheminert, Valco Instruments Houston, Texas, USA) with a flow split capillary (150 mm × 25 µm I.D.) was placed before the injection valve. Both pump and injection valve were connected to this piece, and the third entrance of the T was connected to the mobile phase reservoir through a fused silica capillary (150 mm × 75 µm I.D.) achieving a continuous recycling of the organic solvent. The flow rate was estimated by measuring the volume of mobile phase collected at the outlet of the capillary column over 5 min. The pump flow was varied from 5 to 50 µL/min, which corresponded to flow rates ranging from 100 to 1000 nL/min inside the capillary column. Best chiral separation conditions were established with a capillary flow rate of 200 nL/min. UV detection of lacosamide and colchicine was performed at 210 and 243 nm, respectively. The capillary was washed with MeOH between injections to ensure repeatability. Data acquisition was achieved with Unimicro Trisep™ Workstation 2003 (Shanghai, China).
3. Results and Discussion
3.1. Development of a Nano-LC Methodology for the Enantiomeric Separation of Lacosamide and Colchicine
Amylose-based CSPs can be used both in normal and reversed phase mode [8]. Thus, both modes were evaluated to achieve the enantioseparation of the target drugs. In reversed phase mode, different combinations of MeOH, ACN, and water were tested as the mobile phase at a capillary flow rate of 200 nL/min: MeOH/water 80/20 and 50/50 (v/v), ACN/water 80/20 and 50/50 (v/v). However, under these conditions no separation of the enantiomers was achieved and peaks corresponding to the drugs appeared beyond 20 min. In normal phase mode, usually an alkane, such as n-hexane, is used as the mobile phase mixed with organic modifiers such as ethanol and 2-propanol. Indeed, 2-propanol generally shows better selectivity as it provides lower interaction with the stationary phase through hydrogen bonding and does not compete with the analytes for the active sites [20,21]. Therefore, several combinations of n-hexane with 2-propanol (90/10, 80/20 and 70/30 (v/v)) were evaluated as the mobile phase at a capillary flow rate of 200 nL/min. With n-hexane/2-propanol 90/10 (v/v), lacosamide enantiomers were not separated (Figure 2A), whereas chiral discrimination was observed for colchicine, but the solvent signal overlapped with that of its first eluting enantiomer (Figure 2B). On the other hand, chiral discrimination for both drugs was observed with the combinations of n-hexane/2-propanol 80/20 and 70/30 (v/v) (Figure 2). With the combination n-hexane/2-propanol 80/20 (v/v), both enantiomers of lacosamide were separated within 10.0 min, however, with n-hexane/2-propanol 70/30 (v/v), the retention time for lacosamide enantiomers increased, eluting at 17.0 and 20.8 min (Figure 2A). Conversely, colchicine enantiomers eluted faster within 6.0 min with the combination n-hexane/2-propanol 70/30 (v/v), but the first enantiomer coeluted with the solvent signal (Figure 2B). Thus, since the enantiomeric separation of both lacosamide and colchicine was successfully achieved with the combination of n-hexane/2-propanol 80/20 (v/v), it was chosen as the most suitable mobile phase composition in both cases.
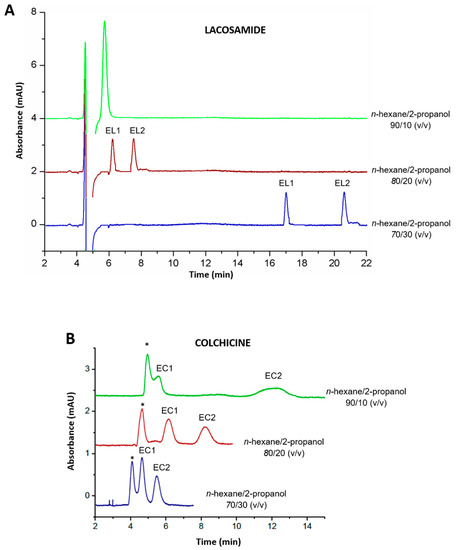
Figure 2.
Chromatograms corresponding to the separation of (A) a racemic mixture of lacosamide enantiomers (10.0 µg/mL each enantiomer) and (B) a racemic mixture of colchicine enantiomers (12.5 µg/mL each enantiomer) with different proportions of n-hexane/2-propanol in the mobile phase (90/10, 80/20 and 70/30 (v/v)). Experimental conditions: capillary flow 200 nL/min, λ: 210 nm (lacosamide) and 243 nm (colchicine), room temperature. * Peak corresponding to the solvent signal; EL1: first eluting enantiomer of lacosamide; EL2: second eluting enantiomer of lacosamide; EC1: first eluting enantiomer of colchicine; EC2: second eluting enantiomer of colchicine.
Since the addition of basic and/or acid additives in the mobile phase may have an important role in the chromatographic separation [22], the incorporation of a basic additive in the mobile phase was also evaluated. Usually, additives such as amines or acids are added in low proportion (0.05–0.5%) to the mobile phase in order to improve peak shape by minimizing peak tailing and/or broadening. Among the basic additives, DEA is the most widely used [8]. Therefore, the addition of DEA to the mobile phase in different proportions (0.1, 0.2, 0.3, 0.4, and 0.5%) was tested in order to improve separation efficiency and the shape of the peaks. It was observed that when adding a 0.4 or a 0.5% of DEA, the peak of the first eluting enantiomer was affected by the signal of the solvent. Therefore, these proportions of DEA were discarded. Among the proportions in the range from 0.0 to 0.3% of DEA, peak shapes slightly improved with a 0.2% of DEA in comparison to the other proportions evaluated. Thus, a 0.2% of DEA was selected to be added to the mobile phase composition.
In order to establish the elution order under the chromatographic conditions determined, lacosamide enantiomers were injected separately, as well as a standard solution enriched in one of the drug enantiomers. It was observed that the enantiomeric impurity ((S)-lacosamide) migrated before the active principle ((R)-enantiomer), which is the most desirable situation, with retention times of 6.3 and 7.5 min, respectively, and a resolution value of 1.6 (Figure 3A). On the other hand, for colchicine, the standard solution of (S)-colchicine was injected separately to evaluate the elution order. It was observed that the enantiomeric impurity ((R)-colchicine) also eluted before (S)-colchicine, at 6.0 and 8.3 min, respectively, with a resolution of 2.3 (Figure 3B).
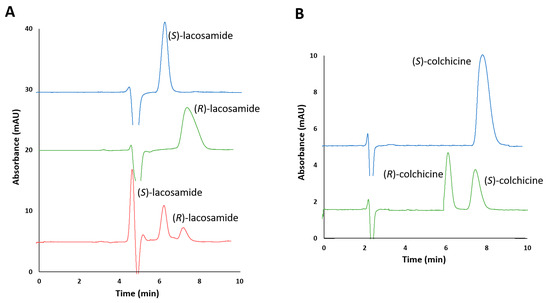
Figure 3.
(A) Chromatograms corresponding to (S)-lacosamide pure enantiomer standard solution (1000 µg/mL), (R)-lacosamide pure enantiomer standard solution (1000 µg/mL) and racemic standard solution containing 250 and 50 µg/mL of (S)-lacosamide and (R)-lacosamide, respectively. (B) Chromatograms corresponding to (S)-colchicine pure enantiomer standard solution (500 µg/mL) and racemic standard solution of colchicine containing 250 µg/mL of each enantiomer. Experimental conditions: mobile phase n-hexane/2-propanol (80/20, v/v) with 0.2% of diethylamine, capillary flow rate 200 nL/min, λ: 210 nm (lacosamide) and 243 nm (colchicine), room temperature.
3.2. Analytical Characteristics of the Nano-LC Method Developed
The analytical characteristics of the nano-LC methodology developed were evaluated according to the ICH guidelines [4], including selectivity, linearity, matrix effects, LOD, LOQ, relative limit of detection (RLOD), accuracy, and precision.
To evaluate the method selectivity, the lacosamide and colchicine pharmaceutical formulations were analyzed under the separation conditions established. No interfering peaks due to the drug excipients were found at the retention times established for the different drug enantiomers (Figure 4). Only the peaks of the majority enantiomer and one corresponding to the solvent were observed in the chromatograms. However, the peak of the solvent did not overlap with those of the enantiomers. Additionally, in the case of the colchicine formulation, the peak of dicycloverine was not detected at the wavelength of 243 nm, as this drug presents other maximum absorption wavelengths. Thus, selectivity of the method was appropriate.
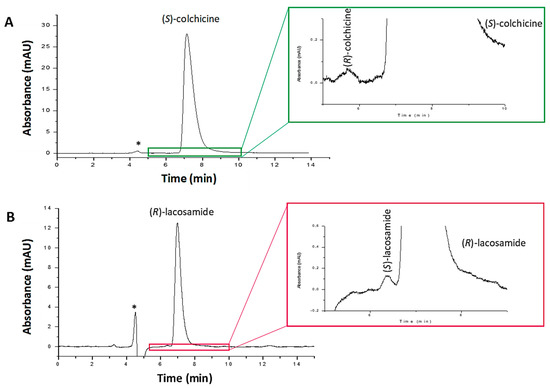
Figure 4.
Chromatograms corresponding to (A) colchicine pharmaceutical formulation solution containing 1000 μg/mL of (S)-colchicine and spiked with (R)-colchicine standard (1.0 μg/mL), and (B) lacosamide pharmaceutical formulation solution containing 1700 μg/mL of (R)-lacosamide and spiked with (S)-lacosamide standard (1.7 μg/mL). Experimental conditions: mobile phase n-hexane/2-propanol (80/20, v/v) with 0.2% of diethylamine, capillary flow rate 200 nL/min, room temperature, λ: 210 nm (lacosamide) and 243 nm (colchicine). * Peak corresponding to the solvent signal.
For linearity, external standard calibration curves were performed using eight concentration levels. Accordingly, the peak areas obtained were plotted against the analyte concentration (µg/mL). Good linearity was achieved, with R2 values ≥0.995 and ≥0.999 for lacosamide and colchicine enantiomers, respectively (Table 1). Additionally, confidence intervals for the slopes of both drug enantiomers did not include the zero value (linearity test) for a 95% confidence level. Moreover, it was confirmed by the analysis of variance (ANOVA) test that experimental data properly followed a linear model in the concentration range studied for both drug enantiomers (p-values > 0.05). To evaluate the presence of matrix effects, standard addition calibration curves were also performed. For this purpose, eight known amounts of lacosamide and colchicine enantiomers were added to their corresponding sample solutions of the pharmaceutical formulations containing a constant concentration of (R)-lacosamide or (S)-colchicine, as appropriate. The peak areas obtained were then plotted against the analyte concentration (µg/mL). To assess the matrix effects, the confidence intervals for the slopes obtained with both calibration methods (external standard calibration and standard addition calibration) were compared. For a 95% confidence level, the confidence intervals of the slopes of both calibration methods overlapped for each enantiomer (Table 1), so no statistically significant differences were observed among the slopes of both calibration methods. These results confirm the absence of matrix interferences, so the content of lacosamide and colchicine in their respective pharmaceutical formulations can be quantified using the external standard calibration method. Moreover, according to the European Pharmacopoeia [23], it has to be confirmed through the response relative factor (RRF) if the enantiomers have an equivalent response. The RRF is obtained by the quotient of the slopes of both enantiomers (slopeimpurity/slopeactive principle), and for an equivalent response the ratio has to be between 0.8 and 1.2. The RRF for lacosamide was 1.0 and 0.9 for colchicine. Therefore, it can be affirmed that the method responds in an equivalent way to both enantiomers in the two drugs.

Table 1.
Analytical characteristics of the nano-LC methodology developed for the enantiomeric determination of lacosamide and colchicine in pharmaceutical formulations using an amylose-based chiral stationary phase.
LODs and LOQs were estimated as the minimum concentration, yielding a S/N of 3 and 10 times, respectively. For (R)- and (S)-lacosamide, LODs were 2.0 and 1.7 µg/mL, and LOQs 6.7 and 5.7 µg/mL, respectively. For colchicine, the LOD was 1.0 µg/mL and the LOQ 3.5 µg/mL for both enantiomers (Table 1). Moreover, according to the ICH guidelines, the RLOD was assessed. The RLOD indicates the minimum concentration of enantiomeric impurity that can be detected as a function of the amount of active principle analyzed, which is measured as the LOD for the minor enantiomer divided by the concentration of the major enantiomer injected × 100. This parameter has to be at least of 0.1% according to the ICH regulations [4]. The RLOD of both drugs was successfully achieved, enabling to detect the 0.1% of their respective enantiomeric impurities, as shown in Figure 4. For lacosamide, the RLOD was achieved with a nominal value of 1700 µg/mL of (R)-lacosamide pharmaceutical formulation solution, whereas for colchicine a nominal value of 1000 µg/mL of (S)-colchicine pharmaceutical formulation solution was used. Therefore, according to the ICH guidelines, the nano-LC chiral method developed is sensitive enough to detect the enantiomeric impurity in the presence of high concentrations of the active principle, so it can be successfully applied to the quality control of the enantiomeric impurity of these drugs.
The accuracy was expressed as the recovery calculated from six solutions of a pharmaceutical formulation containing 50 µg/mL of (R)-lacosamide or (S)-colchicine (based on its labeled content), which were spiked with 5 and 50 µg/mL of (S)- and (R)-lacosamide, respectively, or with 8 and 100 µg/mL of (R)- and (S)-colchicine, respectively. Good recovery values were obtained for both drugs, including in all cases the 100% value (Table 1). Precision was assessed in terms of instrumental repeatability, method repeatability and intermediate precision. For the instrumental repeatability, six consecutive injections of the pharmaceutical formulation solution containing 50 µg/mL of (R)-lacosamide or (S)-colchicine and spiked with 5 or 8 µg/mL of (S)-lacosamide or (R)-colchicine, respectively, were performed. The RSD values obtained for lacosamide and colchicine enantiomers peak areas were lower than 7.5 and 2.0%, respectively (Table 1). For method repeatability, three replicates of a pharmaceutical formulation sample solution containing 50 µg/mL of (R)-lacosamide or (S)-colchicine and spiked with 5 or 8 µg/mL of (S)-lacosamide or (R)-colchicine, respectively, were injected in triplicate in the same day. As shown in Table 1, RSD values were lower than 8.0 and 2.5% for lacosamide and colchicine enantiomers peak areas, respectively. Finally, intermediate precision was assessed by the analysis of three replicates of the pharmaceutical formulation solution containing 50 µg/mL of (R)-lacosamide or (S)-colchicine and spiked with 5 or 8 µg/mL of (S)-lacosamide or (R)-colchicine, respectively, injected in triplicate in three consecutive days. In this case, RSD values for lacosamide enantiomer peak areas were lower than 10.5%, whereas RSD values for colchicine enantiomers were lower than 2.5% (Table 1). Therefore, the method showed good precision for both drugs, although better precision was achieved for colchicine than for lacosamide enantiomers, as RSD values achieved for colchicine were lower (Table 1). In summary, the method showed good analytical performance as all its parameters accomplished the ICH guidelines. Therefore, the nano-LC method developed is suitable for the quality control of these chiral drugs. Nonetheless, this method has only been tested for two pharmaceutical formulations of colchicine and lacosamide, so for other types of drugs the reliability of the procedure must be evaluated.
3.3. Analysis of Pharmaceutical Formulations
To prove the feasibility of the chiral methodology developed by nano-LC for the enantiomeric determination of lacosamide and colchicine, the method was applied to the analysis of different pharmaceutical formulations of these drugs. In the case of the lacosamide injectable pharmaceutical formulation, a content of 10,053 ± 6 µg/mL of (R)-lacosamide was quantified (corresponding to 101 ± 2% of the content indicated on its label). Therefore, this result agrees with the labeled amount of the injectable. On the other hand, the colchicine pharmaceutical formulation presented a content of 0.499 ± 0.001 mg of (S)-colchicine per capsule (corresponding to 99.9 ± 0.3% of the labeled content). Therefore, the content determined agreed with the labeled amount. Regarding the enantiomeric impurities, neither (S)-lacosamide nor (R)-colchicine were detected in their respective pharmaceutical formulations. Therefore, their concentrations were below the LOD of the method or were not present in the samples.
4. Conclusions
The use of an amylose-based CSP lab-packed in a capillary column enabled, under the optimized chromatographic conditions in normal phase mode, the separation of lacosamide and colchicine enantiomers by nano-LC with values for the enantiomeric resolution of 1.6 and 2.3 in less than 8 and 9 min, respectively. This is the first time that the enantiomeric separation of lacosamide was achieved by nano-LC. In this sense, a fast-chiral methodology was developed based on the use of 20 nL sample and 1.8 µL of mobile phase per analysis. Through the evaluation of the analytical characteristics of the method, its good analytical performance was confirmed, enabling to detect the enantiomeric impurities of both drugs up to a 0.1% with respect to the active enantiomer, fulfilling the ICH regulation requirements. Moreover, its feasibility was proved by its successful application to the analysis of pharmaceutical formulations. Therefore, the chiral methodology proposed in this work by nano-LC is suitable to ensure the quality control of lacosamide and colchicine in pharmaceutical formulations and to guarantee the absence of their enantiomeric impurities.
Author Contributions
Conceptualization, M.L.M. and M.Á.G.; methodology, N.C. and Z.J.; software, N.C.; validation, N.C. and M.Á.G.; formal analysis, N.C. and M.L.M.; investigation, N.C.; resources, M.L.M. and Z.J.; data curation, N.C. and M.Á.G.; writing—original draft preparation, N.C.; writing—review and editing, N.C., M.Á.G. and M.L.M.; visualization, M.Á.G. and M.L.M.; supervision, M.L.M. and M.Á.G.; project administration, M.L.M.; funding acquisition, M.L.M. and M.Á.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Spanish Ministry of Economy and Competitiveness (research projects CTQ2013-48740-P and CTQ2016-76368-P) and the University of Alcalá (project CCG19/CC-068).
Acknowledgments
Authors thank M. Montoya and J. Laloma for technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fortuna, A.; Alves, G.; Falcão, A. Chiral chromatographic resolution of antiepileptic drugs and their metabolites: A challenge from the optimization to the application. Biomed. Chromatogr. 2014, 28, 27–58. [Google Scholar] [CrossRef] [PubMed]
- Casado, N.; Valimaña-Traverso, J.; García, M.A.; Marina, M.L. Enantiomeric determination of drugs in pharmaceutical formulations and biological samples by electrokinetic chromatography. Crit. Rev. Anal. Chem. 2019, 1, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.R.; Maia, A.S.; Cass, Q.B.; Tiritan, M.E. Enantioseparation of chiral pharmaceuticals in biomedical and environmental analyses by liquid chromatography: An overview. J. Chromatogr. B 2014, 968, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Impurities in New Dug Products Q3B (R2). ICH Harmonized Tripartite Guidelines; International Conference of Harmonization (ICH): Brussels, Belgium, 2006. [Google Scholar]
- European Medicines Agency (EMA). Note for Guidance: Investigation of Chiral Active Substances. 1994. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/investigation-chiral-active-substances_en.pdf (accessed on 15 January 2020).
- U.S. Food and Drug Administration (FDA). Guidance, Compliance and Regulatory Information: Development of New Stereoisomeric Drugs. 1992. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs (accessed on 15 January 2020).
- Sánchez-López, E.; Castro-Puyana, M.; Marina, M.L. Electrophoresis. Capillary Electrophoresis: Chiral Separations. In Encyclopedia of Analytical Science; Worsfold, P., Poole, C., Townshend, A., Miró, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 334–345. [Google Scholar]
- Padró, J.M.; Keunchkarian, S. State-of-the-art and recent developments of immobilized polysaccharide-based chiral stationary phases for enantioseparations by high-performance liquid chromatography (2013–2017). Microchem. J. 2018, 140, 142–157. [Google Scholar] [CrossRef]
- Teixeira, J.; Tiritan, M.E.; Pinto, M.M.M.; Fernandes, C. Chiral stationary phases for liquid chromatography: Recent developments. Molecules 2019, 24, 865. [Google Scholar] [CrossRef] [PubMed]
- Filippou, O.; Bitas, D.; Samanidou, V. Green approaches in sample preparation of bioanalytical samples prior to chromatographic analysis. J. Chromatogr. B 2017, 1043, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Fanali, S. Nano-liquid chromatography applied to enantiomers separation. J. Chromatogr. A 2017, 1486, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Fanali, C.; Asensio-Ramos, M.; Hernández-Borges, J.; Rocco, A.; Fanali, S. Nanoliquid Chromatographic Separations. In Extreme Chromatography; Byrdwell, W.C., Holcapek, M., Eds.; AOCS Press: Urbana, IL, USA, 2011; pp. 301–380. [Google Scholar]
- Valarmathi, R.; Banu, S.F.; Akilandeswari, S.; Senthamarai, R.; Dhharshini, C.S.D. A review on new antiepileptic drug–Lacosamide and its analytical methods. Int. J. Chem. Pharm. Sci. 2013, 2, 181–186. [Google Scholar]
- Parmar, M.D.; Nimavat, K.S.; Vyas, K.B.; Rao, D.V.N.S.; Pande, R.A. A stability-indicating liquid chromatographic method for the quantification of new anti-epileptic drug lacosamide and its intermediates. Int. J. Pharm. Res. Sch. 2012, 1, 40–47. [Google Scholar]
- Chakravarthy, V.K.; Shankar, D.G. HPLC method for determination of lacosamide S (−) enantiomer in bulk and pharmaceutical formulation. Rasayan J. Chem. 2011, 4, 744–752. [Google Scholar]
- Roesner, M.; Capraro, H.G.; Jacobson, A.E.; Atwell, L.; Brossi, A.; Lorio, M.A.; Williams, T.H.; Sik, R.H.; Chignell, C.F. Biological effects of modified colchicines. Improved preparation of 2-demethylcolchicine, 3-demethylcolchicine, and (+)-colchicine and reassignment of the position of the double bond in dehydro-7-deacetamidocolchicines. J. Med. Chem. 1981, 24, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, R.; Ali, I. Resolution of racemic mixtures of hyoscyamine and colchicine on impregnated silica gel layers. Chromatographia 1993, 35, 679–680. [Google Scholar] [CrossRef]
- Menéndez-López, N.; Valimaña-Traverso, J.; Castro-Puyana, M.; Salgado, A.; García, M.A.; Marina, M.L. Enantiomeric separation of the antiuremic drug colchicine by electrokinetic chromatography. Method development and quantitative analysis. J. Pharm. Biomed. Anal. 2017, 138, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shao, H.; Luo, R.; Wang, Q.; Sánchez-López, E.; Fanali, S.; Marina, M.L.; Jiang, Z. A facile and efficient single-step approach for the fabrication of vancomycin functionalized polymer-based monolith as chiral stationary phase for nano-liquid chromatography. J. Chromatogr. A 2018, 1557, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Fernández, V.; Dominguez-Vega, E.; Chankvetadze, B.; Crego, A.L.; García, M.A.; Marina, M.L. Evaluation of new cellulose-based chiral stationary phases Sepapak-2 and Sepapak-4 for the enantiomeric separation of pesticides by nano liquid chromatography and capillary electrochromatography. J. Chromatogr. A 2012, 1234, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Antonucci, V.; Biba, M.; Gong, X.; Ge, Z. Simultaneous enantioseparation of a basic active pharmaceutical ingredient compound and its neutral intermediate using reversed phase and normal phase liquid chromatography with a new type of polysaccharide stationary phase. J. Pharm. Biomed. Anal. 2010, 51, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Gumustas, M.; Ozkan, S.A.; Chankvetadze, B. Separation and elution order of the enantiomers of some β-agonists using polysaccharide-based chiral columns and normal phase eluents by high-performance liquid chromatography. J. Chromatogr. A 2016, 1467, 297–305. [Google Scholar] [CrossRef] [PubMed]
- European Pharmacopoeia Commision. European Pharmacopoeia, 4th ed.; The European Pharmacopoeia Convention Inc.: Strasbourg, France, 2004; pp. 3843–3849, Supplement 4.6. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).