A Comparative Study of Rat Urine 1H-NMR Metabolome Changes Presumably Arising from Isoproterenol-Induced Heart Necrosis Versus Clarithromycin-Induced QT Interval Prolongation



Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Strategy
2.2. Animals and Protocol Experiment
2.3. Blood Collection and Serum Biochemical Analysis
2.4. Electrocardiogram and QT Interval Determination
2.5. Histopathological Examinations
2.6. Samples Preparation for 1H-NMR
2.7. Acquisition of 1H-NMR Urine Spectra and Data Processing
2.8. Multivariate Data Analysis, Metabolites Identification and Statistical Tests
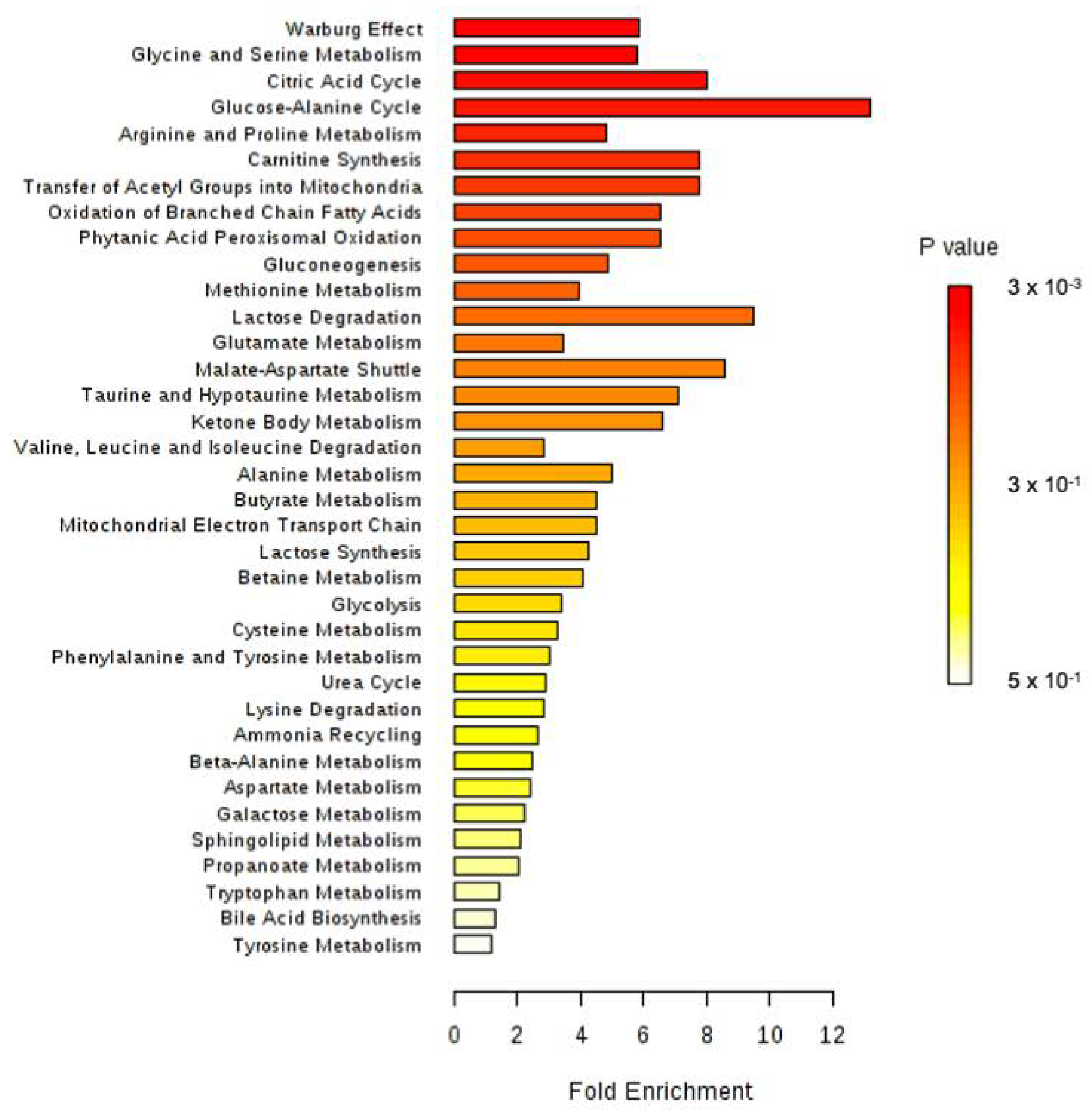
2.9. Enrichment Analysis
3. Results
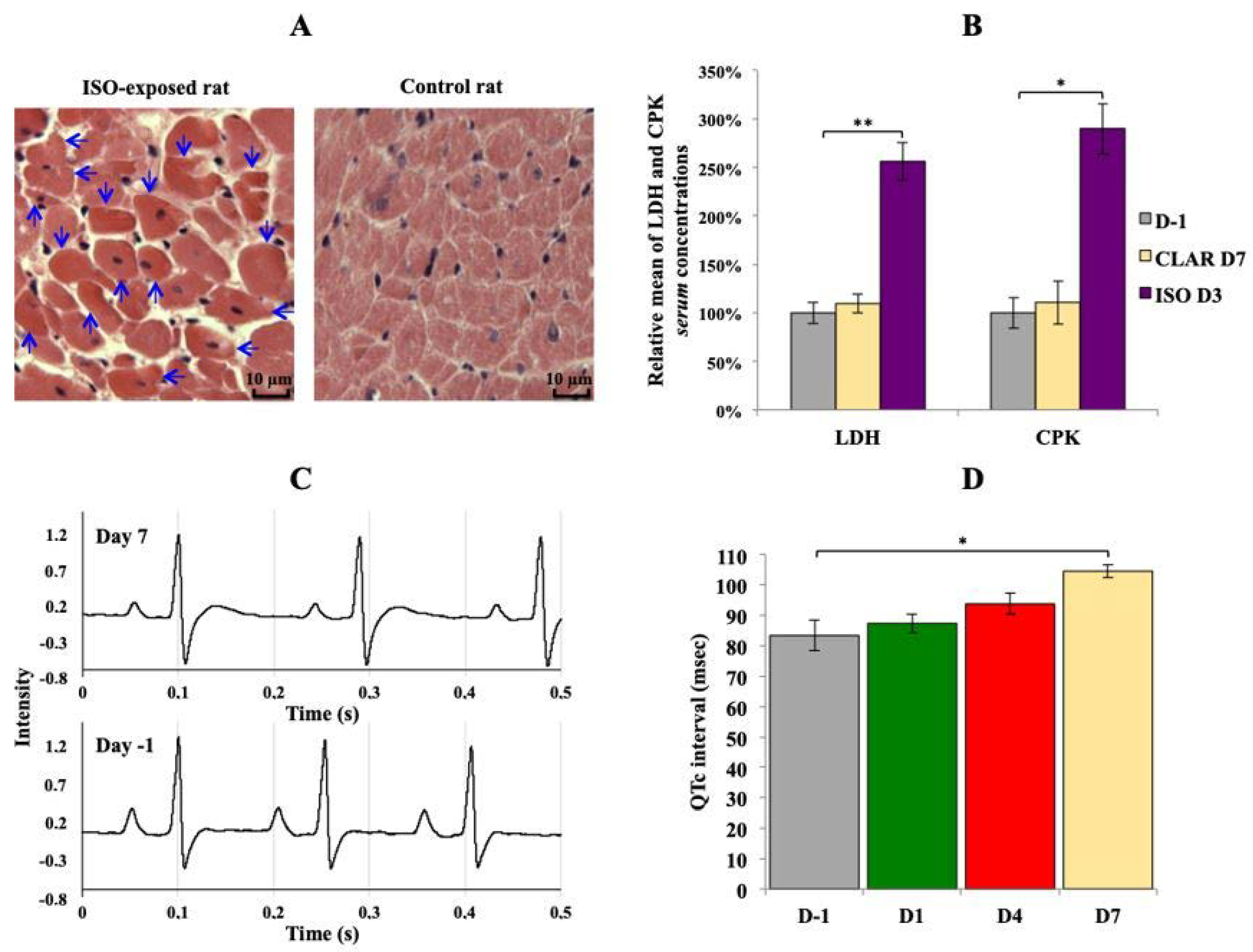
3.1. Validation of Drug-Induced Heart Adverse Effects Specificity
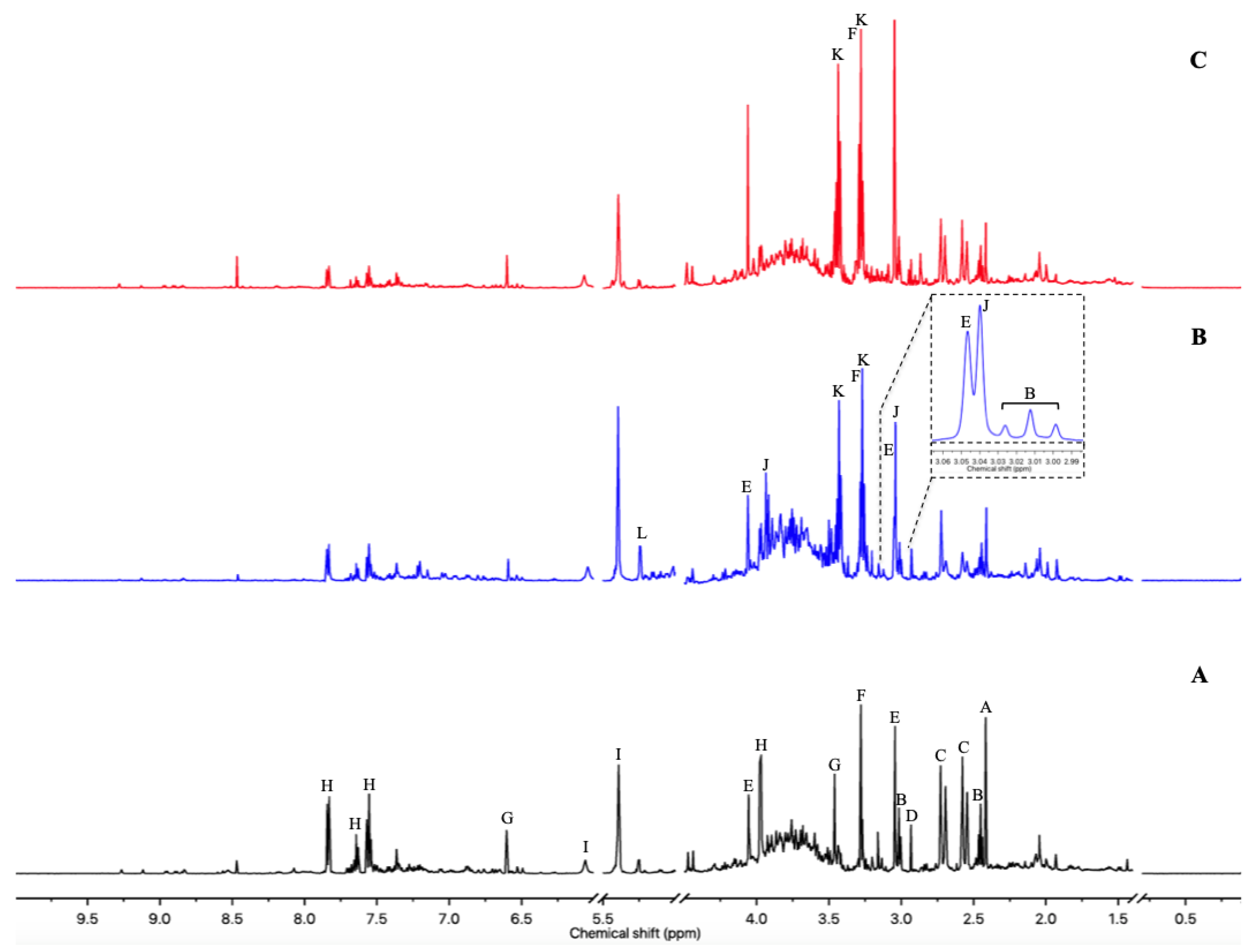
3.2. Urine Metabolome Changes Induced by Isoproterenol Exposure
3.3. Urine Metabolome Changes Induced by Acute Clarithromycin Exposure
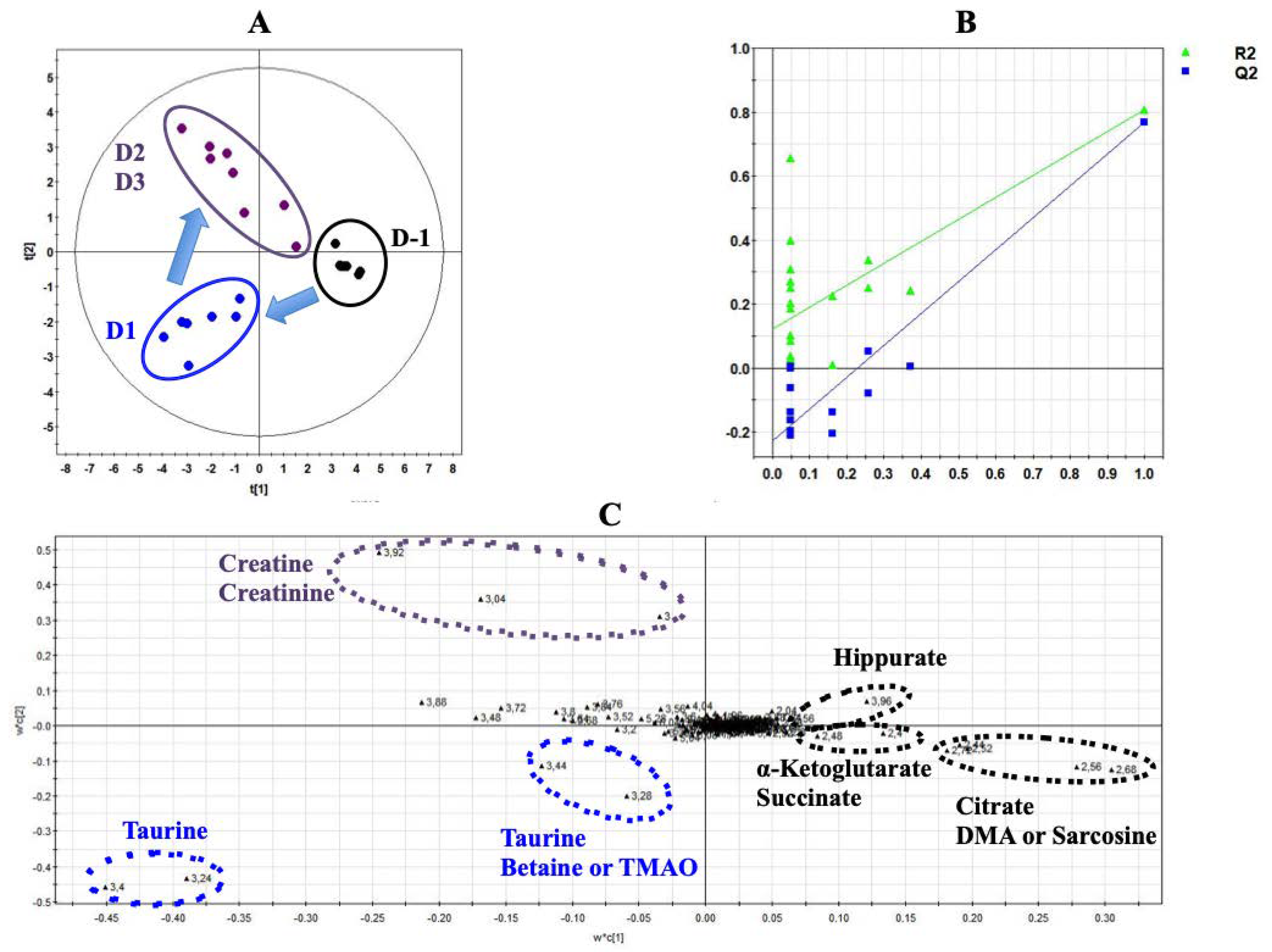
3.4. Multivariate Analysis of Both Experiments Combination
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gwathmey, J.K.; Tsaioun, K.; Hajjar, R.J. Cardionomics: A new integrative approach for screening cardiotoxicity of drug candidates. Expert Opin. Drug Metab. Toxicol. 2009, 5, 647–660. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; LaMantia, G.; Colombo, N.; Civelli, M.; De Giacomi, G.; Rubino, M.; Veglia, F.; Fiorentini, C.; Cipolla, C.M. Anthracycline-Induced Cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Finet, J.E.; Tang, W.H.W. Protecting the heart in cancer therapy. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Florescu, M.; Cinteza, M.; Vinereanu, D. Chemotherapy-induced Cardiotoxicity. Maedica 2013, 8, 59–67. [Google Scholar] [PubMed]
- Steinherz, L.J.; Steinherz, P.G.; Tan, C.T.; Heller, G.; Murphy, M.L. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA 1991, 2662, 1672–1677. [Google Scholar] [CrossRef]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA A Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef]
- Pai, V.B.; Nahata, M.C. Cardiotoxicity of chemotherapeutic agents: Incidence, treatment and prevention. Drug Saf. 2000, 22, 263–302. [Google Scholar] [CrossRef]
- Fung, M.; Thornton, A.; Mybeck, K.; Wu, J.H.-H.; Hornbuckle, K.; Muniz, E. Evaluation of the Characteristics of Safety Withdrawal of Prescription Drugs from Worldwide Pharmaceutical Markets-1960 to 1999. Drug Inf. J. 2001, 35, 293–317. [Google Scholar] [CrossRef]
- Polak, S.; Pugsley, M.K.; Stockbridge, N.; Garnett, C.; Wiśniowska, B. Early Drug Discovery Prediction of Proarrhythmia Potential and Its Covariates. AAPS J. 2015, 17, 1025–1032. [Google Scholar] [CrossRef][Green Version]
- Podrid, P.J. Proarrhythmia, a serious complication of antiarrhythmic drugs. Curr. Cardiol. Rep. 1999, 1, 289–296. [Google Scholar] [CrossRef]
- Zipes, D. Proarrhythmic effects of antiarrhythmic drugs. Am. J. Cardiol. 1987, 59, E26–E31. [Google Scholar] [CrossRef]
- Kelleni, M.T.; AbdelBasset, M. Drug Induced Cardiotoxicity: Mechanism, Prevention and Management. In Cardiotoxicity; Tan, W., Ed.; IntechOpen: London, UK, 2018; Available online: http://www.intechopen.com/books/cardiotoxicity/drug-induced-cardiotoxicity-mechanism-prevention-and-management (accessed on 31 January 2020).
- Varga, Z.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Siegl, P.; Corsini, A.; Herrmann, J.; Lerman, L.O.; Benghozi, R. Drug attrition during pre-clinical and clinical development: Understanding and managing drug-induced cardiotoxicity. Pharmacol. Ther. 2013, 138, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Shockcor, J.; Holmes, E. Metabonomic Applications in Toxicity Screening and Disease Diagnosis. Curr. Top. Med. Chem. 2002, 2, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, F.; Riefke, B.; Schlotterbeck, G.; Ross, A.; Senn, H.; Amberg, A. NMR and MS Methods for Metabonomics. Methods Mol. Biol. Clifton NJ 2010, 691, 385–415. [Google Scholar]
- Lindon, J.; Holmes, E.; Nicholson, J. Metabonomics Techniques and Applications to Pharmaceutical Research & Development. Pharm. Res. 2006, 23, 1075–1088. [Google Scholar] [CrossRef]
- Nicholson, J.; Wilson, I.D. Understanding ’Global’ Systems Biology: Metabonomics and the Continuum of Metabolism. Nat. Rev. Drug Discov. 2003, 2, 668–676. [Google Scholar] [CrossRef]
- Hasim, A.; Ali, M.; Mamtimin, B.; Ma, J.-Q.; Li, Q.-Z.; Abudula, A. Metabonomic signature analysis of cervical carcinoma and precancerous lesions in women by 1H NMR spectroscopy. Exp. Ther. Med. 2012, 3, 945–951. [Google Scholar] [CrossRef]
- Nicholson, J.; Connelly, J.; Lindon, J.C.; Holmes, E. Metabonomics: A platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 2002, 1, 153–161. [Google Scholar] [CrossRef]
- Robertson, D.G. Metabonomics in Toxicology: A Review. Toxicol. Sci. 2005, 85, 809–822. [Google Scholar] [CrossRef]
- Deidda, M.; Mercurio, V.; Cuomo, A.; Noto, A.; Mercuro, G.; Dessalvi, C.C. Metabolomic Perspectives in Antiblastic Cardiotoxicity and Cardioprotection. Int. J. Mol. Sci. 2019, 20, 4928. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ju, L.; Hou, Z.; Deng, H.; Zhang, Z.; Wang, L.; Yang, Z.; Yin, J.; Zhang, Y. Screening, Verification, and Optimization of Biomarkers for Early Prediction of Cardiotoxicity Based on Metabolomics. J. Proteome Res. 2015, 14, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Yadav, C.H.; Najmi, A.K.; Akhtar, M.; Khanam, R. Cardioprotective role of H₃R agonist imetit on isoproterenol-induced hemodynamic changes and oxidative stress in rats. Toxicol. Mech. Methods. 2015, 25, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Kmecova, J.; Klimas, J. Heart rate correction of the QT duration in rats. Eur. J. Pharmacol. 2010, 641, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, H.; Hanada, E.; Yamamoto, K.; Sawada, Y.; Iga, T. Pharmacokinetic-Pharmacodynamic Analysis of the Electrocardiographic Effects of Terfenadine and Quinidine in Rats. Biol. Pharm. Bull. 1996, 19, 1189–1196. [Google Scholar] [CrossRef]
- Ohtani, H.; Taninaka, C.; Hanada, E.; Kotaki, H.; Sato, H.; Sawada, Y.; Iga, T. Comparative Pharmacodynamic Analysis of Q-T Interval Prolongation Induced by the Macrolides Clarithromycin, Roxithromycin, and Azithromycin in Rats. Antimicrob. Agents Chemother. 2000, 44, 2630–2637. [Google Scholar] [CrossRef] [PubMed]
- Cox, I.; Gaudard, M. Discovering Partial Least Squares with JMP; SAS Institute: Charlotte, NC, USA, 2013. [Google Scholar]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Ellinger, J.; Chylla, R.A.; Ulrich, E.L.; Markley, J.L. Databases and Software for NMR-Based Metabolomics. Curr. Metab. 2013, 1, 28–40. [Google Scholar] [CrossRef]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Bartlett, M.S. Properties of sufficiency and statistical tests. Proc. R. Soc. Lond. Ser.-Math. Phys. Sci. 1937, 160, 268–282. [Google Scholar]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Mladěnka, P.; Applova, L.; Patočka, J.; Costa, V.M.; Remião, F.; Pourová, J.; Mladěnka, A.; Karlíčková, J.; Jahodář, L.; Vopršalová, M.; et al. Comprehensive review of cardiovascular toxicity of drugs and related agents. Med. Res. Rev. 2018, 38, 1332–1403. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Pugsley, M.K.; Fermini, B.; Curtis, M.; Koerner, J.; Accardi, M.; Authier, S. Cardiac voltage-gated ion channels in safety pharmacology: Review of the landscape leading to the CiPA initiative. J. Pharmacol. Toxicol. Methods 2017, 87, 11–23. [Google Scholar] [CrossRef]
- Mythili, S.; Malathi, N. Diagnostic markers of acute myocardial infarction. Biomed. Rep. 2015, 3, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.L. Cardiac energetics. Physiol. Rev. 1978, 58, 174–254. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 2018, 176, 4302–4318. [Google Scholar] [CrossRef]
- Sheeran, F.L.; Pepe, S. Energy deficiency in the failing heart: Linking increased reactive oxygen species and disruption of oxidative phosphorylation rate. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2006, 1757, 543–552. [Google Scholar] [CrossRef]
- Asnani, A.; Shi, X.; Farrell, L.; Lall, R.; Sebag, I.A.; Plana, J.C.; Gerszten, R.E.; Scherrer-Crosbie, M. Changes in Citric Acid Cycle and Nucleoside Metabolism Are Associated with Anthracycline Cardiotoxicity in Patients with Breast Cancer. J. Cardiovasc. Transl. Res. 2019, 1–8. [Google Scholar] [CrossRef]
- Ali, S.E.; Farag, M.A.; Holvoet, P.; Hanafi, R.; Gad, M.Z. A Comparative Metabolomics Approach Reveals Early Biomarkers for Metabolic Response to Acute Myocardial Infarction. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Laborde, C.M.; Mourino-Alvarez, L.; Posada-Ayala, M.; Alvarez-Llamas, G.; Serranillos-Reus, M.G.; Moreu, J.; Vivanco, F.; Padial, L.R.; Barderas, M.G. Plasma metabolomics reveals a potential panel of biomarkers for early diagnosis in acute coronary syndrome. Metabolomics 2013, 10, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Wei, D.; Wang, J.-S.; Guo, P.; Li, M.; Yang, M.; Kong, L. Study of the Cardiotoxicity of Venenum Bufonis in Rats using an 1H NMR-Based Metabolomics Approach. PLoS ONE 2015, 10, e0119515. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Lou, Z.; Liao, W.; Zhu, Z.; Dong, X.; Zhang, W.; Li, W.; Chai, Y. Potential Biomarkers in Mouse Myocardium of Doxorubicin-Induced Cardiomyopathy: A Metabonomic Method and Its Application. PLoS ONE 2011, 6, e27683. [Google Scholar] [CrossRef] [PubMed]
- Hoff, R.T.; Koh, C.-K. Isoproterenol Induced Insulin Resistance Leading to Diabetic Ketoacidosis in Type 1 Diabetes Mellitus. Case Rep. Endocrinol. 2018, 2018, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Klein, J.; Neumann, S.; Eszlinger, M.; Paschke, R. Isoproterenol inhibits resistin gene expression through a GS -protein-coupled pathway in 3T3-L1 adipocytes. FEBS Lett. 2001, 500, 60–63. [Google Scholar] [CrossRef]
- Issad, T.; Combettes, M.; Ferre, P. Isoproterenol Inhibits Insulin-Stimulated Tyrosine Phosphorylation of the Insulin Receptor Without Increasing its Serine/Threonine Phosphorylation. JBIC J. Biol. Inorg. Chem. 1995, 234, 108–115. [Google Scholar] [CrossRef]
- Yang, J.; Hodel, A.; Holman, G. Insulin and Isoproterenol Have Opposing Roles in the Maintenance of Cytosol pH and Optimal Fusion of GLUT4 Vesicles with the Plasma Membrane. J. Biol. Chem. 2002, 277, 6559–6566. [Google Scholar] [CrossRef]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and creatinine metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arter. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef]
- Burg, M.B.; Ferraris, J.D. Intracellular organic osmolytes: Function and regulation. J. Biol. Chem. 2008, 283, 7309–7313. [Google Scholar] [CrossRef]
- Huxtable, R.; Bressler, R. Effect of taurine on a muscle intracellular membrane. Biochim. Biophys. Acta 1973, 323, 573–583. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Jong, C.J.; Kc, R.; Azuma, J. Physiological roles of taurine in heart and muscle. J. Biomed. Sci. 2010, 17. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.W.; Azuma, J.; Madura, J. Mechanisms underlying taurine-mediated alterations in membrane function. Amino Acids 1995, 8, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, X.; Hu, P.; Liang, Q.; Liang, X.-P.; Wang, Y.-M.; Luo, G.-A. Metabolomic profiling of rat serum associated with isoproterenol-induced myocardial infarction using ultra-performance liquid chromatography/time-of-flight mass spectrometry and multivariate analysis. Talanta 2009, 79, 254–259. [Google Scholar] [CrossRef]
- Al Makdessi, S.; Andrieu, J.-L.; Herilier, H.; Faucon, G. Effect of isoproterenol on the metabolism of myocardial fatty acids. J. Mol. Cell. Cardiol. 1987, 19, 141–149. [Google Scholar] [CrossRef]
- Ohta, H.; Azuma, J.; Awata, N.; Hamaguchi, T.; Tanaka, Y.; Sawamura, A.; Kishimoto, S.; Sperelakis, N. Mechanism of the protective action of taurine against isoprenaline induced myocardial damage. Cardiovasc. Res. 1988, 22, 407–413. [Google Scholar] [CrossRef]
- Bkaily, G.; Jazzar, A.; Normand, A.; Simon, Y.; Al-Khoury, J.; Jacques, D. Taurine and cardiac disease: State of the art and perspectives. Can. J. Physiol. Pharmacol. 2020, 98, 67–73. [Google Scholar] [CrossRef]
- Jensen, B.C.; Parry, T.L.; Huang, W.; Beak, J.Y.; Ilaiwy, A.; Bain, J.R.; Newgard, C.B.; Muehlbauer, M.J.; Patterson, C.; Johnson, G.L.; et al. Effects of the kinase inhibitor sorafenib on heart, muscle, liver and plasma metabolism in vivo using non-targeted metabolomics analysis. Br. J. Pharmacol. 2017, 174, 4797–4811. [Google Scholar] [CrossRef]
- Yoon, C.; Kim, H.K.; Mishchenko, N.P.; Vasileva, E.A.; Fedoreyev, S.A.; Stonik, V.A.; Han, J. Spinochrome D Attenuates Doxorubicin-Induced Cardiomyocyte Death via Improving Glutathione Metabolism and Attenuating Oxidative Stress. Mar. Drugs 2018, 17, 2. [Google Scholar] [CrossRef]
- Dallons, M.; Schepkens, C.; Dupuis, A.; Tagliatti, V.; Colet, J.-M. New Insights About Doxorubicin-Induced Toxicity to Cardiomyoblast-Derived H9C2 Cells and Dexrazoxane Cytoprotective Effect: Contribution of In Vitro1H-NMR Metabonomics. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Li, Q.; Su, D.; O’Rourke, B.; Pogwizd, S.M.; Zhou, L. Mitochondria-derived ROS bursts disturb Ca2+ cycling and induce abnormal automaticity in guinea pig cardiomyocytes: A theoretical study. Am. J. Physiol. Circ. Physiol. 2014, 308, H623–H636. [Google Scholar] [CrossRef]
- Xie, L.-H.; Chen, F.; Karagueuzian, H.S.; Weiss, J. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 2008, 104, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; O’Rourke, B. Excitation-contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 2007, 102, 369–392. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liu, J.; Wei, C.; Li, K.; Xie, W.; Wang, Y.; Cheng, H. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 2007, 77, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Aon, M.A.; Liu, T.; O’Rourke, B. Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J. Mol. Cell. Cardiol. 2011, 51, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Cortassa, S.; Wei, A.-C.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. Modeling Cardiac Action Potential Shortening Driven by Oxidative Stress-Induced Mitochondrial Oscillations in Guinea Pig Cardiomyocytes. Biophys. J. 2009, 97, 1843–1852. [Google Scholar] [CrossRef]
- Bussing, R.; Gende, A. Severe hypoglycemia from clarithromycin-sulfonylurea drug interaction. Diabetes Care 2002, 25, 1659–1661. [Google Scholar] [CrossRef]
- Schelleman, H.; Bilker, W.B.; Brensinger, C.M.; Wan, F.; Hennessy, S. Anti-Infectives and the Risk of Severe Hypoglycemia in Users of Glipizide or Glyburide. Clin. Pharmacol. Ther. 2010, 88, 214–222. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Chen, Y.-T.; Kuo, S.-C.; Chen, T.-L.; Chang, F.-Y. Rapid hypoglycemia onset associated with antimicrobial use in patients with diabetes: A nationwide population-based case-crossover study. Eur. J. Intern. Med. 2016, 34, e14–e15. [Google Scholar] [CrossRef]
- Campagna, M.; Locci, E.; Piras, R.; Noto, A.; Lecca, L.I.; Pilia, I.; Cocco, P.; D’Aloja, E.; Scano, P. Metabolomic patterns associated to QTc interval in shiftworkers: An explorative analysis. Biomarkers 2016, 21, 607–613. [Google Scholar] [CrossRef]
- Zhang, Y.; Tocchetti, C.G.; Krieg, T.; Moens, A.L. Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radic. Biol. Med. 2012, 53, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Wood, M. The tolerance and toxicity of clarithromycin. J. Hosp. Infect. 1991, 19, 39–46. [Google Scholar] [CrossRef]
- Di Poggio, M.B.; Anfosso, S.; Audenino, D.; Primavera, A. Clarithromycin-induced neurotoxicity in adults. J. Clin. Neurosci. 2011, 18, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Duquesne, M.; Declèves, A.-E.; De Prez, E.; Nortier, J.; Colet, J.-M. Interest of metabonomic approach in environmental nephrotoxicants: Application to aristolochic acid exposure. Food Chem. Toxicol. 2017, 108, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Ryu, S.H.; Kim, S.; Lee, H.W.; Lim, M.-S.; Seong, S.J.; Kim, S.; Yoon, Y.-R.; Kim, K.-B. Pattern Recognition Analysis for Hepatotoxicity Induced by Acetaminophen Using Plasma and Urinary1H NMR-Based Metabolomics in Humans. Anal. Chem. 2013, 85, 11326–11334. [Google Scholar] [CrossRef]
- Lim, S.R.; Hyun, S.-H.; Lee, S.G.; Kim, J.-Y.; Kim, S.-H.; Park, S.-J.; Moon, K.-S.; Sul, D.; Kim, N.H.; Choi, H.-K. Potential urinary biomarkers of nephrotoxicity in cyclophosphamide-treated rats investigated by NMR-based metabolic profiling. J. Biochem. Mol. Toxicol. 2016, 31, e21871. [Google Scholar] [CrossRef]
- Lu, C.; Wang, Y.-M.; Sheng, Z.; Liu, G.; Fu, Z.; Zhao, J.; Zhao, J.; Yan, X.; Zhu, B.; Peng, S. NMR-based metabonomic analysis of the hepatotoxicity induced by combined exposure to PCBs and TCDD in rats. Toxicol. Appl. Pharmacol. 2010, 248, 178–184. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
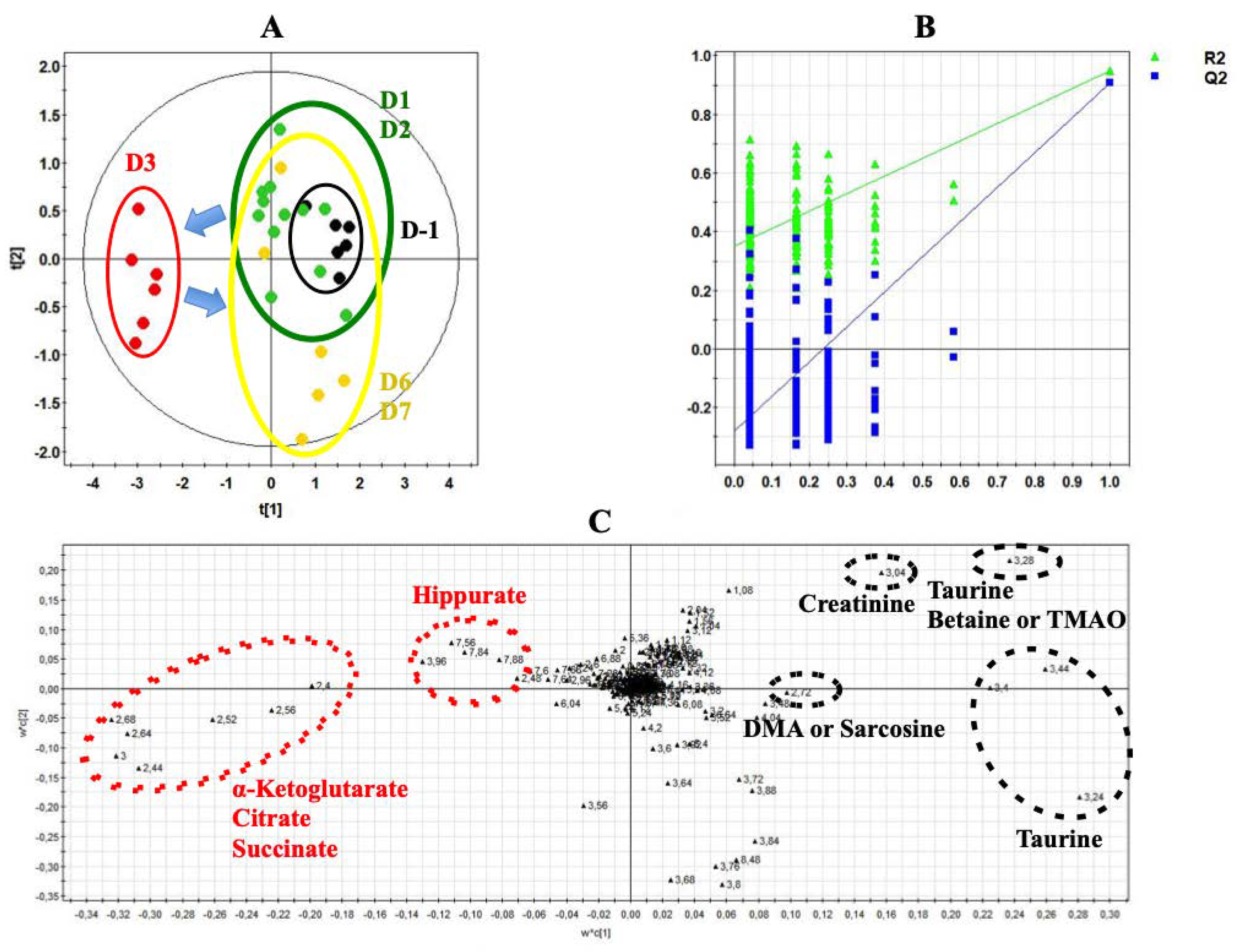{kind=link}
{kind=link}
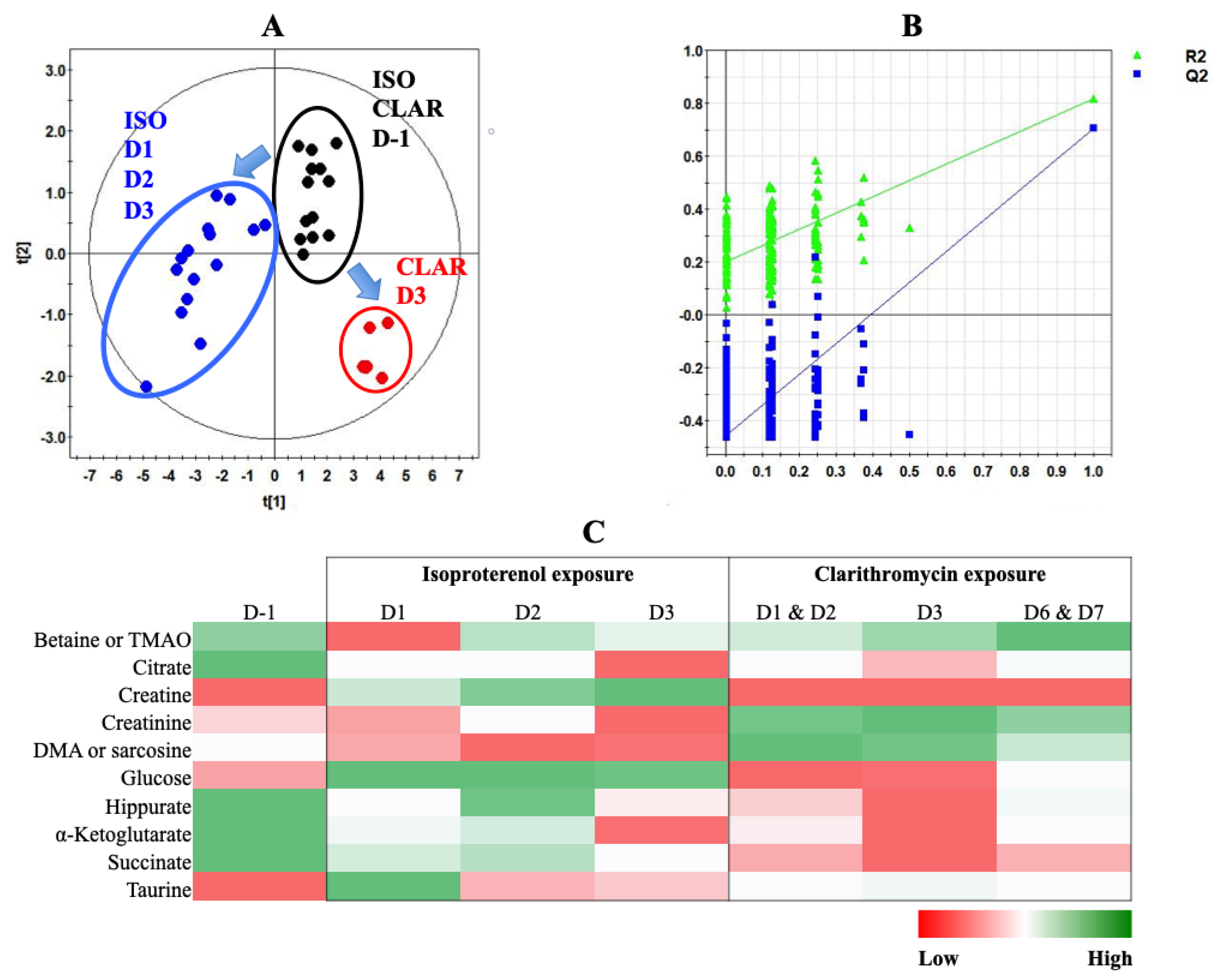{kind=link}
| Metabolites (VIP) | Chemical Shifts | Day 1 | Days 2 and 3 |
|---|---|---|---|
| Betaine or TMAO (0.86) | 3.28 (s) | ↓ * | ↓ |
| Citrate (4.44) | 2.56 (d) 2.68 (d) | ↓ * | ↓ * |
| Creatine (2.46) | 3.04 (s) 3.92 (s) | ↑ * | ↑ * |
| Creatinine (2.46) | 3.05 (s) 4.05 (s) | ↓ | ↓ |
| DMA or sarcosine (2.64) | 2.72 (s) | ↓ | ↓ |
| Glucose (1.63) | 3.23 (dd) 3.39 (m) 3.45 (m) 3.52 (dd) 3.72 (m) 3.82 (m) 3.88 (dd) 5.22 (d) | ↑ * | ↑ * |
| Hippurate (1.76) | 3.96 (d) 7.54 (m) 7.62 (m) 7.83 (dd) | ↓ | ↓ |
| α-Ketoglutarate (2.78) | 2.44 (t) 3.00 (t) | ↓ * | ↓ * |
| Succinate (1.95) | 2.40 (s) | ↓ * | ↓ * |
| Taurine (6.56) | 3.24 (t) 3.40 (t) | ↑ * | ↑ |
| Unknown (3.10) | 3.88 | ↑ | ↑ |
| Unknown (2.24) | 3.72 | ↑ | ↑ |
| Unknown (1.54) | 3.52 3.64 3.68 3.76 3.80 3.84 | ↑ | ↑ |
| Metabolites (VIP) | Chemical Shifts | Days 1 and 2 | Day 3 | Days 6 and 7 |
|---|---|---|---|---|
| Betaine or TMAO (3.42) | 3.28 (s) | ↓ | ↓ | ↑ |
| Citrate (4.72) | 2.56 (d) 2.68 (d) | ↓ ** | ↓ | ↓ ** |
| Creatinine (2.35) | 3.05 (s) 4.05 (s) | ↑ ** | ↑ | ↑ ** |
| DMA or sarcosine (1.62) | 2.72 (s) | ↑ * | ↑ | ↑ |
| Hippurate (1.71) | 3.96 (d) 7.54 (m) 7.62 (m) 7.83 (dd) | ↓ * | ↓ | ↓ ** |
| α-Ketoglutarate (4.80) | 2.44 (t) 3.00 (t) | ↓ ** | ↓ | ↓ ** |
| Succinate (2.98) | 2.40 (s) | ↓ ** | ↓ | ↓ ** |
| Taurine (3.81) | 3.24 (t) 3.40 (t) | ↑ ** | ↑ | ↑ ** |
| Unknown (1.07) | 2.48 | ↓ | ↑ | ↓ |
| Unknown (4.79) | 2.64 | ↓ | ↓ | ↑ |
| Unknown (1.12) | 3.72 3.76 3.84 3.88 | ↓ | ↓ | ↑ |
| Unknown (0.92) | 8.48 | ↓ | ↓ | ↑ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dallons, M.; Delcourt, M.; Schepkens, C.; Podrecca, M.; Colet, J.-M. A Comparative Study of Rat Urine 1H-NMR Metabolome Changes Presumably Arising from Isoproterenol-Induced Heart Necrosis Versus Clarithromycin-Induced QT Interval Prolongation. Biology 2020, 9, 98. https://doi.org/10.3390/biology9050098
Dallons M, Delcourt M, Schepkens C, Podrecca M, Colet J-M. A Comparative Study of Rat Urine 1H-NMR Metabolome Changes Presumably Arising from Isoproterenol-Induced Heart Necrosis Versus Clarithromycin-Induced QT Interval Prolongation. Biology. 2020; 9(5):98. https://doi.org/10.3390/biology9050098
Chicago/Turabian StyleDallons, Matthieu, Manon Delcourt, Corentin Schepkens, Manuel Podrecca, and Jean-Marie Colet. 2020. "A Comparative Study of Rat Urine 1H-NMR Metabolome Changes Presumably Arising from Isoproterenol-Induced Heart Necrosis Versus Clarithromycin-Induced QT Interval Prolongation" Biology 9, no. 5: 98. https://doi.org/10.3390/biology9050098
APA StyleDallons, M., Delcourt, M., Schepkens, C., Podrecca, M., & Colet, J.-M. (2020). A Comparative Study of Rat Urine 1H-NMR Metabolome Changes Presumably Arising from Isoproterenol-Induced Heart Necrosis Versus Clarithromycin-Induced QT Interval Prolongation. Biology, 9(5), 98. https://doi.org/10.3390/biology9050098