Simple Summary
Phaeodactylum tricornutum is rich in bioactive substances, including fucoxanthin and polyunsaturated fatty acids. Epigenetic editing mediated by the human RNA demethylase FTO has been shown to significantly increase yields in rice and potato. Our study aimed to improve the traits of P. tricornutum by applying FTO-mediated epigenetic editing. We successfully constructed transgenic algal strains. Transgenic P. tricornutum exhibits significantly reduced RNA m6A modification levels and faster growth, and also produces markedly higher levels of lipids, proteins, and carotenoids compared to the wild type. Transcriptome analysis revealed that the expression of numerous genes was upregulated. The Kyoto Encyclopedia of Genes and Genomes analysis demonstrated the upregulated expression of multiple key enzymes involved in long-chain fatty acid synthesis, carotenoid synthesis, and amino acid synthesis in transgenic P. tricornutum. These results indicate that exogenous FTO can promote the expression of numerous genes by decreasing overall m6A methylation levels in cells, ultimately enhancing the associated metabolic pathways. These findings suggest that FTO has the potential to serve as a new tool for the epigenetic editing of microalgae.
Abstract
Phaeodactylum tricornutum is rich in bioactive substances, rendering it valuable in nutrition and medicine. Epigenetic editing mediated by human RNA demethylase FTO can significantly increase the yields of rice and potato and offers significant potential for the genetic breeding of microalgae. This study aimed to enhance the production of certain metabolites in P. tricornutum via FTO-mediated epigenetic editing. Phenotypic analysis revealed that transgenic P. tricornutum exhibits significantly reduced RNA m6A modification levels and faster growth, producing markedly higher levels of lipids, proteins, and carotenoids than the wild type. Transcriptome analysis revealed 1009 upregulated genes and 378 downregulated genes. KEGG analysis demonstrated the upregulated expression of multiple key enzymes involved in long-chain fatty acid synthesis (e.g., ACSL, fabF, and fabG), carotenoid synthesis (e.g., crtQ, PDS, and PSY1), and amino acid synthesis (e.g., dapF, glyA, and aroK) in transgenic P. tricornutum, consistent with our phenotypic results. These results indicate that FTO can promote growth and increase the bioactive compound content in P. tricornutum by regulating the m6A modification of RNA, and further suggest that FTO has the potential to serve as a new tool for the epigenetic editing of microalgae.
1. Introduction
Phaeodactylum tricornutum is an important planktonic alga that serves as a food source for many aquatic invertebrates, fish, crustaceans, and mollusks. It has three major morphotypes, namely, fusiform, triradiate, and oval. This alga is highly nutritious and medicinal because it is rich in fucoxanthin, polyunsaturated fatty acids (PUFAs), and other bioactive substances. Fucoxanthin has potent antioxidant properties that surpass those of beta-carotene, lutein, and other carotenoids [1]. It also scavenges intracellular oxygen free radicals, protects cellular and DNA integrity, significantly enhances cellular regenerative capacity, and delays aging. Moreover, it inhibits the proliferation of various cancerous cells [2,3,4,5], prevents obesity and diabetes [6], and improves cardiovascular function markers [7,8]. Fucoxanthin is widely applied to the food, skincare, and healthcare industries.
P. tricornutum is rich in PUFAs, include eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), which are essential fatty acids for humans. They can prevent and treat cardiovascular diseases, and have anti-inflammatory, anti-aging, anti-coagulation, and immune system-regulatory effects. They have been applied clinically as auxiliary therapies for various cancer, skin, and geriatric diseases [9,10]. The lipid content accounts for 20~30% of the dry weight, and EPA can reach up to 30% of the total fatty acid content, which renders P. tricornutum an excellent candidate for EPA production [11,12,13,14]. Furthermore, this microalga offers advantages such as rapid growth, low levels of other PUFAs, ease of harvesting, and high-purity recovery [15]. P. tricornutum also exhibits the advantages of genetic engineering breeding, such as a compact genome (~27 Mb) and ease of genetic transformation [16,17,18,19]. Consequently, P. tricornutum is widely studied. The genome and proteome of P. tricornutum have been fully sequenced [19,20], and molecular tools for reverse genetics and cell biology have matured [21,22]. The CRISPR technology has also been applied to edit the genome in this species [23]. These developments have facilitated molecular investigation using P. tricornutum as a model organism.
The concept of RNA epigenetics posits that reversible chemical modifications of RNA, such as those found in DNA and histones, play crucial regulatory roles in gene expression [24]. This hypothesis was substantiated by the findings that fat mass and obesity-associated (FTO) demethylate RNA m6A [25], and that RNA methylation modifications are reversible. This significantly contributed to gene expression regulation and opened new avenues in RNA epigenetics. The established obesity gene FTO is mainly found in animals and serves as an RNA demethylase, regulating growth and development [25,26]. However, homologous proteins are not found in higher plants [27].
Epigenetics is the study of reversible and heritable changes in gene function without altering DNA sequences. Epigenetic phenomena include DNA methylation, genomic imprinting, maternal effects, gene silencing, nuclear dominance, non-coding RNAs, and histone PTM and RNA methylation [28]. Currently, RNA methylation is one of the most actively studied research areas. The RNA m6A modification by methyltransferases is dynamically reversible and can be removed by demethylases. It regulates gene expression through alternative splicing and mRNA nuclear export, stability, translation, and transcription [29,30]. This modification is found in fungi, animals, higher plants, and algae. It has mostly been studied in animals, where it plays important regulatory roles in biological processes such as stem cell differentiation, tissue development, brain memory, immune responses, and disease mechanisms. m6A is the most prevalent post-transcriptional modification in eukaryotes, accounting for ~80% of all RNA methylation events [29]. In contrast, few studies have investigated RNA m6A modifications in plants. Thus, the understanding of its regulatory role in plant organ development, cell division, development, and stress adaptation remains limited. Nevertheless, RNA m6A modification plays an important role in the regulation of plant growth and stress adaptation [31]. RNA demethylases, such as mRNA adenosine methylase (MTA) and FKBP12 (FK506-binding protein12 KD)-interacting protein 37 KD (FIP37), regulate apical meristem development in Arabidopsis thaliana [32,33]. Both ALKBH9b and and10b can demethylate the m6A modification of RNA, thus influencing flowering and nutrient uptake in Arabidopsis [34,35].
A method was proposed to enhance crop quality and resistance to adverse conditions through m6A editing [36]. This approach elicited a remarkable response that led to increased plant yield and biomass. The introduction of FTO into rice and potatoes facilitates the demethylation of RNA m6A modifications, resulting in substantial enhancements in crop yield, biomass, and stress tolerance [27]. Specifically, FTO overexpression in rice and potatoes resulted in a ~50% increase in yield. A comprehensive investigation of its molecular mechanisms determined that FTO-mediated demethylation of m6A facilitates chromatin remodeling and transcriptional activation, thereby resulting in the respective upregulation of ~11,000 and 7000 genes in leaves and roots, while simultaneously activating multiple signaling pathways. Consequently, this study introduced a novel form of epigenetic editing breeding technology that paved the way for an entirely new paradigm in plant breeding [27].
Epigenetic modifications are strongly associated with life activities in microalgae, which are lower plants. Numerous studies have investigated epigenetic modifications of DNA in microalgae. Four DNA methyltransferases (DNMTs 3, 4, 5, and 6) have recently been identified through bioinformatics analysis of P. tricornutum. A gene knockout of DNMT5a showed that its loss correlated with the global depletion of DNA methylation and the overexpression of young transposable elements [37]. The genome-wide methylation map of P. tricornutum was extensively investigated, revealing that ~6% of its genome exhibits mosaic methylation profiles that are strongly associated with gene expression regulation [38]. Several DNA methyltransferases have been identified in the cyanobacterium Synechocystis sp., including slr0214, M.Ssp6803I, and M.Ssp6803V. In particular, slr0214, M.Ssp6803II, M.Ssp6803III, and M.Ssp6803IV play crucial roles in Synechocystis sp. growth [39,40]. However, research on RNA methylation modifications is scarce. Multiple RNA methyltransferases have been identified in Alexandrium tamutum, Amphidinium carterae, Cylindrotheca closterium, and Tetraselmis suecica. These enzymes exhibit homology with ALKBH9b, ALKBH10b, MTB, and FIP37 in higher plants [41].
In summary, epigenetic modifications play critical roles in the activities of microalgae. However, the sparse investigations there are in this area lack depth. The application of FTO as an epigenetic editing tool in monocotyledonous (rice) and dicotyledonous (potato) plants suggests that this technology has broad applicability across plant species. Therefore, this technology holds significant potential for the genetic breeding of microalgae. The aim of this study was to utilize the demethylation capability of FTO protein to remove m6A modifications from RNA in microalgae, thereby activating specific metabolic pathway genes and enhancing the production of target metabolites.
2. Materials and Methods
2.1. Strains and Growth Conditions
P. tricornutum strain CCAP1055 was obtained from the Algae Culture Collection at the Ocean University of China. P. tricornutum cells were cultured in f/2 medium, which was prepared by filtering seawater through a 0.22-micron filters and subsequently sterilizing it at 121 °C. The final medium was obtained by adding the sterilized f/2 stock solution at a ratio of 1:1000. The diatom strain was inoculated into fresh culture medium at a ratio of 1:10 (v/v), and grown at 20 °C under a 16/8 h light/dark cycle in f/2 medium [42]. The intensity of the light was 70 μmol photons m−2s−1. A s’olid f/2 medium was prepared by adding 1% (w/v) agar to the liquid medium. P. tricornutum cells were harvested by centrifugation at 4000× g for 10 min during the exponential growth phase.
2.2. Plasmid Construction
The original human FTO cDNA sequence was obtained from GenBank Accession no. NP_001073901.1). Optimized FTO cDNA was obtained based on the codon preference of P. tricornutum through codon optimization technology and was referred to as PtFTO. Fragments of PtFTO were synthesized (Sangon Biotech Co., Ltd., Shanghai, China) and subsequently subcloned into the PUC-19 vector. An EcoRI-XbaI fragment encompassing PtFTO from PUC-19 was cloned into the polylinker region of the pPha-T1 plasmid, which enabled diatom selection for positive transformants using zeocin. The constructed vector was confirmed as being error-free by PCR amplification and DNA sequencing. The forward primer sequence 5′-GGTACCATGAAGCGTACCCCCACCGCCGAAG-3′ and the reverse 5′-TCTAGAGGGCTTGGCTTCGAGGAGCTGTC-3′ were used for PCR verification.
2.3. Generation of Transgenic P. tricornutum
P. tricornutum cells were grown for 5 days in f/2-Si liquid medium. Cells were harvested by centrifugation at 4000× g for 10 min and resuspended in a fresh f/2-Si liquid medium at a density of 5 × 108 cells/mL. Cells were bombarded with Biolistic PDS-1000/He (Bio-Rad, Hercules, CA, USA) at a bombardment pressure of 1500 psi. Then, 3 mg gold microcarriers (Bio-Rad) were coated with 5 μg of plasmid in the presence of 50 μL CaCl2 (2.5 mol/L) and 20 μL of spermidine (0.1 mol/L), as outlined by the manufacturer’s instructions [43]. After bombardment, the cells were recovered in an f/2 plate for 24 h and were replated on selective plates containing zeocin (100 μg/mL). The plates were incubated at 20 °C under a 16/8 h light/dark cycle (70 μmol photons m−2s−1) for 2 weeks. The transformation was verified using PCR and sequencing techniques. The forward primer sequence 5′-GGTACCATGAAGCGTACCCCCACCGCCGAAG-3′ and the reverse sequence 5′-TCTAGAGGGCTTGGCTTCGAGGAGCTGTC-3′ were used for PCR verification.
2.4. Total RNA m6A Modification Level
RNA m6A levels in P. tricornutum were determined using an enzyme-linked immunosorbent assay (ELISA). Total RNA was extracted from P. tricornutum cells using an E.Z.N.A.® Plant RNA Kit (Omega Biotek, Norcross, GA, USA). All RNA samples were treated with DNase I to eliminate genomic DNA contamination. The quality of the extracted RNA was spectrophotometrically assessed to confirm that the experimental requirements were met (OD, 260/280 > 1.9; 260/230 > 1.7). PolyAt mRNA was then purified using a Hieff NGS® mRNA Isolation Master Kit (Yeasen Biotechnology, Shanghai, China). The global m6A levels of mRNA were detected using an EpiQuik m6A RNA Methylation Quantification Kit (Epigentek Group Inc., Farmingdale, NY, USA). All experiments proceeded as described by the manufacturer and sample analysis included 200 ng of poly(A) mRNA. Absorbance was measured at 450 nm using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA), and a standard curve was generated to calculate the m6A level, as described by the manufacturer’s protocol. All the assays were repeated at least three times.
2.5. Lipid Analysis
The relative contents of neutral lipids in the microalgae were determined by Neutral Red staining. Then, 8-day-old wild-type and transgenic P. tricornutum cells were collected for Nile Red staining. We dissolved 1 mg of Nile Red in 10 mL of acetone to prepare a stock solution with a concentration of 0.1 mg/mL. The cells were resuspended in 20% dimethyl sulfoxide (DMSO) and incubated in a 40 °C water bath for 10 min. Subsequently, 20 μL of Nile red stock solution was added into the 2 mL of cell suspension and incubated in the dark for 15 min at room temperature. The excitation and emission wavelengths for detection were set to 530 and 580 nm, respectively. The fluorescence intensities represented the relative contents of neutral lipids in the stained cells. All the assays were repeated at least three times.
Microalgal samples were harvested by centrifugation at 4000× g for 10 min, lyophilized at −40 °C for 24 h, and subsequently weighed to determine the dry weight. Total lipid extraction was performed using the solvent extraction method [44]. The freeze-dried samples were resuspended in a methanol/chloroform solution (2:1, v/v) and ultrasonicated for 1 h. A saturated NaCl solution was added to facilitate phase separation. The mixture was centrifuged at 10,000× g for 5 min, and the chloroform layer was collected. Chloroform was removed from the collected solution using a rotary evaporator. The residue was dried in a 60 °C oven for 3–4 h and subsequently weighed to determine the total lipid content. All the assays were repeated at least three times.
The lyophilized samples were thoroughly ground and then mixed with 5 mL of 0.5 mol·L−1 NaOH methanol. The mixture was ultrasonicated for 1 h and then sealed under N2 and reacted at 65 °C for 2 h. After cooling, 5 mL boron trifluoride–methanol (BF–M) solution (25% w/w in methanol) was added to the mixture, which was esterified at 65 °C for 20 min; then, fatty acids were extracted by shaking with hexane. Subsequently, hexane was added to the mixture, followed by shaking to facilitate the extraction. A saturated NaCl solution was used to promote phase separation, and the hexane phase was collected for fatty acid analysis. The fatty acid samples were analyzed by gas chromatography–mass spectrometry (GC–MS) (Agilent Technologies Inc., Santa Clara, CA, USA) following the method previously described [45]. All the assays were repeated at least three times.
2.6. Determination of Total Soluble Protein
The total protein was determined using a BCA protein assay kit (Solarbio Inc., Beijing, China). The 8-day-old wild-type and transgenic P. tricornutum cells were collected by centrifugation at 4000× g for 5 min. Then, the cells were resuspended in a buffer and lysed using an ultrasonic homogenizer (Ningbo Scientz Biotechnology Co., Ltd., Ningbo, China). The lysates were centrifuged at 10,000× g for 5 min at 2–4 °C, and the sedimentary impurities were eliminated. A 20 μL aliquot of the sample was added to 200 μL BCA working solution. The mixture was incubated at 37 °C for 30 min; then, the absorbance was measured at 562 nm. Protein concentrations were calculated based on a standard curve generated using bovine serum albumin (BSA). All the assays were repeated at least three times.
2.7. Determination of Total Carotenoids
The 8-day-old wild-type and transgenic P. tricornutum cells were collected by centrifugation. The cells were resuspended with DMSO and then incubated at 60 °C in the dark for 1 h until the algal cells became completely decolorized. The extract was centrifuged at 12,000× g for 5 min, and the supernatant was collected. The optical density (OD) of the extract was detected at 480, 649, and 665 nm using a microplate reader. Carotenoid and total chlorophyll concentrations were estimated using the Wellburn formula [46] as follows:
where Ca and Cb represent the concentrations of chlorophyll a and b, respectively; A665, A649, and A480 represent OD values at wavelengths of 665, 649, and 480 nm, respectively; and TCC represents the total carotenoid concentration. All the assays were repeated at least three times.
Ca = 12.19A665 − 3.45A649
Cb = 21.99A649 − 5.32A665
TCC = (1000A480 − 2.14Ca − 70.16Cb)/220
2.8. Determination of Fucoxanthin
The 8-day-old wild-type and transgenic P. tricornutum cells were collected by centrifugation. After freeze-drying the cells for 48 h, 10 mL of 90% ethanol was added per 1 g of microalgae powder. The extract was obtained by ultrasonic-assisted extraction at 40 °C for 1 h. The extract was centrifuged at 12,000× g for 5 min, and the supernatant was collected. The supernatant was filtered through a 0.22 μm filter. The content of fucoxanthin in the extract was analyzed using high-performance liquid chromatography (HPLC). The entire process was conducted in the dark. All the assays were repeated at least three times.
2.9. Transcriptome Sampling and Sequencing
The 8-day-old wild-type and transgenic P. tricornutum cells were collected by centrifugation at 4000× g for 5 min. For the transcriptome analysis, both the wild-type and transgenic line 9# group consist of three sequencing samples each. Total RNA was extracted from microalgae using an E.Z.N.A. ® Plant RNA Kit (Omega Biotek, Norcross, GA, USA). Genedenovo Biotechnology Co., Ltd. (Guangzhou, China) provided services for cDNA library construction, sequencing, and data analysis. Transcriptome analysis was performed using reference genome-based read mapping. The P. tricornutum reference genome (GCF_000150955.2) was obtained from the National Center for Biotechnology Information (NCBI) database. Bioinformatic analysis was performed using Omicsmart, a real-time interactive online platform for data analysis (http://www.omicsmart.com) (accessed on 20 November 2024). The reads were further filtered by fastp [47] (version 0.18.0). Bowtie2 [48] (version 2.2.8) was used to map reads to the ribosome RNA (rRNA) database, and the reads mapped to rRNA were subsequently removed. The paired-end clean reads were mapped to the reference genome using HISAT2 2.1.0 [49]. For differential expression analysis, the input data used for DESeq2 [50] software (version 1.40.0) are read counts, which are used to calculate p-values. Subsequently, multiple testing correction is applied to obtain the FDR. The FPKM values estimated by RSEM are utilized for calculating fold changes. Differentially expressed genes (DEGs) were assessed with up-regulation or down-regulation fold changes of ≥±2 and p < 0.05. All analyses were performed using the R package (version 3.30.3). Gene Ontology (GO) terms were assigned by using Blast2GO (version 3.3.5) [51]. GO classification was conducted with the OmicShare tools (http://www.omicshare.com/tools) (accessed on 20 November 2024), and the categorization results were represented as three independent hierarchies: molecular function, biological process, and cellular component. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation was performed by comparison against the KEGG database [52]. A corrected p ≤ 0.05 was set as the threshold to identify significantly enriched GO terms or KEGG pathways. The raw data were deposited in the National Genomics Data Center (NGDC) Genome Sequence Archive under the accession number CRA023747.
3. Results
3.1. Introduction of PtFTO into P. tricornutum
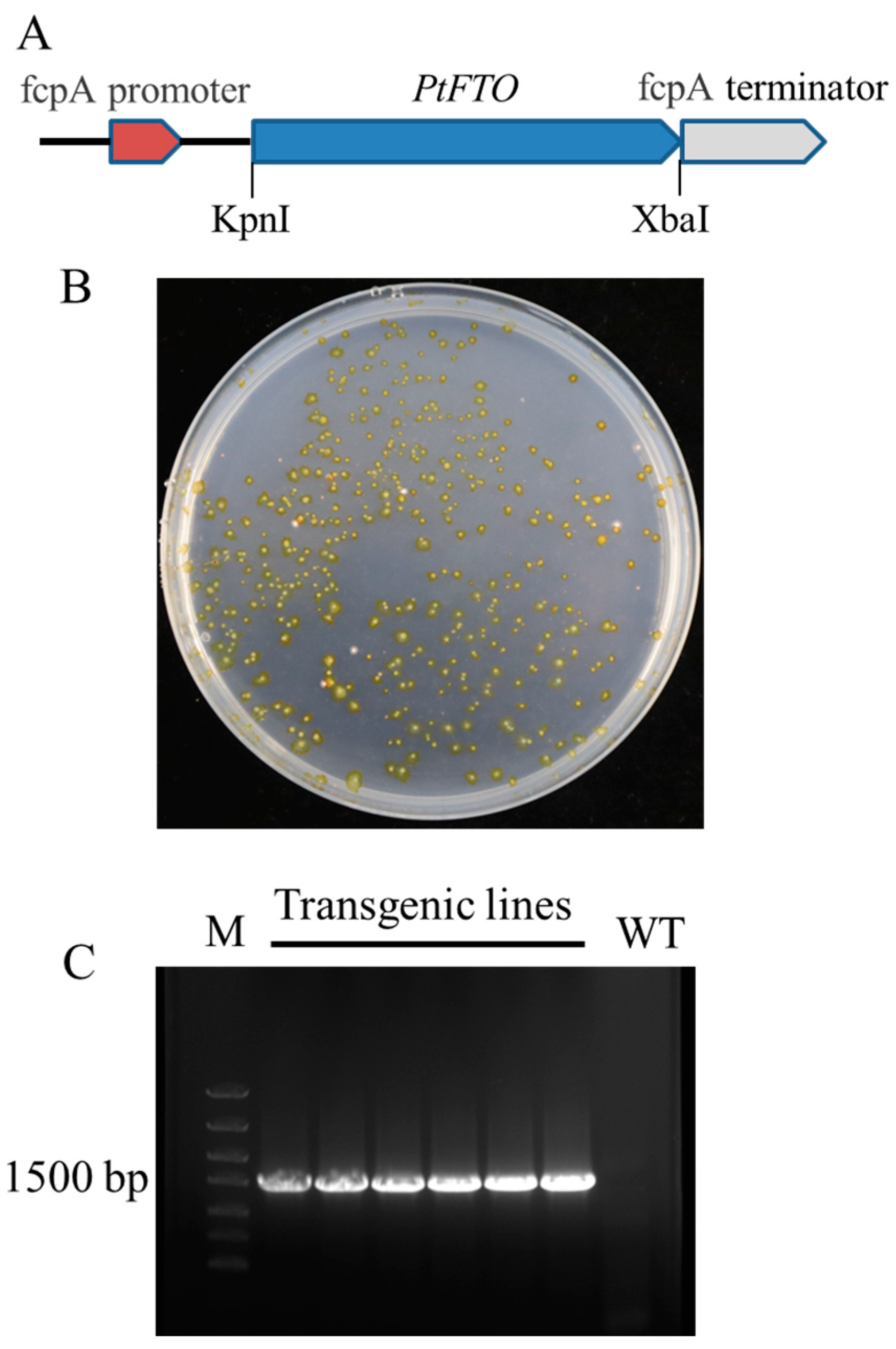
The original sequence of human FTO cDNA was obtained from GenBank (accession no. NP_001073901.1). The optimized FTO cDNA was obtained using codon optimization technology according to the codon bias of P. tricornutum and was referred to as PtFTO (Table S1). The PtFTO spans 1518 base pairs and was resynthesized accordingly. PtFTO fragments were cloned into the pPha-T1 plasmid and placed under the control of the promoter fcpA. The constructed vector was introduced into P. tricornutum using a gene gun. Resistant colonies were obtained in a zeocin-selective medium (Figure 1). The transformation was verified by PCR and sequencing techniques. The PCR products were analyzed by agarose gel electrophoresis, and a 1.5-kb target fragment was successfully detected in the transgenic strains, whereas no band was present in the wild-type strain (Figure 1 and Figure S1). These results confirm that PtFTO was introduced into P. tricornutum.
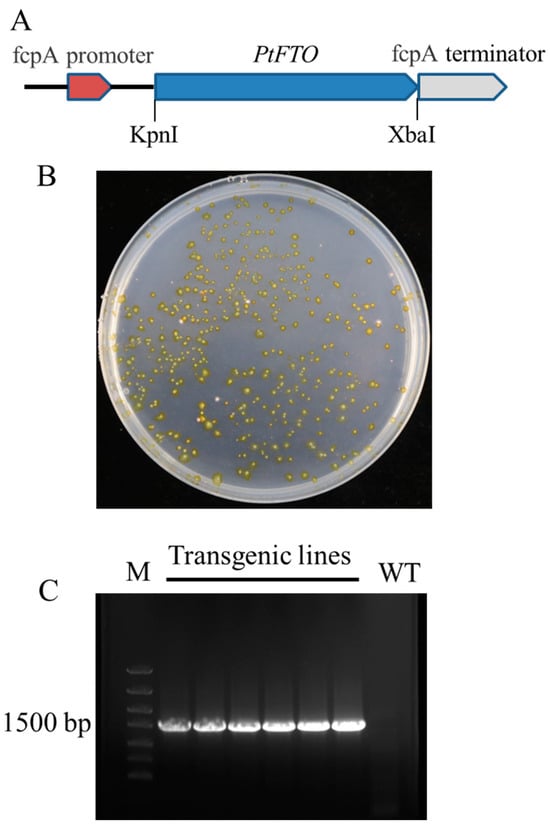
Figure 1.
Generation of transgenic P. tricornutum with PtFTO. (A) Construction of transforming plasmids. PtFTO fragment was inserted between fcpA promoter A and fcpA terminator of pPha-T1. (B) Screening transformants through zeomycin resistance (100 μg/mL). Transgenic positive clones were visible after 2 weeks, and transformants were kept in F/2 medium containing 100 μg/mL zeomycin. (C) PCR verification of transgenic P. tricornutum with PtFTO. Specific fragments of 1518 bp were amplified from total DNA of transformants. Marker (M), transgenic lines with PtFTO, and wild type (WT). No PtFTO products were detected in WT.
3.2. Increased Biomass Production in Transgenic P. tricornutum
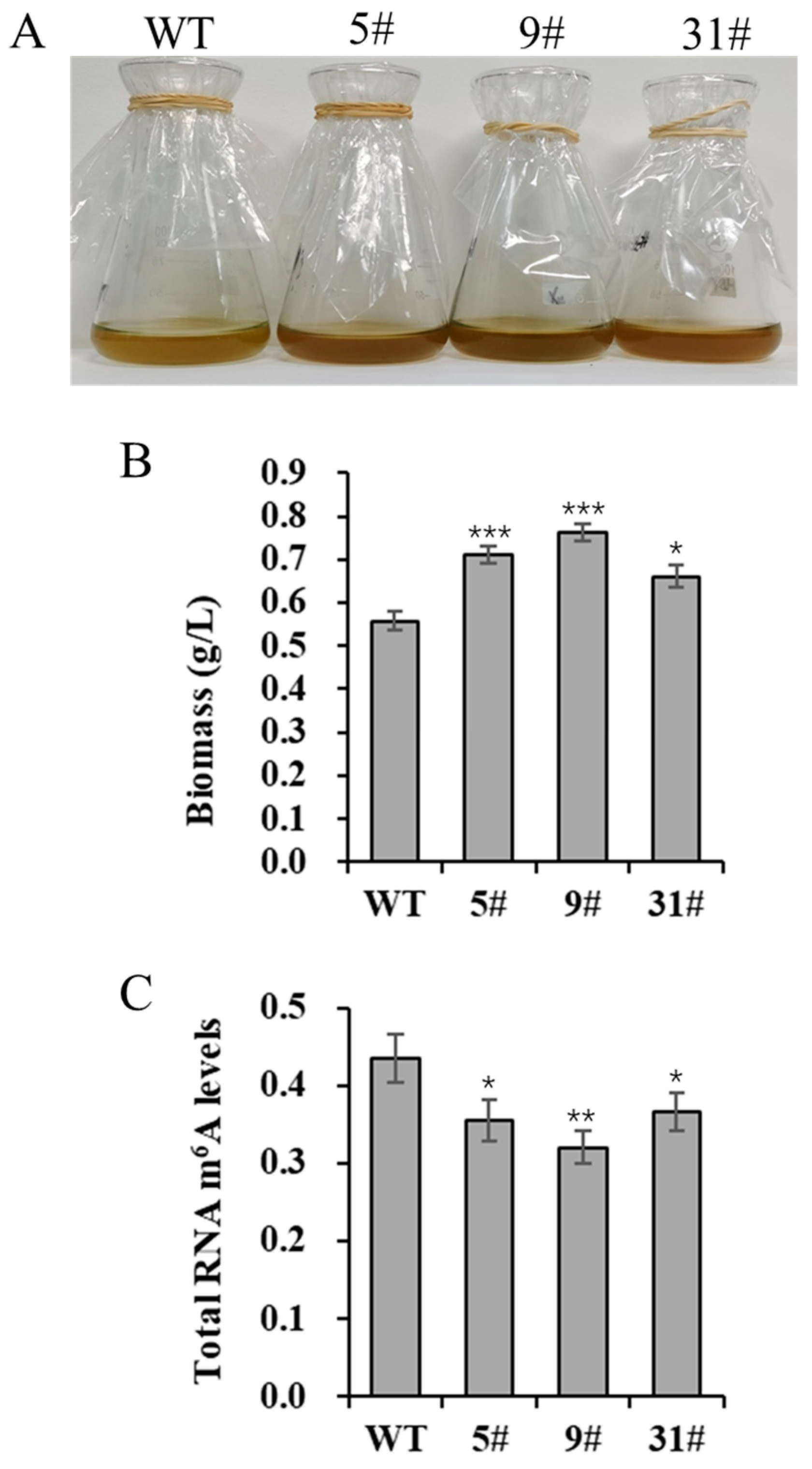
We generated a series of primary transgenic lines via genetic transformation. Initially, independent transgenic lines were screened based on growth and neutral lipid content. Several transgenic lines (5#, 9#, and 31#) were selected for further analysis. To investigate the effects of PtFTO overexpression on the growth characteristics of P. tricornutum, we analyzed the biomass of both the transgenic and wild-type lines. The results showed that the biomass of transgenic P. tricornutum was higher than that of the wild type after 8 days of cultivation. Specifically, the biomass significantly increasing, rising by 27.4% and 36.9% in lines 5# and 9#, respectively, whereas the increase in the transgenic line 31# was slight (Figure 2A,B). These results suggest that PtFTO overexpression affects the growth and biomass accumulation of transgenic P. tricornutum.
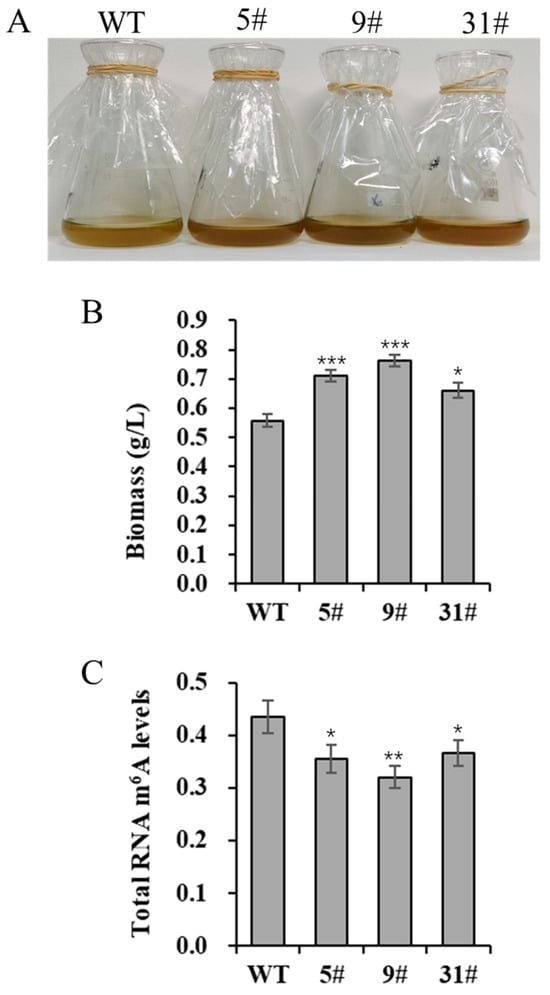
Figure 2.
Biomass and total RNA m6A modification level of transgenic P. tricornutum. Eight-day-old P. tricornutum cells (WT, transgenic line 5#, 9#, and 31#) were used for all assays. (A) Growth phenotypic characteristics of P. tricornutum. (B) Fresh weight of P. tricornutum. (C) RNA m6A modification level of P. tricornutum. Each value represents mean ± SD (n = 3). Asterisks in (B,C) represent significant differences (t test, * p < 0.05; ** p < 0.005; *** p < 0.001). Results of significant analyses were compared with WT in statistical analysis.
3.3. Total m6A Modification of Transgenic P. tricornutum Is Significantly Decreased
m6A is the most abundant mRNA modification in higher eukaryotes and is reversible. In animals, the FTO gene encodes an mRNA demethylase that mediates RNA m6A demethylation. Therefore, the overexpression of FTO in P. tricornutum may affect m6A modification levels in the cells. In this study, the total m6A levels in RNA were detected by ELISA. The total m6A modification levels of the transgenic lines (5#, 9#, and 31#) were, respectively, 18.3%, 26.3%, and 15.7% lower than those in the wild-type cells. The extent of reduction in strain 9# was the most significant (Figure 2C). These results demonstrate that the expression of PtFTO can significantly decrease the total m6A modification level in transgenic P. tricornutum cells.
3.4. Transgenic P. tricornutum Exhibit Higher Lipid and Protein Contents
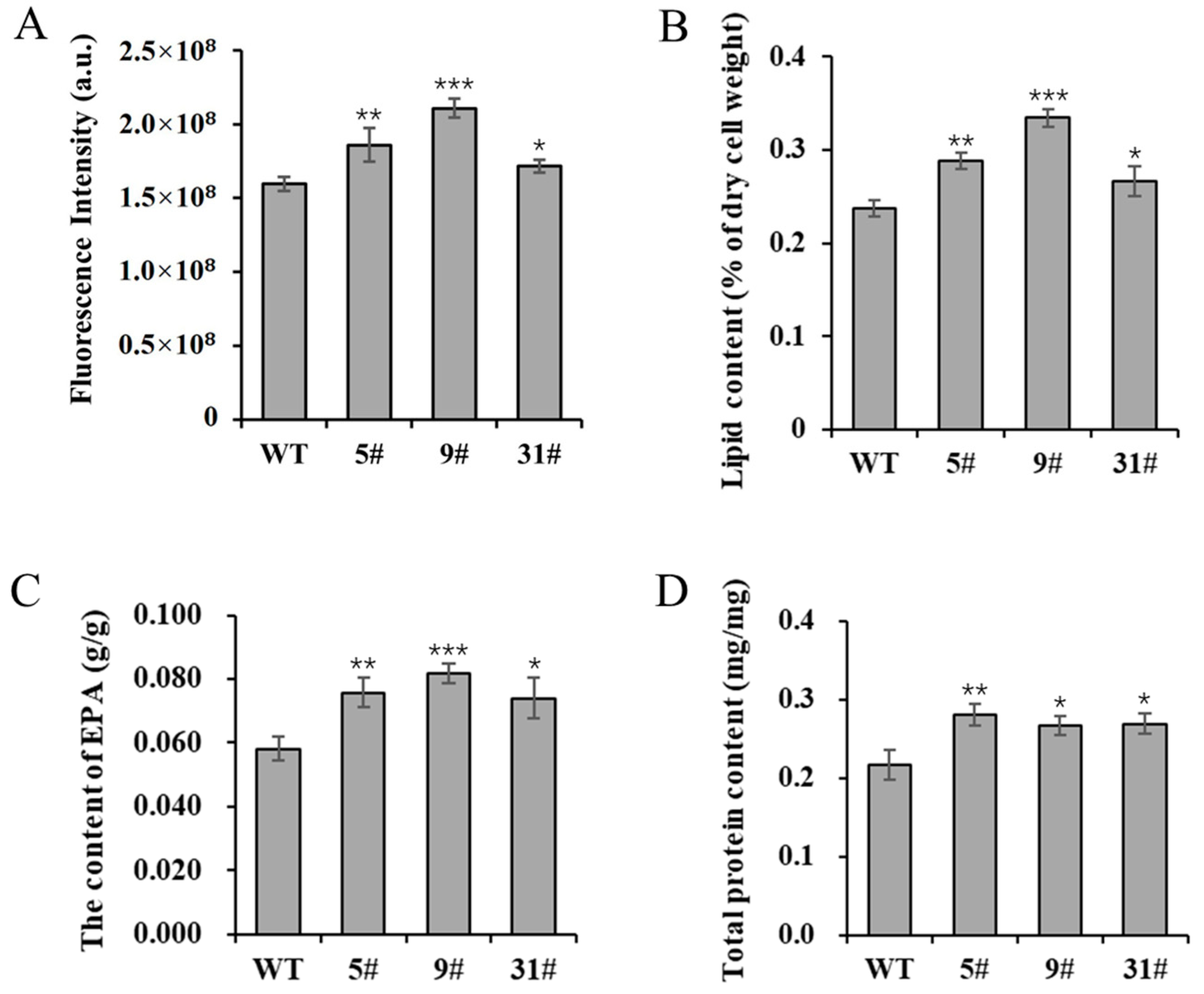
The expression of PtFTO in P. tricornutum can reduce the amounts of m6A modification, thus potentially enhancing the expression of numerous genes in a way that subsequently affects cellular metabolic activities. In the present study, the neutral lipid content of transgenic P. tricornutum was determined using Nile Red staining. The results showed that the NR fluorescence intensity of transgenic lines 5# and 9# was enhanced by 16.5% and 32% compared with the wild-type variant, whereas line 31# showed a slight increase, rising by 7.4% (Figure 3A). These findings indicated a significant increase in neutral lipid levels. Total lipids were extracted from transgenic P. tricornutum and wild-type plants under normal culture conditions. The total lipid contents of transgenic lines 5#, 9#, and 31# were increased by 21.3%, 40.8%, and 12.4%, respectively, compared with the wild type (Figure 3B). F. tricornutum is an excellent candidate for EPA production. The EPA contents of transgenic lines 5#, 9#, and 31# were significantly increased, rising by 30.3%, 40.6%, and 27.2%, respectively (Figure 3C). Additionally, the total protein contents of transgenic lines 5#, 9#, and 31# were also increased by 29.7%, 23.2%, and 24.2%, respectively (Figure 3D). Collectively, these results demonstrate that overexpressed PtFTO promotes the accumulation of lipids and long-chain saturated fatty acids.
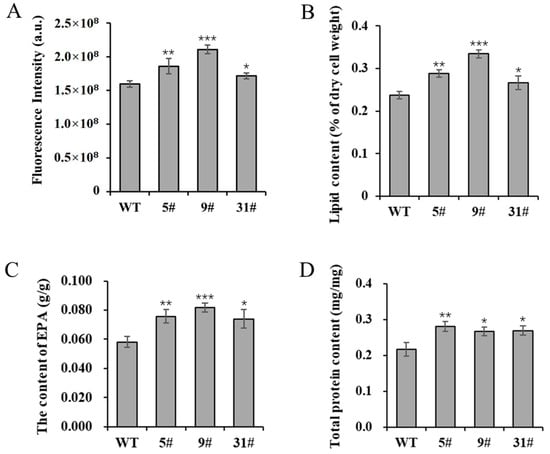
Figure 3.
The lipid and protein contents of transgenic P. tricornutum. The 8-day-old P. tricornutum cells (WT, transgenic line 5#, 9#, and 31#) were used to all assays. (A) Neutral lipid content determined by Nile red staining. (B) Total lipid content in dry cell weight determined by gravimetry. (C) EPA content in dry cell weight determined by GC–MS. (D) Total protein content determined by BCA protein assay kit. Each value represents mean ± SD (n = 3). Asterisks in (A–D) represent significant differences (t test, * p < 0.05; ** p < 0.005; *** p < 0.001). The results of significant analyses were compared with WT in the statistical analysis.
3.5. Transgenic P. tricornutum Exhibit High Carotenoid Contents
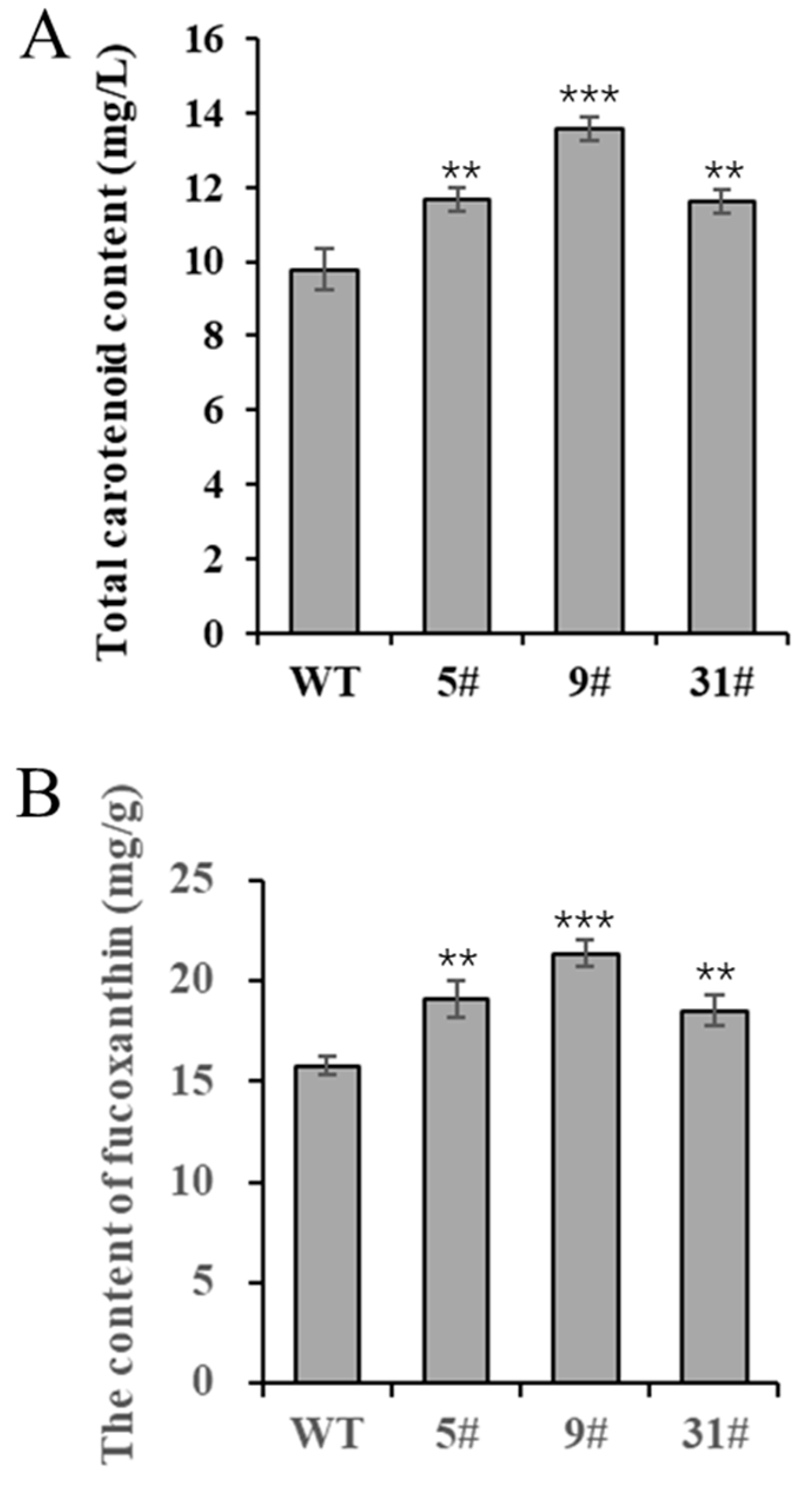
P. tricornutum is rich in carotenoids, especially fucoxanthin, and is a high-quality algal species used for the production of fucoxanthin. In this study, we measured the carotenoid content of transgenic P. tricornutum. Carotenoid levels were significantly higher in the three transgenic lines (particularly in line 9#) compared with the wild type, increasing by 19.2%, 38.7%, and 18.8%, respectively (Figure 4A). The trend of the fucoxanthin content in transgenic P. tricornutum was similar, with line 9# producing considerably significantly higher levels compared to the wild type, increasing by 21.1%, 35.4%, and 17.2%, respectively (Figure 4B). These data demonstrate that PtFTO expression can significantly increase the carotenoid content of P. tricornutum.
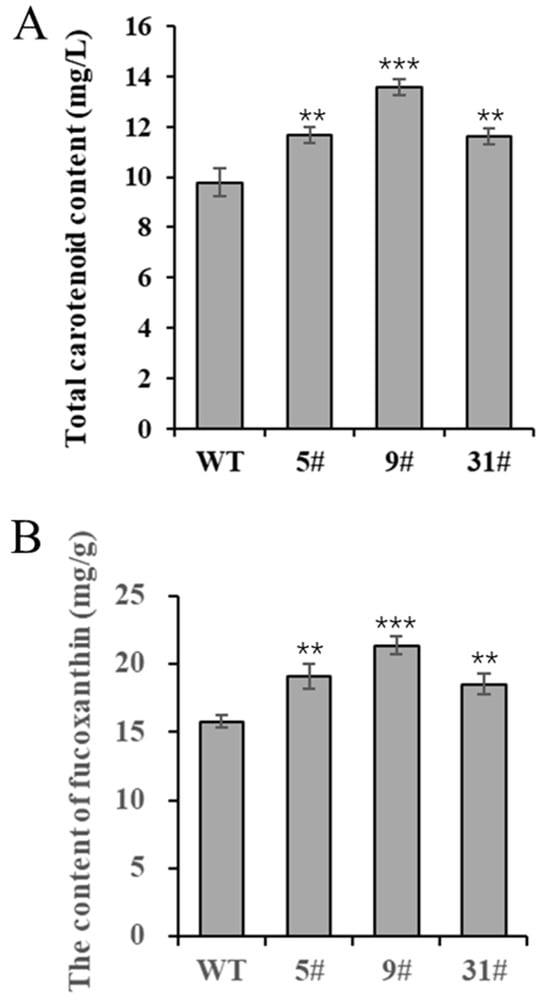
Figure 4.
Determination of carotenoids in transgenic P. tricornutum. Eight-day-old P. tricornutum cells (WT, transgenic line 5#, 9#, and 31#) were used in all assays. (A) Total carotenoid content as determined by spectrophotometer. (B) Fucoxanthin content determined by HPLC. Each value represents mean ± SD (n = 3). Asterisks in (A,B) represent significant differences (t test, ** p < 0.005; *** p < 0.001). The results of significant analyses were compared with WT in the statistical analysis.
3.6. Transcriptome Analysis of Transgenic P. tricornutum
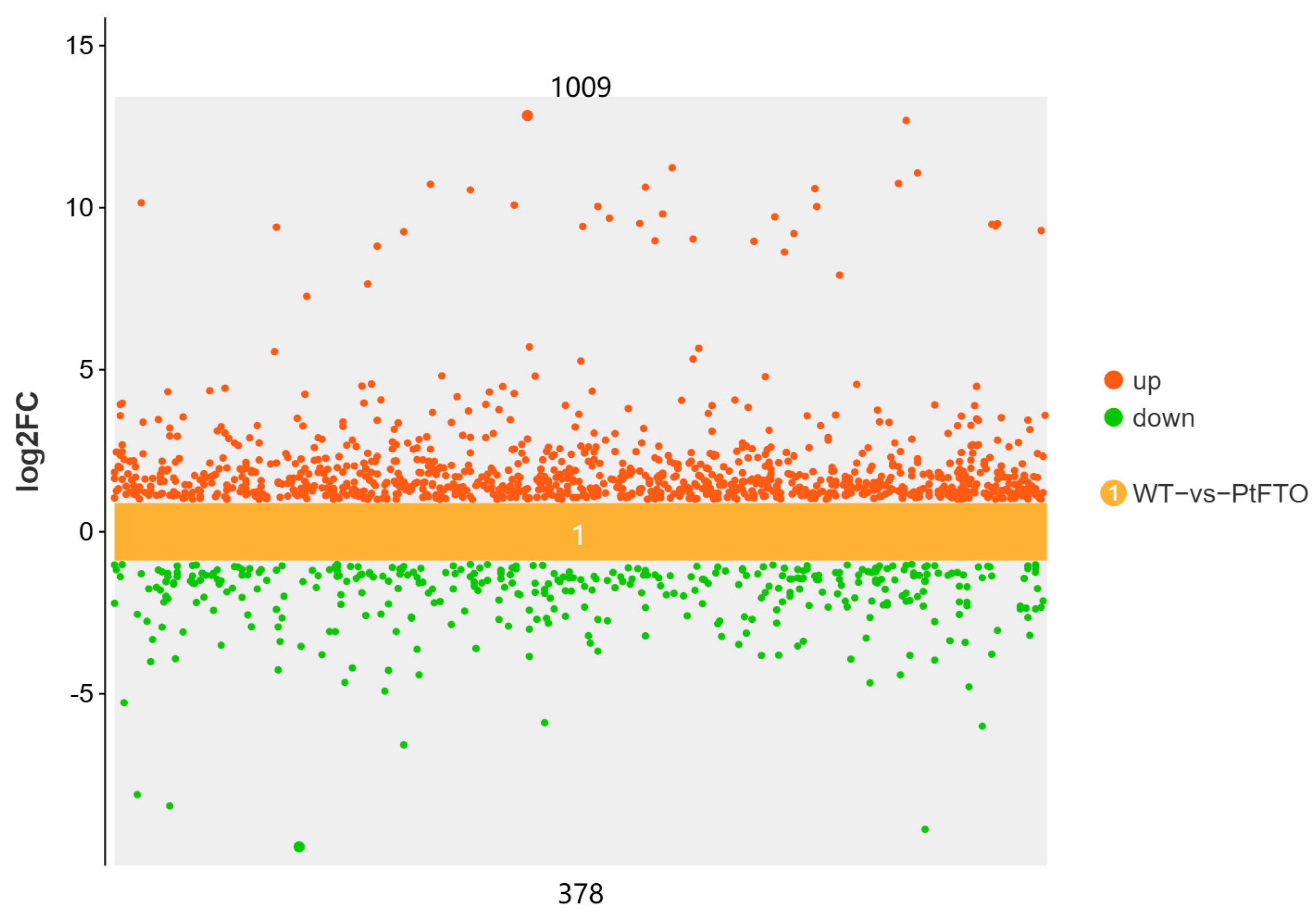
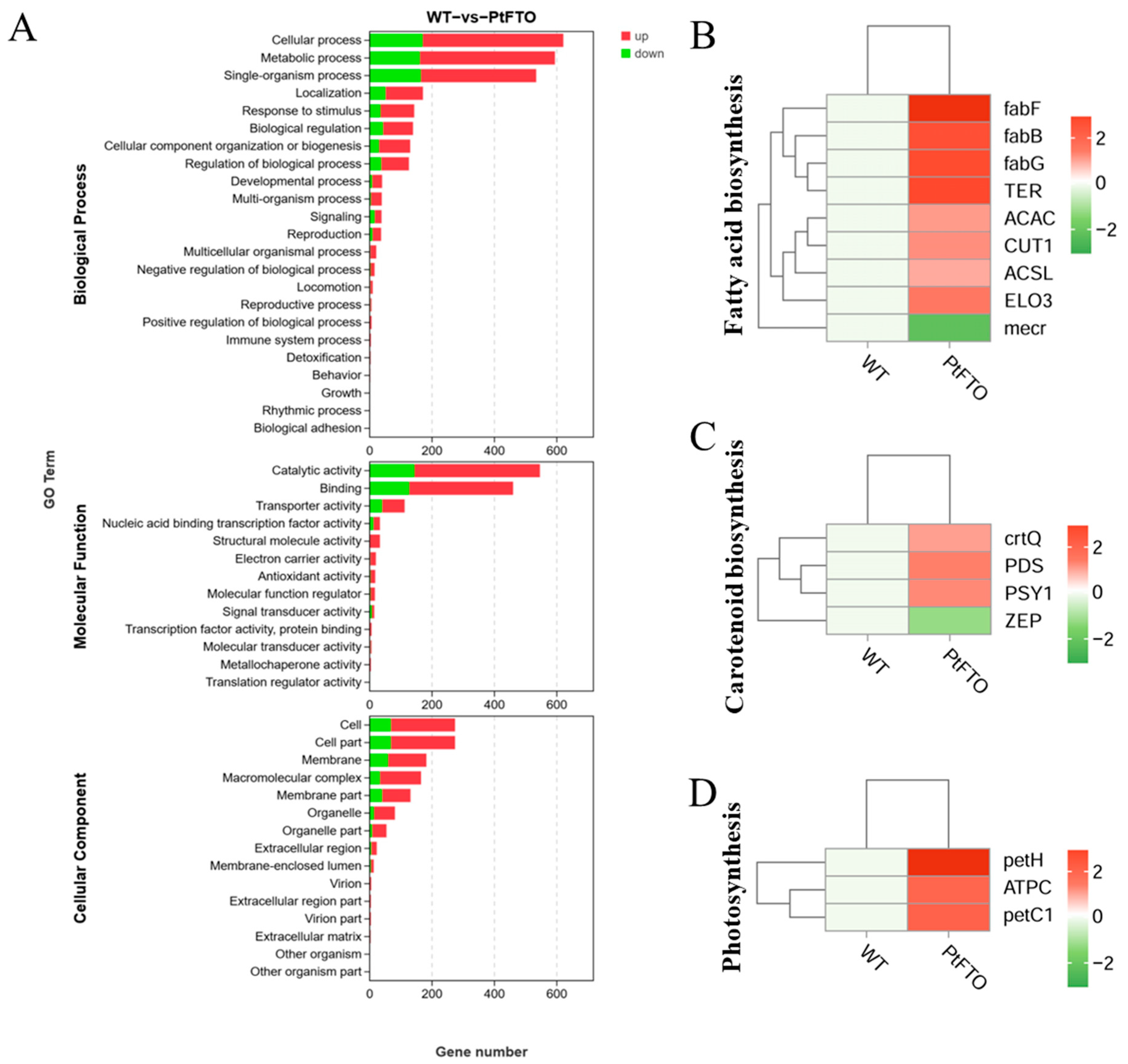
To study the influence of the PtFTO gene on the expression of various genes in transgenic microalgae, transcriptome analysis technology was used to analyze the differences between transgenic P. tricornutum and wild type. Transgenic line 9#, which exhibited the most pronounced phenotypical change, was selected for transcriptome analysis. Based on the screening criteria of p < 0.05 and up-regulation or down-regulation fold changes ≥ ±1.5, we identified 2103 DEGs between transgenic and wild-type P. tricornutum groups, among which 1009 and 378 genes were, respectively, upregulated and downregulated (Figure 5). Based on GO analysis, all DEGs were classified into 51 GO functional terms under the three main ontologies: biological processes, cellular components, and molecular functions. In the majority of GO functional terms, significantly more genes were upregulated than downregulated (Figure 6A). These results demonstrate that the overexpression of PtFTO in P. tricornutum activated the expression of numerous genes, potentially resulting in significant changes in characteristics.
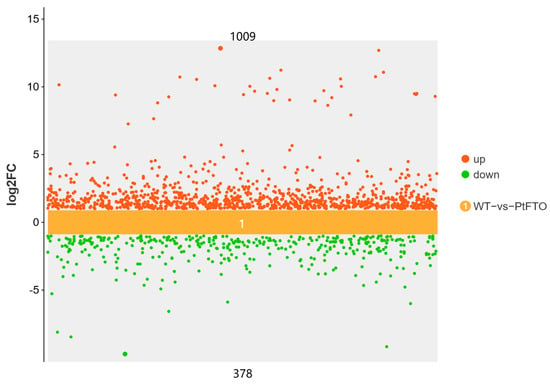
Figure 5.
Scatter plots of DEGs between transgenic and wild-type P. tricornutum groups. Red dots represent up-regulated genes (1009) and green dots represent down-regulated genes (378). DEGs were enriched with up-regulation or down-regulation fold changes ≥ ±2 and p < 0.05.
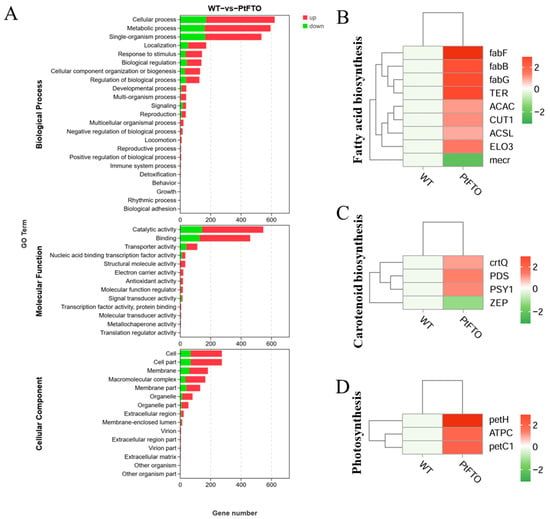
Figure 6.
Transcriptome analysis between transgenic and wild-type P. tricornutum groups. (A) GO analysis of DEGs between transgenic and wild-type P. tricornutum groups. All DEGs were classified into 51 GO functional terms under the three main ontologies: biological processes, cellular components, and molecular functions. The numerical labels on the bars of column chart represent the counts of up-regulated and down-regulated genes, respectively. KEGG analysis mapped fatty acid biosynthesis (B), carotenoid biosynthesis (C), and photosynthesis (D). Red bars represent up-regulated genes and green bars represent down-regulated genes. DEGs were enriched with up-regulation or down-regulation fold changes of ≥±2 and p < 0.05.
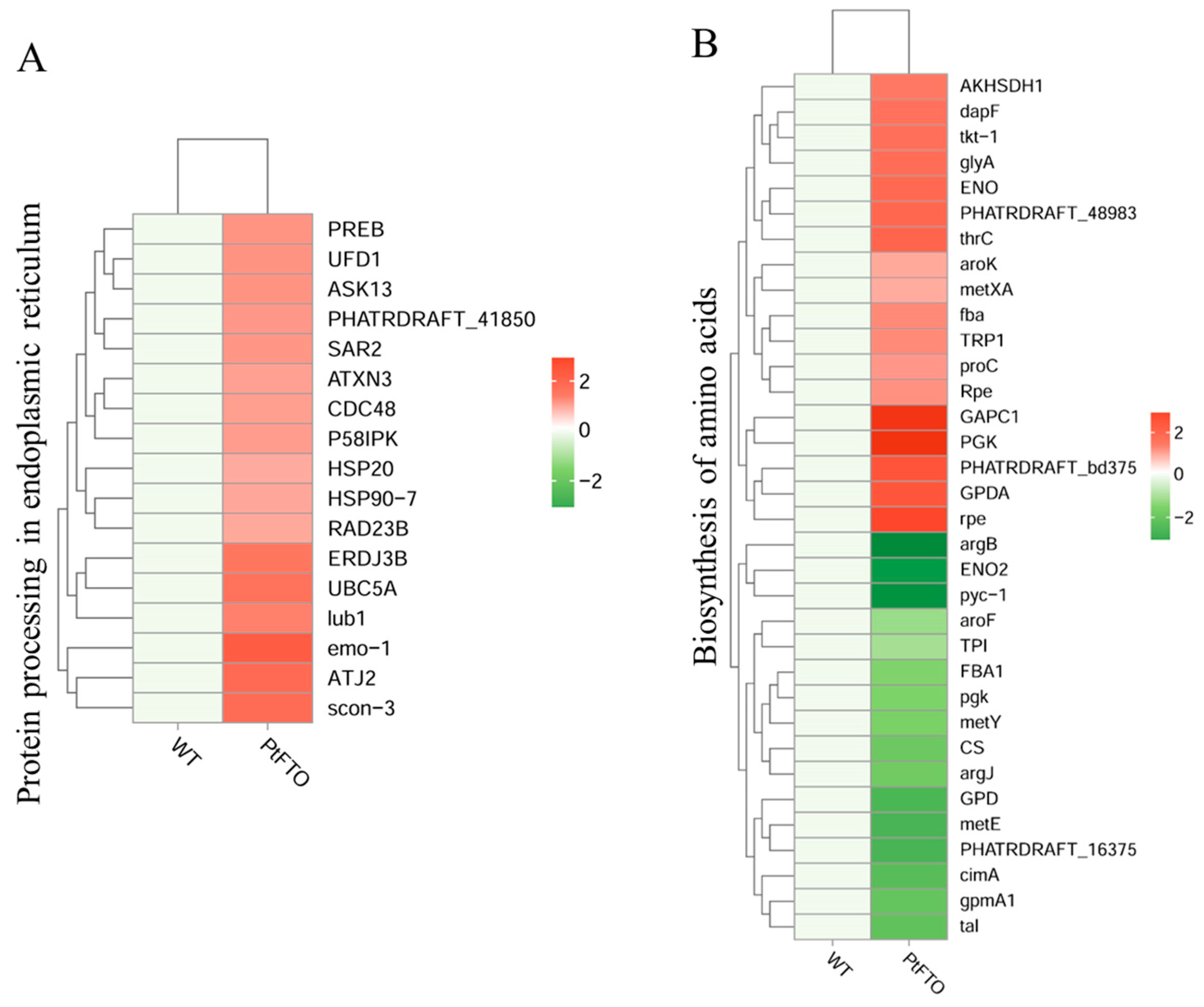
KEGG pathway analysis mapped multiple pathways, including the biosynthesis of amino acids, protein processing in the endoplasmic reticulum, fatty acid biosynthesis, carotenoid biosynthesis, and photosynthesis. The expression of the long-chain fatty acid synthase genes ACSL, fabF, and fabG in the fatty acid biosynthesis pathway of transgenic P. tricornutum was significantly upregulated compared with the wild-type variant. (Figure 6B). This indicated that overexpressed exogenous PtFTO in P. tricornutum modulated the expression of these genes, thus increasing the synthesis of polyunsaturated fatty acids (PUFAs) in transgenic P. tricornutum. Expression of the key enzymes zeta-carotene desaturase (crtQ), phytoene desaturase (PDS), and phytoene synthase (PSY1) in the carotenoid biosynthesis pathway of transgenic P. tricornutum was significantly upregulated compared with the wild type (Figure 6C). Additionally, the activation of photosynthetic pathways and carbohydrate metabolism might account for the accelerated growth of transgenic P. tricornutum (Figure 6D). Furthermore, many key genes that correlated positively with amino acid synthesis (e.g., dapF, glyA, and aroK) were significantly upregulated (Figure 7). These results are consistent with the upregulated lipid, carotenoid, and protein contents.
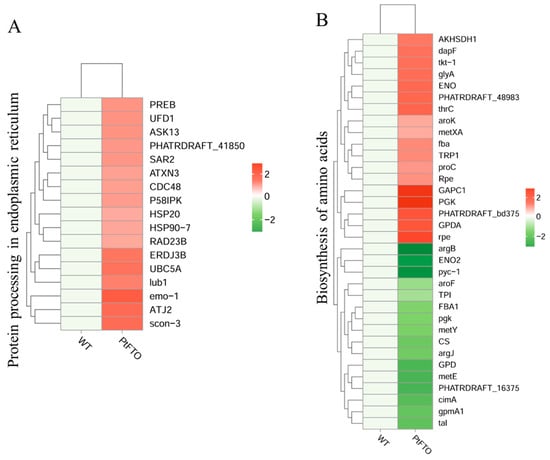
Figure 7.
The DEGS involved in amino acid synthesis. KEGG analysis mapped protein processing in endoplasmic reticulum (A) and biosynthesis of amino acids (B). Red bars represent up-regulated genes, and green bars represent down-regulated genes. DEGs were enriched with up-regulation or down-regulation fold changes of ≥±2 and p < 0.05.
Transcriptome analysis revealed that exogenous RNA demethylase PtFTO plays a broad-spectrum role in P. tricornutum. It promotes the expression of numerous genes by decreasing overall m6A methylation levels in cells, ultimately enhancing the associated metabolic pathways.
4. Discussion
Microalgae comprise a promising biological resource with significant value for the energy, food, and medicine industries, as well as in environmental protection. Genetic breeding technology provides a critical means for the directional improvement and functional expansion of microalgae. Gene editing technology has emerged as a powerful new tool that enables the precise and efficient editing of genes in humans, other animals, and plants, thus facilitating improvements, enhancements, and the development of new characteristics [53,54,55]. Gene editing has become an important means of genetic breeding and it has been applied to various microalgae [56]. Additionally, new genetic breeding techniques are still being explored. The discovery of FTO, an RNA m6A demethylase, holds promise as a tool for epigenetic editing, potentially leading to the development of novel epigenetic breeding technologies aimed at enhancing crop yield. Introducing FTO into rice and potato can demethylate RNA m6A modifications, leading to significant increases in crop yield and biomass [27]. This discovery paves the way for novel directions in plant breeding.
Microalgae are classified as lower plant species. We speculated that FTO also functions in epigenetic editing microalgae. Therefore, we selected P. tricornutum as our research focus. After codon optimization, PtFTO was introduced into P. tricornutum (Figure 1). The phenotypic analysis of the transgenic P. tricornutum revealed a significant reduction in the abundance of RNA m6A modifications (Figure 2C).
RNA m6A is a dynamic and reversible epigenetic modification that regulates gene expression via various mechanisms such as alternative splicing, mRNA export, mRNA stability, translation, and transcriptional regulation [29,30]. The expression of FTO in rice and potatoes mediates m6A demethylation, thereby promoting chromatin accessibility and activating transcription [27]. In this study, transcriptome analysis demonstrated that PtFTO activated the expression of numerous genes in transgenic P. tricornutum, of which 1556 were upregulated (Figure 5). These findings indicate that the mechanism through which PtFTO operates in P. tricornutum is analogous to its function in higher plants.
Further phenotypic analysis revealed that the growth rate of transgenic P. tricornutum was significantly faster than that of the wild type (Figure 2A,B); moreover, the levels of bioactive compounds, such as fatty acids, carotenoids, and proteins, were also markedly increased (Figure 3 and Figure 4). These results showed that PtFTO exerts a comprehensive influence on the physiological activities of P. tricornutum. KEGG analysis revealed that the expression levels of key enzymes involved in long-chain fatty acid synthesis (e.g., ACSL, fabF, and fabG), carotenoid synthesis (e.g., crtQ, PDS, and PSY1), and amino acid synthesis (e.g., dapF, glyA, and aroK) were upregulated in transgenic P. tricornutum (Figure 6B,C and Figure 7). This upregulation might directly contribute to increased levels of fatty acids, carotenoids, and proteins and may be regulated by FTO-mediated m6A demethylation. Additionally, the activation of photosynthetic pathways and carbohydrate metabolism might contribute to the accelerated growth of transgenic P. tricornutum (Figure 6D). Therefore, similar to rice and potatoes, the expression of exogenous FTO in P. tricornutum may induce chromatin openness and transcriptional activation by reducing RNA m⁶A modification levels, thereby activating relevant metabolic pathways.
Based on the potential mechanisms and phenotypic changes associated with PtFTO, compared with gene editing methods, such as CRISPR/Cas, FTO-mediated epigenetic editing revealed more target sites, enabling the activation of more genes and metabolic pathways. This regulation is reversible and flexible. However, the disadvantages are significant. For example, a relatively short duration of editing effect increases the likelihood of trait degeneration. Additionally, achieving precise targeting might be challenging because it depends on RNA metabolism. Therefore, enhancing the editing efficiency and improving the genetic stability of transgenic microalgae strains are key directions for future research.
5. Conclusions
This study expressed PtFTO in P. tricornutum and showed that PtFTO protein reduced m6A methylation levels in transgenic P. tricornutum. Phenotypic analysis revealed significantly faster growth rates and more abundant lipids, proteins, and carotenoids in transgenic compared to wild-type P. tricornutum. Transcriptome analysis revealed that the expression levels of numerous genes in transgenic P. tricornutum are significantly upregulated. The results of our KEGG analysis revealed the upregulated expression of key enzymes involved in long-chain fatty acid (e.g., ACSL, fabF, and fabG), carotenoid (e.g., crtQ, PDS, and PSY1), and amino acid synthesis (e.g., dapF, glyA, and aroK) in transgenic P. tricornutum, which agreed with our phenotypic findings. These results indicated that FTO promotes growth and increases the content of bioactive compounds in P. tricornutum by regulating the RNA m6A modification. This suggests that FTO has the potential to serve as a new tool for the epigenetic editing of microalgae.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biology14040414/s1, Table S1. The cDNA sequence of PtFTO; Figure S1. The original agarose gel electrophoresis image for PCR verification of PtFTO transgene integration in P. tricornutum.
Author Contributions
Conceptualization, X.C. and B.H.; data curation, M.Y. and B.H.; formal analysis, B.H.; funding acquisition, X.C. and B.H.; investigation, Y.Y., M.Y. and Y.Z.; methodology, Y.Y., M.Y., Y.Z., X.C. and B.H.; resources, X.C. and B.H.; supervision, B.H.; validation, Y.Y., M.Y. and B.H.; visualization, B.H.; writing—original draft, Y.Y. and B.H.; writing—review and editing, Y.Y., X.C. and B.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Guangxi Natural Science Foundation (2022GXNSFBA035553, 2024GXNSFAA010509), Guangxi Science and Technology Major Program (AA23073011), Project for Enhancing Young and Middle-aged Teacher’s Research Basis Ability in Colleges of Guangxi (2024KY0324), The Special Fund for Bagui Scholars of Guangxi (05019055).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated and/or analyzed during this study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mikami, K.; Hosokawa, M. Biosynthetic pathway and health benefits of fucoxanthin, an algae-specific xanthophyll in brown seaweeds. Int. J. Mol. Sci. 2013, 14, 13763–13781. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Fucoxanthin and its metabolite fucoxanthinol in cancer prevention and treatment. Mar. Drugs 2015, 13, 4784–4798. [Google Scholar] [CrossRef]
- Takahashi, K.; Hosokawa, M.; Kasajima, H.; Hatanaka, K.; Kudo, K.; Shimoyama, N.; Miyashita, K. Anticancer effects of fucoxanthin and fucoxanthinol on colorectal cancer cell lines and colorectal cancer tissues. Oncol. Lett. 2015, 10, 1463–1467. [Google Scholar] [CrossRef]
- Ye, G.; Wang, L.; Yang, K.; Wang, C. Fucoxanthin may inhibit cervical cancer cell proliferation via downregulation of HIST1H3D. J. Int. Med. Res. 2020, 48, 300060520964011. [Google Scholar] [CrossRef]
- Yu, R.X.; Yu, R.T.; Liu, Z. Inhibition of two gastric cancer cell lines induced by fucoxanthin involves downregulation of Mcl-1 and STAT3. Hum. Cell 2018, 31, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Hosokawa, M.; Sashima, T.; Murakami-Funayama, K.; Miyashita, K. Anti-obesity and anti-diabetic effects of fucoxanthin on diet-induced obesity conditions in a murine model. Mol. Med. Rep. 2009, 2, 897–902. [Google Scholar] [CrossRef]
- Jeon, S.M.; Kim, H.J.; Woo, M.N.; Lee, M.K.; Shin, Y.C.; Park, Y.B.; Choi, M.S. Fucoxanthin-rich seaweed extract suppresses body weight gain and improves lipid metabolism in high-fat-fed C57BL/6J mice. Biotechnol. J. 2010, 5, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Grasa-López, A.; Miliar-García, Á.; Quevedo-Corona, L.; Paniagua-Castro, N.; Escalona-Cardoso, G.; Reyes-Maldonado, E.; Jaramillo-Flores, M.E. Undaria pinnatifida and fucoxanthin ameliorate lipogenesis and markers of both Inflammation and cardiovascular dysfunction in an animal model of diet-induced obesity. Mar. Drugs 2016, 14, 148. [Google Scholar] [CrossRef]
- Troesch, B.; Eggersdorfer, M.; Laviano, A.; Rolland, Y.; Smith, A.D.; Warnke, I.; Weimann, A.; Calder, P.C. Expert opinion on benefits of long-chain omega-3 fatty acids (DHA and EPA) in aging and clinical nutrition. Nutrients 2020, 12, 2555. [Google Scholar] [CrossRef]
- Sawada, Y.; Saito-Sasaki, N.; Nakamura, M. Omega 3 fatty acid and skin diseases. Front. Immunol. 2020, 11, 623052. [Google Scholar] [CrossRef]
- Sharma, K.; Schuhmann, H.; Schenk, P. High lipid induction in microalgae for biodiesel production. Energies 2012, 5, 1532–1553. [Google Scholar] [CrossRef]
- Horst, I.; Parker, B.M.; Dennis, J.S.; Howe, C.J.; Scott, S.A.; Smith, A.G. Treatment of Phaeodactylum tricornutum cells with papain facilitates lipid extraction. J. Biotechnol. 2012, 162, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Molina-Grima, E.; Robles, A.; Giménez, A.; Ibáñez, M. Gram-scale purification of eicosapentaenoic acid (EPA, 20:5n-3) from wet Phaeodactylum tricornutum UTEX 640 biomass. J. Appl. Phycol. 1996, 8, 359–367. [Google Scholar] [CrossRef]
- Fajardo, A.; Esteban, L.; Robles, A.; Acien, G.; Moreno, P.; Molina-Grima, E. Lipid extraction from the microalga Phaeodactylum tricornutum. Eur. J. Lipid Sci. Technol. 2007, 109, 120–126. [Google Scholar] [CrossRef]
- Robles, A.; Esteban, L.; Giménez, A.; Páez, B.; Ibáñez-González, M.; Molina-Grima, E. Lipse-catalyzed esterification of glycerol and polyunsaturated fatty acids from fish and microalgae oils. Prog. Ind. Microbiol. 1999, 70, 379–391. [Google Scholar] [CrossRef]
- Apt, K.E.; Kroth-Pancic, P.G.; Grossman, A.R. Stable nuclear transformation of the diatom Phaeodactylum tricornutum. Mol. Gen. Genet. MGG 1996, 252, 572–579. [Google Scholar] [CrossRef]
- Falciatore, A.; d’Alcalà, M.R.; Croot, P.; Bowler, C. Perception of environmental signals by a marine diatom. Science 2000, 288, 2363–2366. [Google Scholar] [CrossRef]
- Scala, S.; Carels, N.; Falciatore, A.; Chiusano, M.L.; Bowler, C. Genome properties of the diatom Phaeodactylum tricornutum. Plant Physiol. 2002, 129, 993–1002. [Google Scholar] [CrossRef]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef]
- Yang, M.; Lin, X.; Liu, X.; Zhang, J.; Ge, F. Genome annotation of a model diatom Phaeodactylum tricornutum using an integrated proteogenomic pipeline. Mol. Plant 2018, 11, 1292–1307. [Google Scholar] [CrossRef]
- Siaut, M.; Heijde, M.; Mangogna, M.; Montsant, A.; Coesel, S.; Allen, A.; Manfredonia, A.; Falciatore, A.; Bowler, C. Molecular toolbox for studying diatom biology in Phaeodactylum tricornutum. Gene 2007, 406, 23–35. [Google Scholar] [CrossRef] [PubMed]
- De Riso, V.; Raniello, R.; Maumus, F.; Rogato, A.; Bowler, C.; Falciatore, A. Gene silencing in the marine diatom Phaeodactylum tricornutum. Nucleic Acids Res. 2009, 37, e96. [Google Scholar] [CrossRef] [PubMed]
- Stukenberg, D.; Zauner, S.; Dell’Aquila, G.; Maier, U.G. Optimizing CRISPR/Cas9 for the Diatom Phaeodactylum tricornutum. Front. Plant Sci. 2018, 9, 740. [Google Scholar] [CrossRef]
- He, C. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef]
- Yu, Q.; Liu, S.; Yu, L.; Xiao, Y.; Zhang, S.; Wang, X.; Xu, Y.; Yu, H.; Li, Y.; Yang, J.; et al. RNA demethylation increases the yield and biomass of rice and potato plants in field trials. Nat. Biotechnol. 2021, 39, 1581–1588. [Google Scholar] [CrossRef]
- Shen, W.; He, J.; Hou, T.; Si, J.; Chen, S. Common pathogenetic mechanisms underlying aging and tumor and means of Interventions. Aging Dis. 2022, 13, 1063–1091. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Yue, H.; Nie, X.; Yan, Z.; Weining, S. N6-methyladenosine regulatory machinery in plants: Composition, function and evolution. Plant Biotechnol. J. 2019, 17, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Vespa, L.; Vachon, G.; Berger, F.; Perazza, D.; Faure, J.D.; Herzog, M. The immunophilin-interacting protein AtFIP37 from Arabidopsis is essential for plant development and is involved in trichome endoreduplication. Plant Physiol. 2004, 134, 1283–1292. [Google Scholar] [CrossRef]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.C.; Wei, L.H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.; Chen, P.R.; He, C.; Jia, G. ALKBH10B is an RNA N(6)-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell 2017, 29, 2995–3011. [Google Scholar] [CrossRef]
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. USA 2017, 114, 10755–10760. [Google Scholar] [CrossRef]
- Zheng, H.X.; Sun, X.; Zhang, X.S.; Sui, N. m6A editing: New tool to improve crop quality? Trends Plant Sci. 2020, 25, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Hoguin, A.; Yang, F.; Groisillier, A.; Bowler, C.; Genovesio, A.; Ait-Mohamed, O.; Vieira, F.R.J.; Tirichine, L. The model diatom Phaeodactylum tricornutum provides insights into the diversity and function of microeukaryotic DNA methyltransferases. Commun. Biol. 2023, 6, 253. [Google Scholar] [CrossRef]
- Veluchamy, A.; Lin, X.; Maumus, F.; Rivarola, M.; Bhavsar, J.; Creasy, T.; O’Brien, K.; Sengamalay, N.A.; Tallon, L.J.; Smith, A.D.; et al. Insights into the role of DNA methylation in diatoms by genome-wide profiling in Phaeodactylum tricornutum. Nat. Commun. 2013, 4, 2091. [Google Scholar] [CrossRef]
- Scharnagl, M.; Richter, S.; Hagemann, M. The cyanobacterium Synechocystis sp. strain PCC 6803 expresses a DNA methyltransferase specific for the recognition sequence of the restriction endonuclease PvuI. J. Bacteriol. 1998, 180, 4116–4122. [Google Scholar] [CrossRef]
- Hagemann, M.; Gärtner, K.; Scharnagl, M.; Bolay, P.; Lott, S.C.; Fuss, J.; Huettel, B.; Reinhardt, R.; Klähn, S.; Hess, W.R. Identification of the DNA methyltransferases establishing the methylome of the cyanobacterium Synechocystis sp. PCC 6803. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2018, 25, 343–352. [Google Scholar] [CrossRef]
- Ambrosino, L.; Riccardi, A.; Welling, M.S.; Lauritano, C. Comparative transcriptomics to identify RNA writers and erasers in microalgae. Int. J. Mol. Sci. 2024, 25, 8005. [Google Scholar] [CrossRef]
- Guillard, R. The Culture of Marine Invertebrate Animals; Plenum Press: New York, NY, USA; London, UK, 1975. [Google Scholar] [CrossRef]
- Zaslavskaia, L.; Lippmeier, J.; Kroth, P.; Apt, K. Transformation of the diatom Phaeodactylum Tricornutum (Bacillariophyceae) with a variety of selectable marker and reporter genes. J. Phycol. 2000, 36, 379–386. [Google Scholar] [CrossRef]
- Lepage, G.; Roy, C.C. Improved recovery of fatty acid through direct transesterification without prior extraction or purification. J. Lipid Res. 1984, 25, 1391–1396. [Google Scholar] [CrossRef]
- Yang, Z.K.; Niu, Y.F.; Ma, Y.H.; Xue, J.; Zhang, M.H.; Yang, W.D.; Liu, J.S.; Lu, S.H.; Guan, Y.; Li, H.Y. Molecular and cellular mechanisms of neutral lipid accumulation in diatom following nitrogen deprivation. Biotechnol. Biofuels 2013, 6, 67. [Google Scholar] [CrossRef]
- Wellburn, A.R. The spectral determination of chlorophylls a and b, as well as total carotenoids, using various solvents with spectrophotometers of different resolution. J. Plant Physiol. 1994, 144, 307–313. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; Terol, J.; Talon, M.; Robles, M. BLAST2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, F.V.; Link, W.; Ferreira, B.I. CRISPR/Cas9-mediated genome editing: From basic research to translational medicine. J. Cell. Mol. Med. 2020, 24, 3766–3778. [Google Scholar] [CrossRef] [PubMed]
- Piergentili, R.; Del Rio, A.; Signore, F.; Umani Ronchi, F.; Marinelli, E.; Zaami, S. CRISPR-Cas and its wide-ranging applications: From human genome editing to environmental implications, technical limitations, hazards and bioethical issues. Cells 2021, 10, 969. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Doudna, J.A. CRISPR technology: A decade of genome editing is only the beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef]
- Jeon, S.; Lim, J.M.; Lee, H.G.; Shin, S.E.; Kang, N.K.; Park, Y.I.; Oh, H.M.; Jeong, W.J.; Jeong, B.R.; Chang, Y.K. Current status and perspectives of genome editing technology for microalgae. Biotechnol. Biofuels 2017, 10, 267. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).