
Physiological Changes and Time-Course Transcriptomic Analysis of Salt Stress in Chenopodium quinoa

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Measurement of Malondialdehyde Levels
2.3. Measurement of H2O2 Levels and Antioxidant Enzyme Activities
2.4. RNA-Seq and Data Processing
2.5. K-Means Clustering and KEGG Enrichment Analysis of DEGs
2.6. Identification of Transcription Factors from DEGs and Discovery of Cis-Regulatory Elements in the Promoter Regions
2.7. Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction (RT–qPCR)
2.8. Statistical Analysis
3. Results
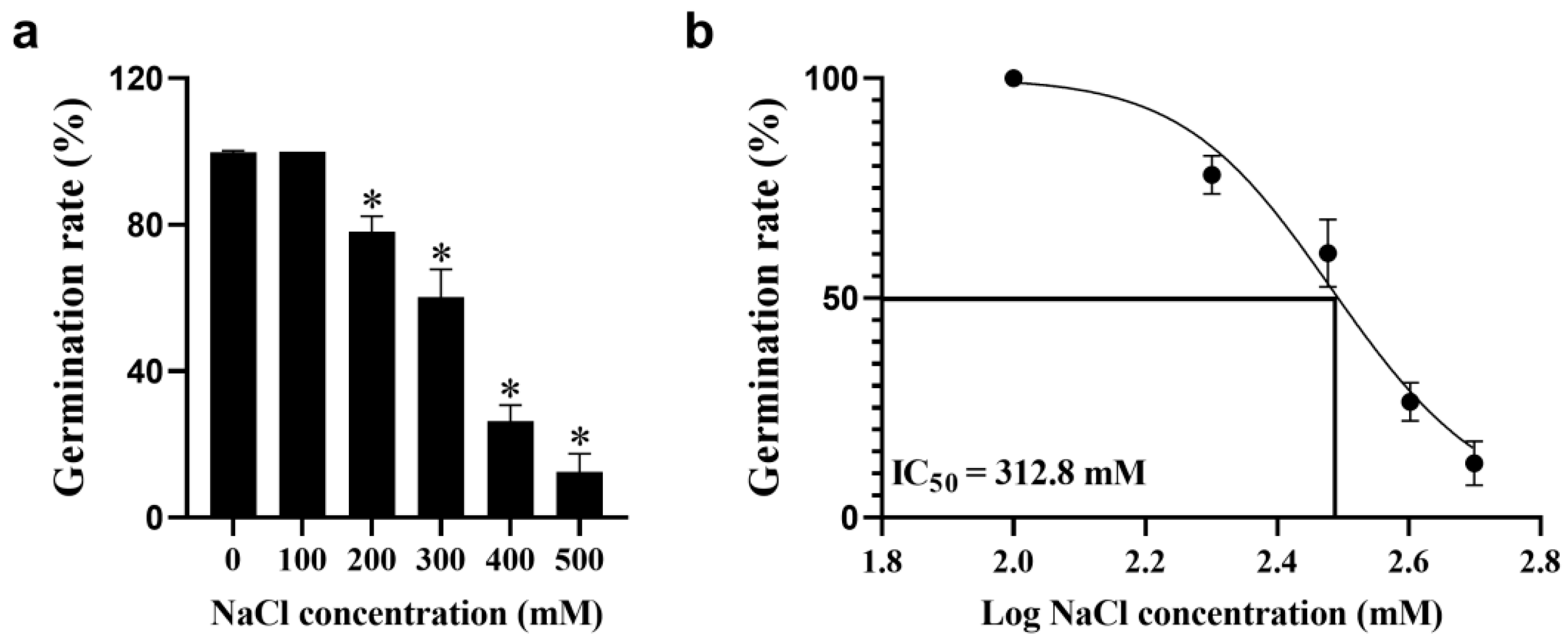
3.1. Seed Germination Assay
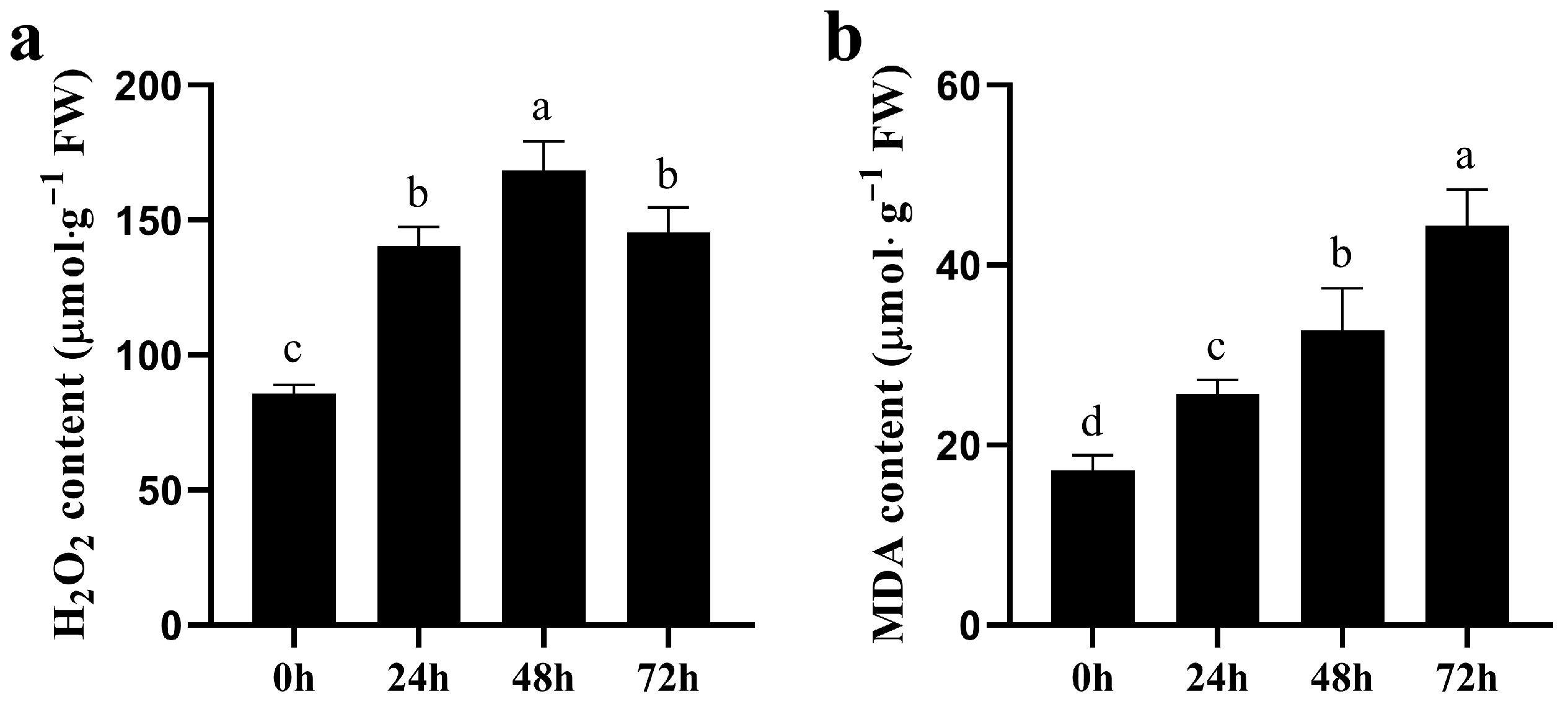
3.2. Cell Membrane Damage and H2O2 Production Under CK and NaCl Stress
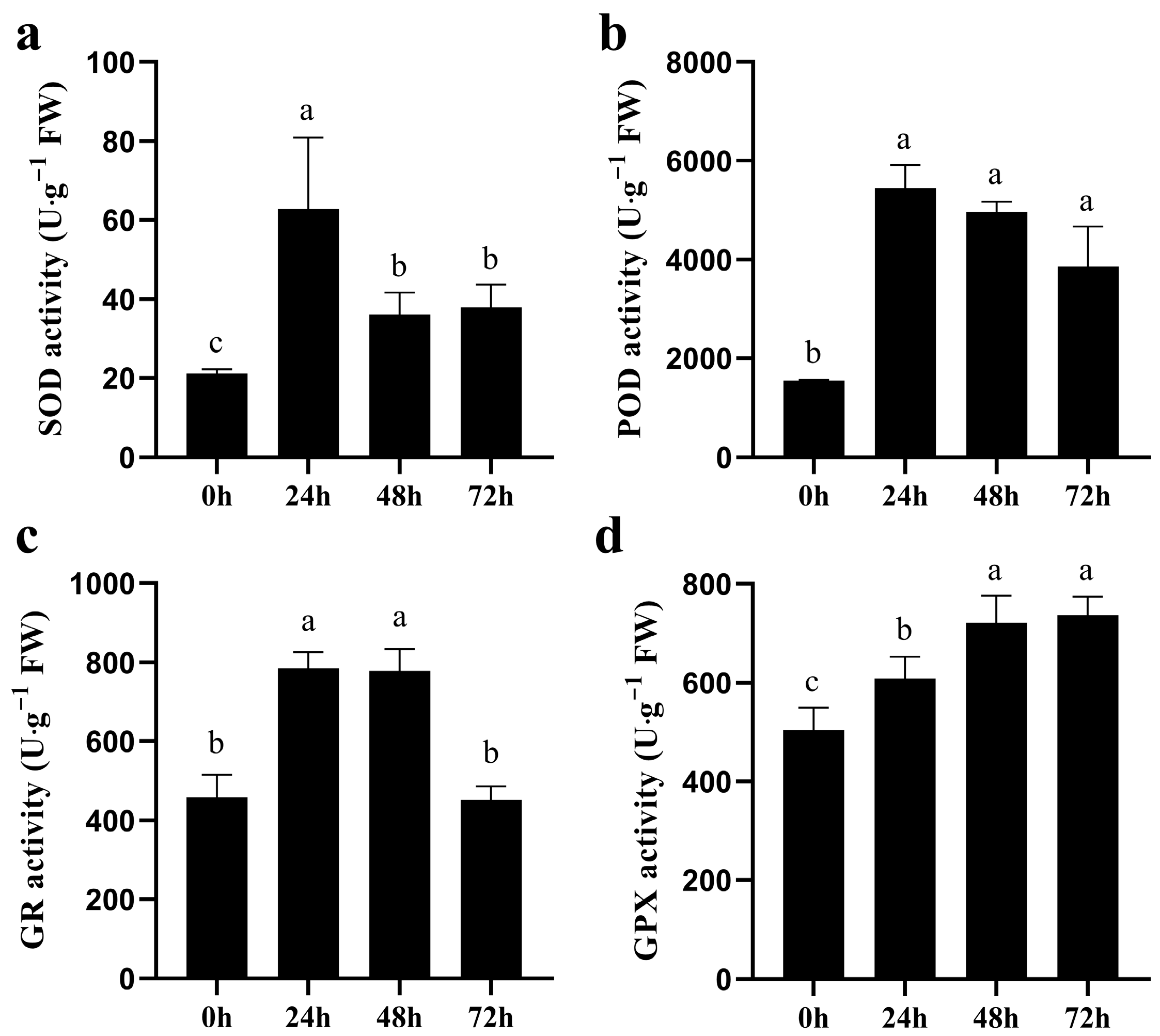
3.3. Activities of Antioxidant Enzymes in Quinoa Under CK and NaCl Stress
3.4. Time-Course Transcriptional Profiles of Seedlings in Quinoa
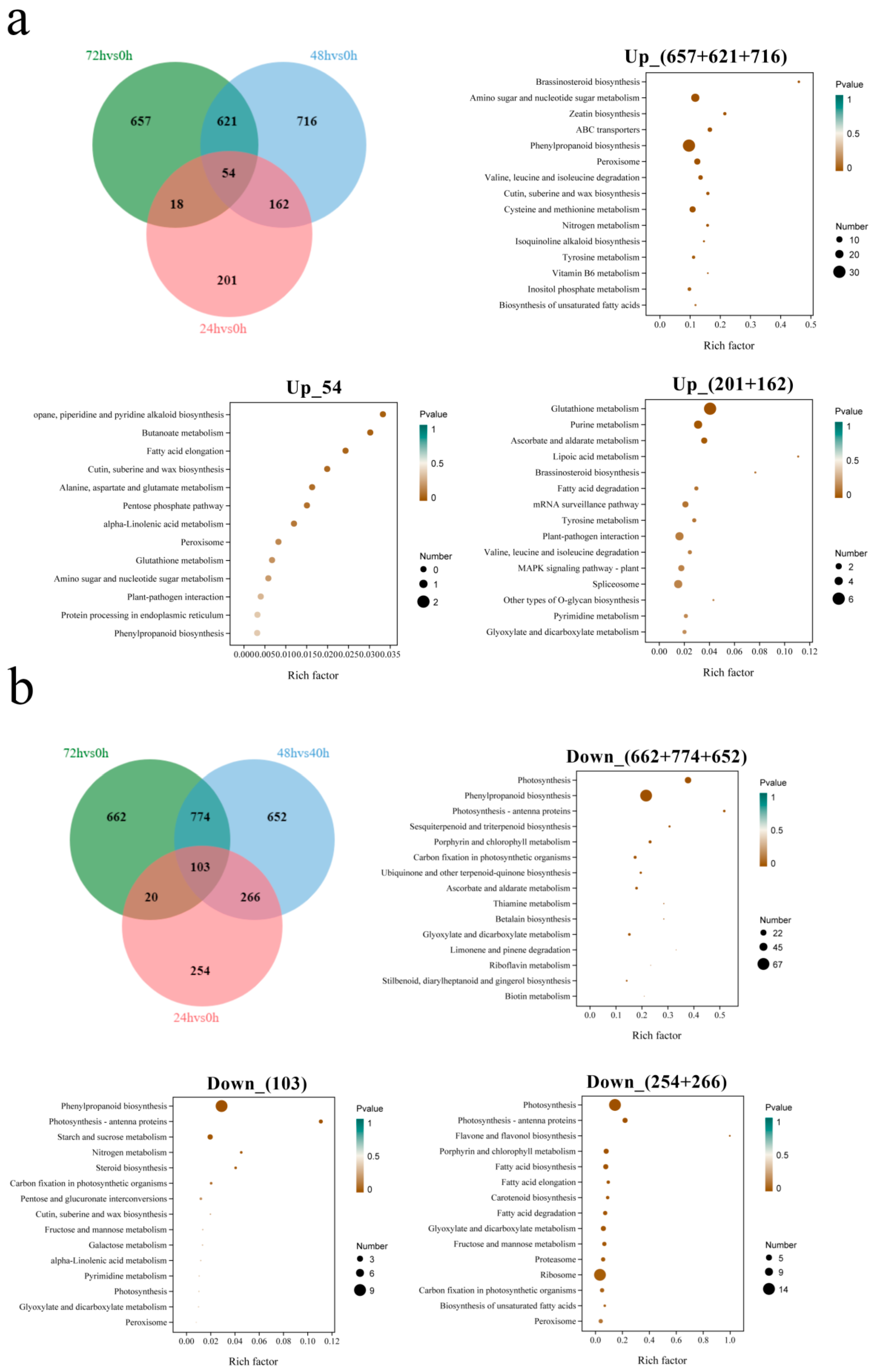
3.5. KEGG Enrichment Analysis Between Groups
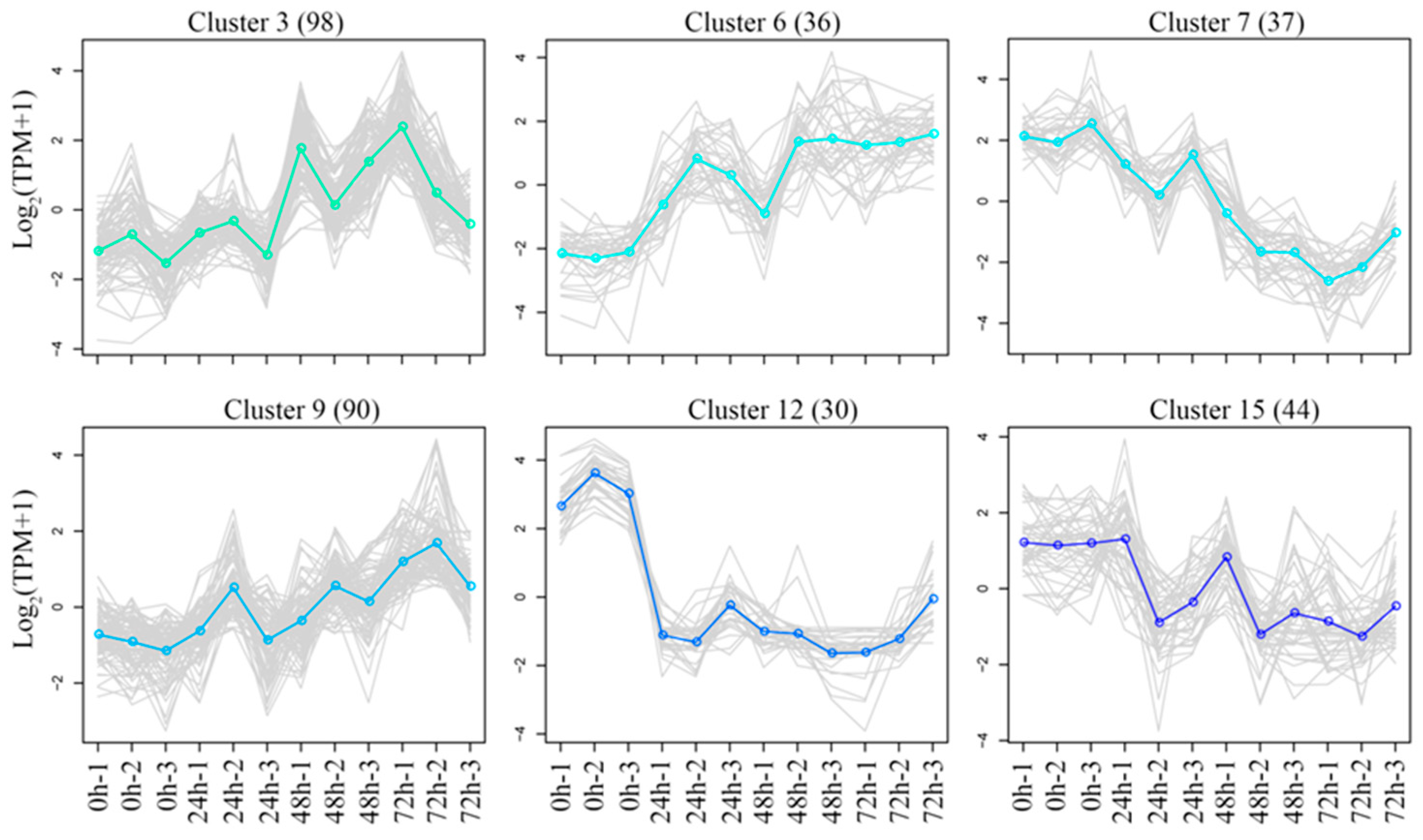
3.6. K-Means Clustering of DGEs
3.7. KEGG Enrichment Analysis of DEGs Under Salt Stress
3.8. Salt Stress Treatment Affected the Expression of Photosynthetic Pathway Genes in Quinoa Seedlings
3.9. Salt Stress Treatment Affects the Expression of of Glutathione Metabolism Genes in Quinoa Seedlings
3.10. Salt Stress Treatment Affects the Expression of Phenylpropanoid Biosynthesis, Starch and Sucrose Metabolism Genes in Quinoa Seedlings
3.11. Transcription Factors in Quinoa Are Involved in Salt Stress Regulation
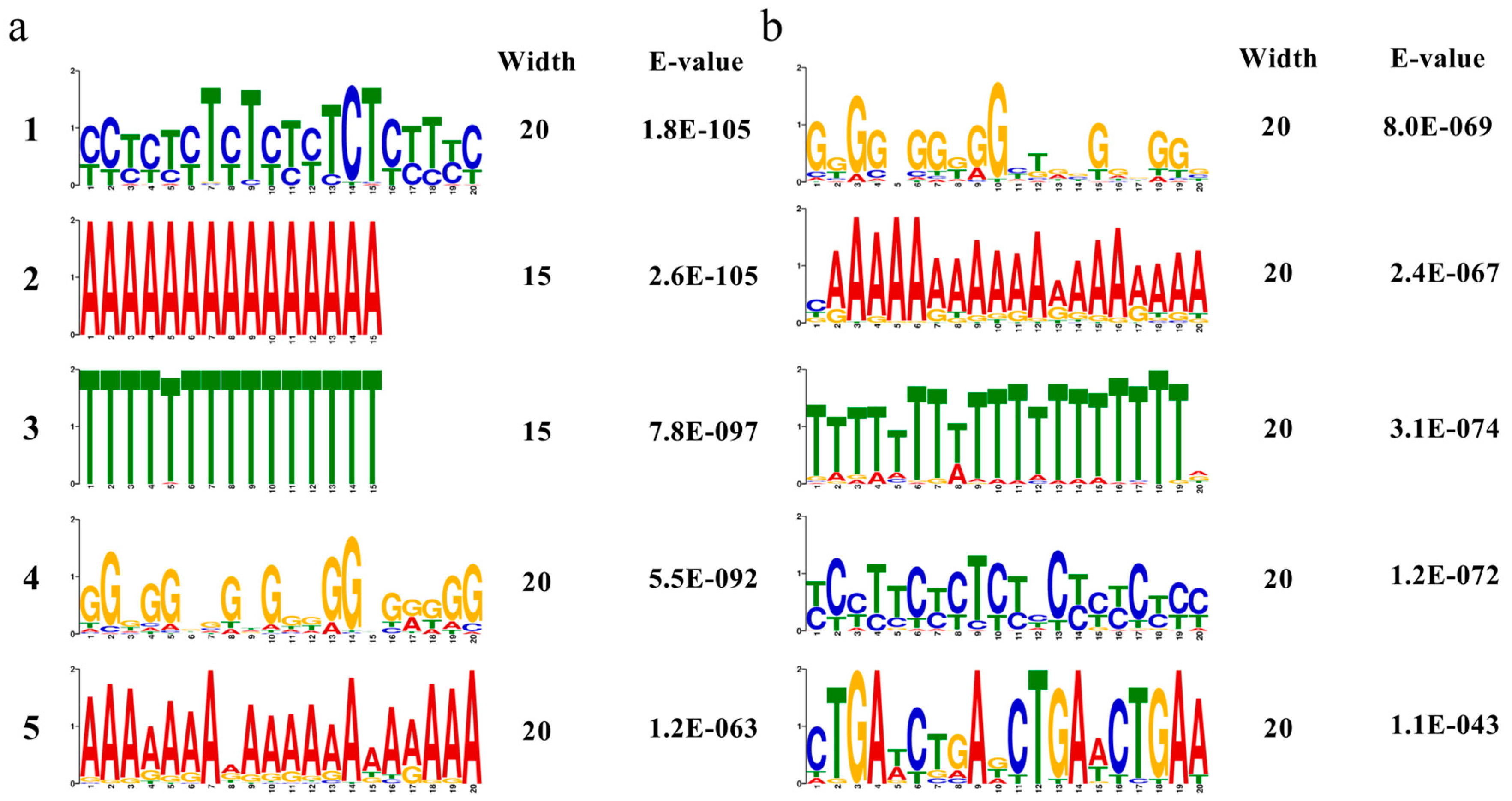
3.12. Prediction and Overrepresented Cis-Elements in the Promoter Regions of Transcription Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, S.; Zhang, Q.; Liu, M.; Zhou, H.; Ma, C.; Wang, P. Regulation of plant responses to salt stress. Int. J. Mol. Sci. 2021, 22, 4609. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhang, L.; Cui, Y.N.; Wang, S.; Bao, A.K. Identification of candidate genes related to salt tolerance of the secretohalophyte Atriplex canescens by transcriptomic analysis. BMC Plant Biol. 2019, 19, 213. [Google Scholar] [CrossRef]
- Shrivastava, P.; Kumar, R. Soil salinity, A serious environmental issue and plant growth promoting bacteria as one of the tools for its alleviation. Saudi J. Biol. Sci. 2015, 22, 123–131. [Google Scholar] [CrossRef]
- Weber, H.; Sciubba, J.D. The effect of population growth on the environment, evidence from European regions. Eur. J. Popul. 2018, 35, 379–402. [Google Scholar] [CrossRef]
- Salwan, R.; Sharma, A.; Sharma, V. Microbes mediated plant stress tolerance in saline agricultural ecosystem. Plant Soil 2019, 442, 1–22. [Google Scholar] [CrossRef]
- Wu, P.; Wu, X.; Wang, Y.; Xu, H.; Owens, G. Towards sustainable saline agriculture, interfacial solar evaporation for simultaneous seawater desalination and saline soil remediation. Water Res. 2022, 212, 118099. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, J.M. The evolution of halophytes, glycophytes and crops, and its implications for food security under saline conditions. New Phytol. 2015, 206, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Chen, A.; Xiao, L.; Muller, H.M.; Ache, P.; Haberer, G.; Zhang, M.; Jia, W.; Deng, P.; Huang, R.; et al. High-quality genome assembly of quinoa provides insights into the molecular basis of salt bladder-based salinity tolerance and the exceptional nutritional value. Cell Res. 2017, 27, 1327–1340. [Google Scholar] [CrossRef]
- Vega-Gálvez, A.; Miranda, M.; Vergara, J.; Uribe, E.; Puente, L.; Martínez, E.A. Nutrition facts and functional potential of quinoa (Chenopodium quinoa willd.), an ancient Andean grain, a review. J. Sci. Food Agric. 2010, 90, 2541–2547. [Google Scholar] [CrossRef]
- Filho, A.M.; Pirozi, M.R.; Borges, J.T.; Pinheiro-Sant’Ana, H.M.; Chaves, J.B.; Coimbra, J.S. Quinoa, nutritional, functional, and antinutritional aspects. Crit. Rev. Food Sci. Nutr. 2017, 57, 618–1630. [Google Scholar] [CrossRef]
- Delatorre-Herrera, J.; Pinto, M. Importance of ionic and osmotic components of salt stress on the germination of four quinua (Chenopodium quinoa Willd.) selections. Chil. J. Agric. Res. 2009, 69, 477–485. [Google Scholar] [CrossRef]
- Hariadi, Y.; Marandon, K.; Tian, Y.; Jacobsen, S.E.; Shabala, S. Ionic and osmotic relations in quinoa (Chenopodium quinoa Willd.) plants grown at various salinity levels. J. Exp. Bot. 2011, 62, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Shabala, L.; Mackay, A.; Tian, Y.; Jacobsen, S.E.; Zhou, D.; Shabala, S. Oxidative stress protection and stomatal patterning as components of salinity tolerance mechanism in quinoa (Chenopodium quinoa). Physiol. Plant. 2012, 146, 26–38. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Qiang, Q.; Li, Z.Z.; Chen, Z.G. Comparative physiological and biochemical mechanisms of drought tolerance in three contrasting cultivars of quinoa (Chenopodium quinoa). An. Jard. Bot. Madr. 2022, 79, e123. [Google Scholar] [CrossRef]
- Slimani, N.; Arraouadi, S.; Hajlaoui, H.; Cid-Samamed, A.; Borgi, M.A.; Snoussi, M. Salt stress enhanced bioactivity of quinoa leaf extracts: An in vitro and in silico study of acetylcholinesterase and tyrosinase inhibition for sustainable drug development. Pharmaceuticals 2025, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Peng, F.; Tedeschi, A.; Xue, X.; Wang, T.; Liao, J.; Zhang, W.; Huang, C. Do halophytes and glycophytes differ in their interactions with arbuscular mycorrhizal fungi under salt stress? A meta-analysis. Bot. Stud. 2020, 61, 13. [Google Scholar] [CrossRef]
- Burrieza, H.P.; Koyro, H.W.; Tosar, L.M.; Kobayashi, K.; Maldonado, S. High salinity induces dehydrin accumulation in Chenopodium quinoa Willd. Cv, Hualhuas Embryos. Plant Soil 2012, 354, 69–79. [Google Scholar] [CrossRef]
- Jarvis, D.E.; Ho, Y.S.; Lightfoot, D.J.; Schmöckel, S.M.; Li, B.; Borm, T.J.; Ohyanagi, H.; Mineta, K.; Michell, C.T.; Saber, N.; et al. The genome of Chenopodium quinoa. Nature 2017, 542, 307–312. [Google Scholar] [CrossRef]
- Hassan, I.; Chen, Y.; Muhammad, W.; Syed, T.R.; Muhammad, S.; Zeeshan, A. Salinity and exogenous H2O2 improve gas exchange, osmoregulation, and antioxidant metabolism in quinoa under drought stress. Physiol. Plant. 2023, 175, e14057. [Google Scholar]
- Bodrug-Schepers, A.; Stralis-Pavese, N.; Buerstmayr, H.; Dohm, J.C.; Himmelbauer, H. Quinoa genome assembly employing genomic variation for guided scaffolding. Theor. Appl. Genet. 2021, 134, 3577–3594. [Google Scholar] [CrossRef]
- Schmöckel, S.M.; Lightfoot, D.J.; Razali, R.; Tester, M.; Jarvis, D.E. Identification of putative transmembrane proteins involved in salinity tolerance in Chenopodium quinoa by integrating physiological data, RNAseq, and SNP analyses. Front. Plant Sci. 2017, 8, 1023. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.W.; Murphy, T.M.; Lin, C.H. Hydrogen peroxide-induced chilling tolerance in mung beans mediated through ABA-independent glutathione accumulation. Funct. Plant Biol. 2003, 30, 955–963. [Google Scholar] [CrossRef]
- Ma, N.L.; Che, L.W.; Abd, K.N.; Mustaqim, M.; Rahmat, Z.; Ahmad, A.; Lam, S.D.; Ismail, M.R. Susceptibility and tolerance of rice crop to salt threat: Physiological and metabolic inspections. PLoS ONE 2018, 13, e0192732. [Google Scholar] [CrossRef]
- Hemeda, H.M.; Klein, B. Effects of naturally occurring antioxidants on peroxidase activity of vegetable extracts. J. Food Sci. 1990, 55, 184–185. [Google Scholar] [CrossRef]
- Gallé, Á.; Bela, K.; Hajnal, Á.; Faragó, N.; Horváth, E.; Horváth, M.; Puskás, L.; Csiszár, J. Crosstalk between the redox signalling and the detoxification: GSTs under redox control? Plant Physiol. Biochem. 2021, 169, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT, a fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kazachkova, Y.; Eshel, G.; Pantha, P.; Cheeseman, J.M.; Dassanayake, M.; Barak, S. Halophytism, what have we learnt from Arabidopsis thaliana relative model systems? Plant Physiol. 2018, 178, 972–988. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0, toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van-de-Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.P.; Shi, H.F.; Yang, Y.Q.; Feng, Y.Q.; Chen, X.; Xiao, F.; Lin, H.H.; Guo, Y. Insights into plant salt stress signaling and tolerance. J. Genet. Genom. 2024, 51, 16–34. [Google Scholar] [CrossRef]
- Kaur, S.; Gupta, A.; Kaur, N. Effect of kinetin on starch and sucrose metabolising enzymes in salt stressed chickpea seedlings. Biol. Plant 2003, 46, 67–72. [Google Scholar] [CrossRef]
- Jacobsen, S.E. The worldwide potential for quinoa (Chenopodium quinoa Willd.). Food Rev. Int. 2003, 19, 167–177. [Google Scholar] [CrossRef]
- Shabala, S. Learning from halophytes, physiological basis and strategies to improve abiotic stress tolerance in crops. Ann. Bot. 2013, 112, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Derbali, W.; Manaa, A.; Goussi, R.; Derbali, I.; Abdelly, C.; Koyro, H.W. Post-stress restorative response of two quinoa genotypes differing in their salt resistance after salinity release. Plant Physiol. Biochem. 2021, 164, 222–236. [Google Scholar] [CrossRef]
- Vita, F.; Ghignone, S.; Bazihizina, N.; Rasouli, F.; Sabbatini, L.; Kiani-Pouya, A.; Kiferle, C.; Shabala, S.; Balestrini, R.; Mancuso, S. Early Responses to salt stress in quinoa genotypes with opposite behavior. Physiol. Plant. 2021, 173, 1392–1420. [Google Scholar] [CrossRef] [PubMed]
- Cimini, S.; Locato, V.; Giacinti, V.; Molinari, M.; De, G.L. A multifactorial regulation of glutathione metabolism behind salt tolerance in rice. Antioxidants 2022, 11, 1114. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Shabala, S.; Cuin, T.A.; Pang, J.; Percey, W.; Chen, Z.; Conn, S.; Eing, C.; Wegner, L.H. Xylem ionic relations and salinity tolerance in barley. Plant J. 2010, 61, 839–853. [Google Scholar] [CrossRef]
- Hussain, S.; Khaliq, A.; Tanveer, M.; Matloob, A.; Hussain, H.A. Aspirin priming circumvents the salinity-induced effects on wheat emergence and seedling growth by regulating starch metabolism and antioxidant enzyme activities. Acta Physiol. Plant. 2018, 40, 68. [Google Scholar] [CrossRef]
- Cha-um, S.; Charoenpanich, A.; Roytrakul, S.; Kirdmanee, C. Sugar accumulation, photosynthesis and growth of two indica rice varieties in response to salt stress. Acta Physiol. Plant. 2009, 31, 477–486. [Google Scholar] [CrossRef]
- Yin, Y.G.; Kobayashi, Y.; Sanuki, A.; Kondo, S.; Fukuda, N.; Ezura, H.; Sugaya, S.; Matsukura, C. Salinity induces carbohydrate accumulation and sugar-regulated starch biosynthetic genes in tomato (Solanum lycopersicum L. Cv. ‘Micro-tom’) fruits in an ABA- and osmotic stress-independent manner. J. Exp. Bot. 2010, 61, 563–574. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG, kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, K.B.; Maldonado, J.; Biondi, S.; Silva, H. RNA-seq analysis of salt-stressed versus non salt-stressed transcriptomes of Chenopodium quinoa landrace R49. Genes 2019, 10, 1042. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, R.; Jiang, Q.; Sun, X.; Zhang, H.; Hu, Z. GmNAC06, a nac domain transcription factor enhances salt stress tolerance in Soybean. Plant Mol. Biol. 2021, 105, 333–345. [Google Scholar] [CrossRef]
- Shen, L.; Zhao, E.; Liu, R.; Yang, X. Transcriptome analysis of eggplant under salt stress, AP2/ERF transcription factor SmERF1 acts as a positive regulator of salt stress. Plants 2022, 11, 2205. [Google Scholar] [CrossRef]
- Fu, M.; Kang, H.K.; Son, S.H.; Kim, S.K.; Nam, K.H. A subset of Arabidopsis rav transcription factors modulates drought and salt stress responses independent of ABA. Plant Cell Physiol. 2014, 55, 1892–1904. [Google Scholar] [CrossRef]
- Van-Zelm, E.; Zhang, Y.; Testerink, C. Salt tolerance mechanisms of plants. Annu. Rev. Plant Biol. 2020, 71, 403–433. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Bases | CG | Q30 | Inputs Reads | Mapped Reads | Multiple Mapped | Uniquely Mapped |
|---|---|---|---|---|---|---|---|
| 0 h-1 | 8,843,742,774 | 44.42% | 94.04% | 59,298,848 | 57,400,456 (96.80%) | 3,921,182 (6.61%) | 53,479,274 (90.19%) |
| 0 h-2 | 10,255,249,580 | 44.35% | 93.61% | 67,999,860 | 65,990,183 (97.04%) | 4,623,856 (6.80%) | 61,366,327 (90.24%) |
| 0 h-3 | 8,680,840,872 | 43.98% | 93.74% | 59,573,372 | 56,359,117 (94.60%) | 3,395,622 (5.70%) | 52,963,495 (88.90%) |
| 24 h-1 | 7,918,433,584 | 44.53% | 93.94% | 52,369,708 | 50,951,859 (97.29%) | 4,434,371 (8.47%) | 46,517,488 (88.83%) |
| 24 h-2 | 8,200,490,347 | 44.56% | 93.09% | 54,658,448 | 52,822,028 (96.64%) | 4,634,431 (8.48%) | 48,187,597 (88.16%) |
| 24 h-3 | 8,226,565,958 | 44.88% | 93.33% | 52,652,994 | 50,838,650 (96.55%) | 5,104,926 (9.70%) | 45,733,724 (86.86%) |
| 48 h-1 | 8,594,627,494 | 43.67% | 93.84% | 58,684,348 | 57,358,612 (97.74%) | 3,938,468 (6.71%) | 53,420,144 (91.03%) |
| 48 h-2 | 8,227,452,486 | 44.32% | 92.79% | 56,137,574 | 54,107,863 (96.38%) | 6,304,612 (11.23%) | 47,803,251 (85.15%) |
| 48 h-3 | 7,261,377,841 | 44.41% | 92.95% | 49,692,458 | 48,213,248 (97.02%) | 4,596,286 (9.25%) | 43,616,962 (87.77%) |
| 72 h-1 | 7,089,095,616 | 44.31% | 92.88% | 48,094,656 | 46,794,677 (97.30%) | 5,274,057 (10.97%) | 41,520,620 (86.33%) |
| 72 h-2 | 7,299,664,280 | 44.05% | 92.27% | 50,057,912 | 48,374,621 (96.64%) | 6,299,653 (12.58%) | 42,074,968 (84.05%) |
| 72 h-3 | 6,901,494,782 | 44.80% | 93.22% | 47,039,728 | 45,724,732 (97.20%) | 5,697,288 (12.11%) | 40,027,444 (85.09%) |
| Group | Number of Expressed Genes (Average TPM > 1) |
|---|---|
| 0 h | 24,350 |
| 24 h | 24,251 |
| 48 h | 24,075 |
| 72 h | 24,292 |
| Up-Regulated | Down-Regulated | |
|---|---|---|
| 24 h vs. 0 h | 435 | 643 |
| 48 h vs. 0 h | 1350 | 1795 |
| 48 h vs. 24 h | 524 | 588 |
| 72 h vs. 0 h | 1553 | 1559 |
| 72 h vs. 24 h | 348 | 503 |
| 72 h vs. 48 h | 22 | 54 |
| KEGG Pathway | ID | p Value | |
|---|---|---|---|
| Cluster 3 | Phenylpropanoid biosynthesis | ko00940 | 4.58 × 10−13 |
| Zeatin biosynthesis | ko00908 | 2.75 × 10−23 | |
| Glutathione metabolism | ko00480 | 0.002475429 | |
| Cluster 6 | Phenylpropanoid biosynthesis | ko00940 | 0.002516425 |
| Cluster 7 | Photosynthesis-antenna proteins | ko00196 | 7.56 × 10−7 |
| Cluster 9 | Phenylpropanoid biosynthesis | ko00940 | 0.001351993 |
| Cysteine and methionine metabolism | ko00520 | 0.001256953 | |
| Cluster 12 | Phenylpropanoid biosynthesis | ko00940 | 0.000255861 |
| Cluster 15 | Phenylpropanoid biosynthesis | ko00940 | 3.21 × 10−8 |
| Ubiquitin mediated proteolysis | ko04120 | 0.000542454 | |
| RNA polymerase | ko03020 | 3.54 × 10−12 | |
| Protein processing in endoplasmic reticulum | ko04141 | 1.25 × 10−4 |
| Discovered Motif | Matched Known Motif in Arabidopsis | E-Value | |
|---|---|---|---|
| Up-regulated | YYHNHYBYYYYYYYHYYYHH | BBRBPC_tnt.BPC5_colamp_a_m1 (BPC5) | 1.54 × 10−3 |
| AAAAAAAAAAAAAAA | ABI3VP1_tnt.VRN1_col_a_m1 (VRN1) | 2.71 × 10−13 | |
| TTTTWTTTTTTTTTT | ABI3VP1_tnt.VRN1_col_a_m1 (VRN1) | 5.17 × 10−7 | |
| NVNNNDNDDNNDDRBDNDDV | MYB_tnt.MYB67_col_a_m1 (MYB67) | 9.79 × 10−3 | |
| VRRRDDDADDVNVWDHRRRA | BBRBPC_tnt.BPC5_colamp_a_m1 (BPC5) | 1.24 × 10−2 | |
| YYHNHYBYYYYYYYHYYYHH | BBRBPC_tnt.BPC5_colamp_a_m1 (BPC5) | 1.54 × 10−3 | |
| Down-regulated | NNRNRNNNDKHNNNNDNDNN | MYB_tnt.MYB60_col_m1 (MYB60) | 2.04 × 100 |
| BRWRRRRDDDDDDDDRRNND | MYB_tnt.MYB60_col_m1 (MYB60) | 3.07 × 10−4 | |
| NDDWNDKWDWWHWWWWHTKD | E2FDP_tnt.DEL2_col_a_m1 (DEL2) | 5.79 × 10−1 | |
| HYHYNHHBHYHHYYBHNNNH | TCP_tnt.TCP1_col_a_m1 (TCP1) | 3.74 × 10−1 | |
| HKGADYBVADYTKADYYKAW | LIM_tnt.WLIM2A_col_a_m1 (WLIM2A) | 9.65 × 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Zhang, Y. Physiological Changes and Time-Course Transcriptomic Analysis of Salt Stress in Chenopodium quinoa. Biology 2025, 14, 416. https://doi.org/10.3390/biology14040416
Li P, Zhang Y. Physiological Changes and Time-Course Transcriptomic Analysis of Salt Stress in Chenopodium quinoa. Biology. 2025; 14(4):416. https://doi.org/10.3390/biology14040416
Chicago/Turabian StyleLi, Peipei, and Yemeng Zhang. 2025. "Physiological Changes and Time-Course Transcriptomic Analysis of Salt Stress in Chenopodium quinoa" Biology 14, no. 4: 416. https://doi.org/10.3390/biology14040416
APA StyleLi, P., & Zhang, Y. (2025). Physiological Changes and Time-Course Transcriptomic Analysis of Salt Stress in Chenopodium quinoa. Biology, 14(4), 416. https://doi.org/10.3390/biology14040416