

Eosinophilic Gastrointestinal Diseases in Inborn Errors of Immunity


 ,
, 
Abstract
1. Inborn Errors of Immunity and Gastrointestinal Manifestations
2. Eosinophilic Gastrointestinal Diseases
3. Material and Methods
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Notarangelo, L.D.; Bacchetta, R.; Casanova, J.L.; Su, H.C. Human Inborn Errors of Immunity: An Expanding Universe. Sci. Immunol. 2020, 5, eabb1662. [Google Scholar] [CrossRef]
- Castagnoli, R.; Delmonte, O.M.; Notarangelo, L.D. Congenital and Acquired Defects of Immunity: An Ever-Evolving Story. Pediatr. Allergy Immunol. 2022, 33, 61–64. [Google Scholar] [CrossRef]
- Castagnoli, R.; Notarangelo, L.D. Updates on New Monogenic Inborn Errors of Immunity. Pediatr. Allergy Immunol. 2020, 31, 57–59. [Google Scholar] [CrossRef]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef]
- Bousfiha, A.; Moundir, A.; Tangye, S.G.; Picard, C.; Jeddane, L.; Al-Herz, W.; Rundles, C.C.; Franco, J.L.; Holland, S.M.; Klein, C.; et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J. Clin. Immunol. 2022, 42, 1508–1520. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, R.; Pala, F.; Bosticardo, M.; Licari, A.; Delmonte, O.M.; Villa, A.; Marseglia, G.L.; Notarangelo, L.D. Gut Microbiota–Host Interactions in Inborn Errors of Immunity. Int. J. Mol. Sci. 2021, 22, 1416. [Google Scholar] [CrossRef]
- Ouahed, J.D. Understanding Inborn Errors of Immunity: A Lens into the Pathophysiology of Monogenic Inflammatory Bowel Disease. Front. Immunol. 2022, 13, 1026511. [Google Scholar] [CrossRef]
- Castagnoli, R.; Lougaris, V.; Giardino, G.; Volpi, S.; Leonardi, L.; Torre, F.L.; Federici, S.; Corrente, S.; Cinicola, B.L.; Soresina, A.; et al. Inborn Errors of Immunity with Atopic Phenotypes: A Practical Guide for Allergists. World Allergy Organ. J. 2021, 14, 100513. [Google Scholar] [CrossRef]
- Licari, A.; Votto, M.; Scudeller, L.; De Silvestri, A.; Rebuffi, C.; Cianferoni, A.; Marseglia, G.L. Epidemiology of Nonesophageal Eosinophilic Gastrointestinal Diseases in Symptomatic Patients: A Systematic Review and Meta-Analysis. J. Allergy Clin. Immunol. Pract. 2020, 8, 1994–2003.e2. [Google Scholar] [CrossRef] [PubMed]
- Licari, A.; Votto, M.; D’Auria, E.; Castagnoli, R.; Caimmi, S.M.E.; Marseglia, G.L. Eosinophilic Gastrointestinal Diseases in Children: A Practical Review. Curr. Pediatr. Rev. 2019, 16, 106–114. [Google Scholar] [CrossRef]
- Dellon, E.S.; Jensen, E.T.; Martin, C.F.; Shaheen, N.J.; Kappelman, M.D. Prevalence of Eosinophilic Esophagitis in the United States. Clin. Gastroenterol. Hepatol. 2014, 12, 589–596.e1. [Google Scholar] [CrossRef]
- Dellon, E.S.; Hirano, I. Epidemiology and Natural History of Eosinophilic Esophagitis. Gastroenterology 2018, 154, 319. [Google Scholar] [CrossRef]
- Dellon, E.S.; Gonsalves, N.; Abonia, J.P.; Alexander, J.A.; Arva, N.C.; Atkins, D.; Attwood, S.E.; Auth, M.K.H.; Bailey, D.D.; Biederman, L.; et al. International Consensus Recommendations for Eosinophilic Gastrointestinal Disease Nomenclature. Clin. Gastroenterol. Hepatol. 2022, 20, 2474–2484.e3. [Google Scholar] [CrossRef]
- Shaheen, N.J.; Mukkada, V.; Eichinger, C.S.; Schofield, H.; Todorova, L.; Falk, G.W. Natural History of Eosinophilic Esophagitis: A Systematic Review of Epidemiology and Disease Course. Dis. Esophagus 2018, 31, doy015. [Google Scholar] [CrossRef] [PubMed]
- Votto, M.; De Filippo, M.; Olivero, F.; Raffaele, A.; Cereda, E.; De Amici, M.; Testa, G.; Marseglia, G.L.; Licari, A. Malnutrition in Eosinophilic Gastrointestinal Disorders. Nutrients 2021, 13, 128. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.M.; Lenti, M.V.; Merli, S.; Licari, A.; Votto, M.; Marseglia, G.L.; Di Sabatino, A. Primary Eosinophilic Gastrointestinal Disorders and Allergy: Clinical and Therapeutic Implications. Clin. Transl. Allergy 2022, 12, e12146. [Google Scholar] [CrossRef]
- Votto, M.; Raffaele, A.; De Filippo, M.; Caimmi, S.; Brunero, M.; Riccipetitoni, G.; Marseglia, G.L.; Licari, A. Eosinophilic Gastrointestinal Disorders in Children and Adolescents: A Single-Center Experience. Dig. Liver Dis. 2022, 54, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Capucilli, P.; Cianferoni, A.; Grundmeier, R.W.; Spergel, J.M. Comparison of Comorbid Diagnoses in Children with and without Eosinophilic Esophagitis in a Large Population. Ann. Allergy. Asthma Immunol. 2018, 121, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Votto, M.; Marseglia, G.L.; De Filippo, M.; Brambilla, I.; Caimmi, S.M.E.; Licari, A. Early Life Risk Factors in Pediatric EoE: Could We Prevent This Modern Disease? Front. Pediatr. 2020, 8, 263. [Google Scholar] [CrossRef]
- O’Shea, K.M.; Aceves, S.S.; Dellon, E.S.; Gupta, S.K.; Spergel, J.M.; Furuta, G.T.; Rothenberg, M.E. Pathophysiology of Eosinophilic Esophagitis. Gastroenterology 2018, 154, 333. [Google Scholar] [CrossRef] [PubMed]
- Licari, A.; Castagnoli, R.; Marseglia, A.; Olivero, F.; Votto, M.; Ciprandi, G.; Marseglia, G.L. Dupilumab to Treat Type 2 Inflammatory Diseases in Children and Adolescents. Pediatr. Drugs 2020, 22, 295–310. [Google Scholar] [CrossRef]
- Votto, M.; Fasola, S.; Cilluffo, G.; Ferrante, G.; La Grutta, S.; Marseglia, G.L.; Licari, A. Cluster Analysis of Clinical Data Reveals Three Pediatric Eosinophilic Gastrointestinal Disorder Phenotypes. Pediatr. Allergy Immunol. 2022, 33, e13746. [Google Scholar] [CrossRef]
- Tran, P.; Gober, L.; Garabedian, E.K.; Fuleihan, R.L.; Puck, J.M.; Sullivan, K.E.; Spergel, J.M.; Ruffner, M.A. Eosinophilic Gastrointestinal Disorders in Patients with Inborn Errors of Immunity: Data from the USIDNET Registry. Front. Immunol. 2022, 13, 987895. [Google Scholar] [CrossRef]
- Yamazaki, S.; Ohtsuka, Y.; Yokokura, T.; Yokota, R.; Honjo, A.; Inage, E.; Baba, Y.; Mori, M.; Suzuki, R.; Iwata, T.; et al. Eosinophilic Gastroenteritis in a Patient with Bruton’s Tyrosine Kinase Deficiency. Pediatr. Int. 2016, 58, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ko, H.M.; Riffle, M.E.; Andreae, D.A.; Cunningham-Rundles, C.; Chehade, M.; Maglione, P.J. Eosinophilic Esophagitis Diagnosed in a Patient with Common Variable Immunodeficiency. J. Allergy Clin. Immunol. Pract. 2016, 4, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Hannouch, K.J.; McGoey, B.A.; Hauk, M.J.; Michelis, M.A.E. Common Variable Immunodeficiency and Eosinophilic Esophagitis Complicated by Atypical Burkitt’s Lymphoma: A Case Report. J. Allergy Clin. Immunol. 2017, 139, AB114. [Google Scholar] [CrossRef]
- Dixit, C.; Thatayatikom, A.; Pappa, H.; Knutsen, A.P. Treatment of Severe Atopic Dermatitis and Eosinophilic Esophagitis with Dupilumab in a 14-Year-Old Boy with Autosomal Dominant Hyper-IgE Syndrome. J. Allergy Clin. Immunol. Pract. 2021, 9, 4167–4169. [Google Scholar] [CrossRef] [PubMed]
- Scott, O.; Sharfe, N.; Dadi, H.; Vong, L.; Garkaby, J.; Abrego Fuentes, L.; Willett Pachul, J.; Nelles, S.; Nahum, A.; Roifman, C.M. Case Report: Eosinophilic Esophagitis in a Patient With a Novel STAT1 Gain-of-Function Pathogenic Variant. Front. Immunol. 2022, 13, 801832. [Google Scholar] [CrossRef]
- Tang, J.; Zhou, X.; Wang, L.; Hu, G.; Zheng, B.; Wang, C.; Lu, Y.; Jin, Y.; Guo, H.; Liu, Z. Eosinophilic Colitis in a Boy with a Novel XIAP Mutation: A Case Report. BMC Pediatr. 2020, 20, 171. [Google Scholar] [CrossRef]
- Arora, M.; Bagi, P.; Strongin, A.; Heimall, J.; Zhao, X.; Lawrence, M.G.; Trivedi, A.; Henderson, C.; Hsu, A.; Quezado, M.; et al. Gastrointestinal Manifestations of STAT3-Deficient Hyper-IgE Syndrome. J. Clin. Immunol. 2017, 37, 695–700. [Google Scholar] [CrossRef]
- Jung, Y.; Rothenberg, M.E. Roles and Regulation of Gastrointestinal Eosinophils in Immunity and Disease. J. Immunol. 2014, 193, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Wang, M.; Pemmaraju, V.R.; Collins, M.H.; Fulkerson, P.C.; Abonia, J.P.; Blanchard, C.; Putnam, P.E.; Rothenberg, M.E. Esophageal Remodeling Develops as a Consequence of Tissue Specific IL-5-Induced Eosinophilia. Gastroenterology 2008, 134, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Furuta, G.T. Eosinophils in Gastrointestinal Disorders: Eosinophilic Gastrointestinal Diseases, Celiac Disease, Inflammatory Bowel Diseases, and Parasitic Infections. Immunol. Allergy Clin. N. Am. 2015, 35, 413–437. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Aguilar, C. XIAP Deficiency Syndrome in Humans. Semin. Cell Dev. Biol. 2015, 39, 115–123. [Google Scholar] [CrossRef]



{kind=link}
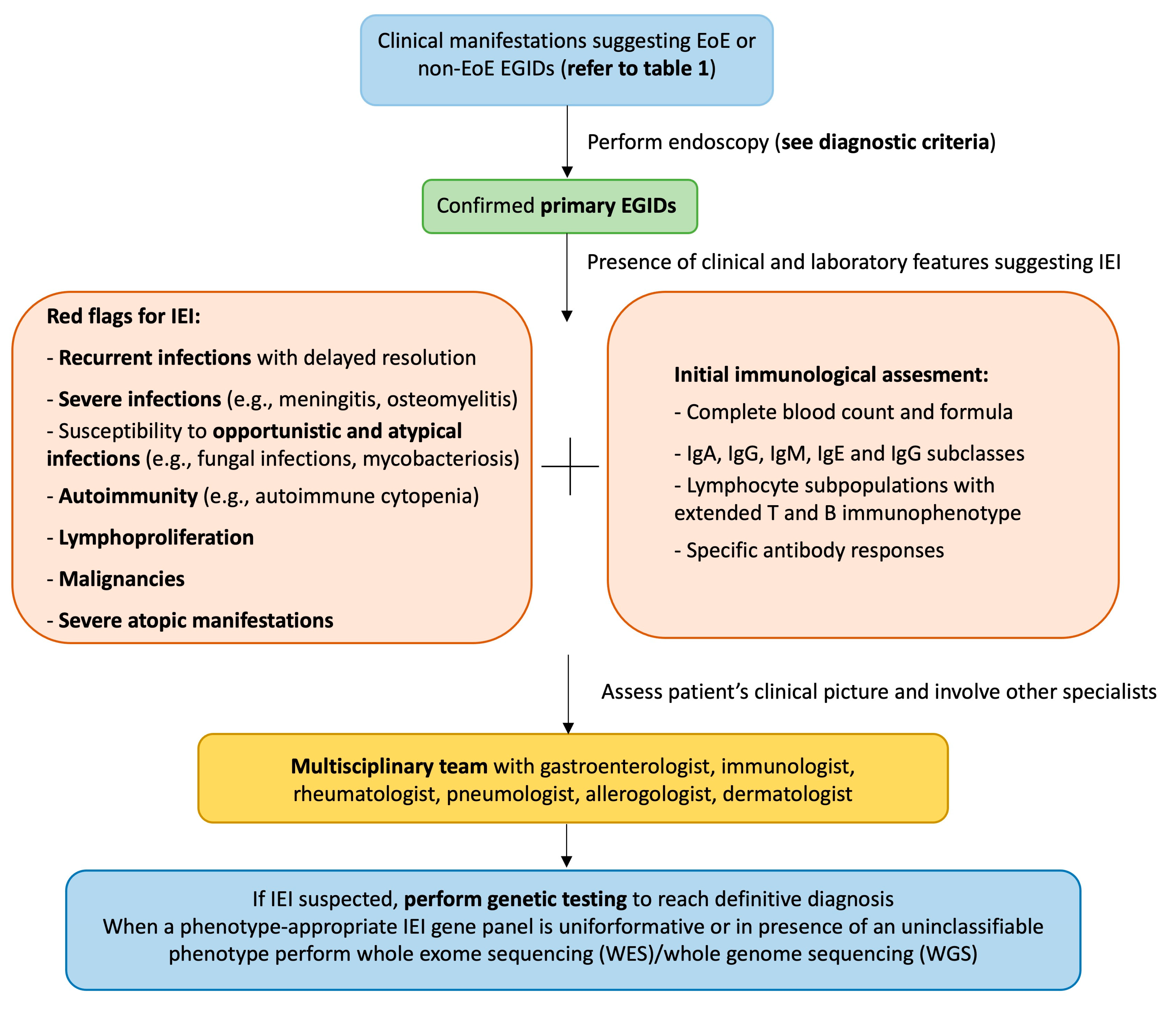{kind=link}
| Symptoms | Diagnosis | Treatments | |
|---|---|---|---|
| Eosinophilic esophagitis (EoE) | Symptoms mainly depend on the patient’s age
|
|
|
Non-EoE EGIDs
| Symptoms mainly depend on the site and the depth of intestinal inflammation
| 30 eos/HPF 52 eos/HPF 100 eos/HPF 84 eos/HPF 64 eos/HPF |
|
| Author, Year [Ref] | Type of Study | IEI | EGID | Age at EGID Diagnosis | Family History | EGID Symptoms | Other Comorbidities | EGID Diagnosis | Complications | EGID Treatment |
|---|---|---|---|---|---|---|---|---|---|---|
| Yamakazi et al., 2016 [24] | Case report | XLA | EoC | 27 years | n.a. | Chronic diarrhea, emaciation | Recurrent infections | >20 eos/HPF | n.a. | Prednisolone |
| Chen et al., 2016 [25] | Case report | CVID | EoE | 28 years | n.a. | Dysphagia, recurrent episodes of esophageal food impaction | Recurrent sinopulmonary infections | n.a. | Esophageal stenosis | Esophageal dilatation, PPI, FED, Oral fluticasone |
| Hannouch et al., 2016 [26] | Case report | CVID | EoE | n.a. | n.a. | Weight loss, food impaction | Burkitt’s lymphoma | n.a. | n.a. | Oral inhaled corticosteroids |
| Dixit et al., 2021 [27] | Case report | STAT3-HIES | EoE | n.a. | n.a. | Abdominal pain, dysphagia | Eczema, recurrent respiratory tract infections, cutaneous and retropharyngeal abscesses, and mycosis. | n.a. | n.a. | Dupilumab |
| Scott et al., 2022 [28] | Case report | STAT1-GOF | EoE | Late adolescence | Mother with choking episodes and CMCC; a daughter with CMCC and recurrent AOM. | Choking episodes, solid and liquid dysphagia | Vaginal candidiasis, scalp fungal infection, Candida esophagitis | 22 eos/HPF | Esophageal stenosis | Balloon dilatation FED Montelukast PPI Slurry budesonide |
| Tang et al., 2020 [29] | Case report | XIAP-deficiency | EoC | Infancy | Mother and sister had the mutation | Abdominal distension, perianal abscess. | Anemia, respiratory tract infections, impaired growth | n.a. | n.a. | n.a. |
| Tran et al., 2022 [23] | Retrospective cohort study | CVID (43.2%), combined immunodeficiencies (21.6%), CGD (8.1%), HIES (6.8%), and ALPS (6.8%). | 61/74 (82,5%) patients with EoE and 13/74 (17.5%) with EoG, EoN, and EoC. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Votto, M.; Naso, M.; Brambilla, I.; Caimmi, S.; De Filippo, M.; Licari, A.; Marseglia, G.L.; Castagnoli, R. Eosinophilic Gastrointestinal Diseases in Inborn Errors of Immunity. J. Clin. Med. 2023, 12, 514. https://doi.org/10.3390/jcm12020514
Votto M, Naso M, Brambilla I, Caimmi S, De Filippo M, Licari A, Marseglia GL, Castagnoli R. Eosinophilic Gastrointestinal Diseases in Inborn Errors of Immunity. Journal of Clinical Medicine. 2023; 12(2):514. https://doi.org/10.3390/jcm12020514
Chicago/Turabian StyleVotto, Martina, Matteo Naso, Ilaria Brambilla, Silvia Caimmi, Maria De Filippo, Amelia Licari, Gian Luigi Marseglia, and Riccardo Castagnoli. 2023. "Eosinophilic Gastrointestinal Diseases in Inborn Errors of Immunity" Journal of Clinical Medicine 12, no. 2: 514. https://doi.org/10.3390/jcm12020514
APA StyleVotto, M., Naso, M., Brambilla, I., Caimmi, S., De Filippo, M., Licari, A., Marseglia, G. L., & Castagnoli, R. (2023). Eosinophilic Gastrointestinal Diseases in Inborn Errors of Immunity. Journal of Clinical Medicine, 12(2), 514. https://doi.org/10.3390/jcm12020514