Abstract
Neurodegenerative diseases (NDs) pose a significant health burden globally, and this burden is increasing with an ageing population. Despite this challenge, restorative treatments for NDs remain elusive. In these conditions, the brain is vulnerable to oxidative stress and inflammation due to a deficiency or reduction in antioxidative enzymes. Oxidative stress and inflammation damage neuronal cells, leading to neurodegeneration. Various studies have explored the neuroprotective effects of flavonoids in different in vitro and animal models, primarily due to their antioxidative and anti-inflammatory properties. Crude extracts and active metabolites of Semecarpus anacardium L. have shown potential in reversing dysregulated oxidative stress and neuroinflammation. S. anacardium L. extract (SAE) and its phytocomponents, such as butein, anacardic acid, and amentoflavone, have been experimentally demonstrated to modulate oxidative stress and neuroinflammation through coordinated activation of Nrf2-mediated antioxidant pathways and suppression of NF-ĸB-driven inflammatory signaling. At a molecular level, flavonoids from SAE induce the expression of p38 MAPK and Nrf2, as well as antioxidant enzymes. Furthermore, inflammatory genes such as NF-ĸB, MAPK, AP-1, iNOS, and COX-2 are suppressed following treatment with SAE. NF-ĸB inhibition leads to neuroprotection via inhibiting the function of caspase-3 and apoptosis. Overall, this review discusses the protective role of SAE and its phytocomponents in mitigating neuronal oxidative stress, inflammation, and degeneration. Furthermore, this review highlights the translational potential of SAE and its phytocomponents as complementary therapeutic candidates for neurodegenerative disorders. However, variability in extract composition and limited pharmacokinetic characterization remain key barriers to clinical translation.
1. Introduction
1.1. Overview of Neurodegenerative Diseases
Neurodegenerative diseases (NDs) are a diverse group of CNS disorders that affect memory, reasoning, emotions, and motor skills [1,2]. The pathology of NDs associated with the dysfunction of the central and peripheral nervous systems and death involves autophagosomal or lysosomal systems, oxidative stress, inflammation, protein misfolding and homeostasis, programmed cell death, genetics, and environmental factors [3]. Common NDs include stroke, brain trauma, prion disease, SCI, ALS, AD, PD, HD, MND, SCA, TBI, and SMA.
1.2. Role of Oxidative Stress and Inflammation in Neurodegeneration
Oxidative damage is the main pathological factor associated with the age-related cognitive changes [4,5]. The brain is particularly susceptible to oxidative stress due to low levels of GSH [6], high polyunsaturated fatty acid (PUFA) content [7], elevated oxygen requirements [8], and a limited antioxidant defense system [9]. Elevated levels of calcium, lipoxygenase, lipid peroxidation, and COX were observed in brain hypoxia, which alters the intracellular microenvironment [10]. Excessive cellular ROS causes oxidative stress, which can activate or repress NF-ĸB signalling depending on the phase and context. NF-ĸB can have both antioxidant and pro-oxidant roles [11] and serves as a regulator of the inflammatory response [12]. Oxidative stress and inflammation contribute cognitively to various disease conditions [13], including neuronal degeneration [14,15]. Various factors inducing neurodegeneration are depicted in Figure 1.
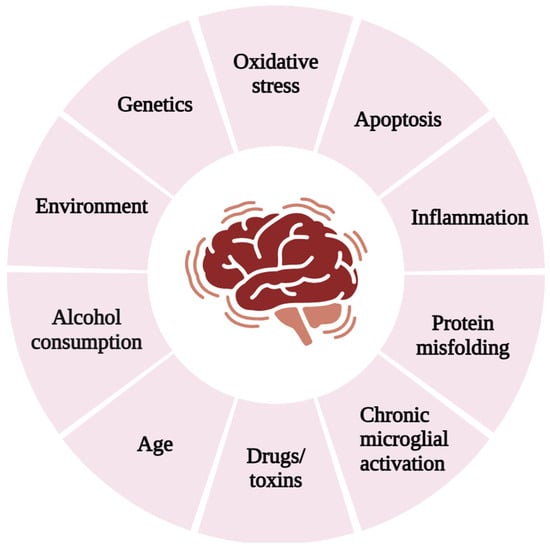
Figure 1.
Risk factors contributing to neurodegenerative diseases.
1.3. Semecarpus anacardium L.: Ethnopharmacological Background and Relevance
S. anacardium L. commonly known as ‘Bhallataka’ or ‘marking nut,’ is an important medicinal plant classified as ‘Upavisha’ (toxic but not lethal for human health) in Ayurveda. Invariably, the S. anacardium L. nut contains a wide range of biflavonoids and bioflavonoids, and its extracts have been traditionally used for their neuroprotective effects. S. anacardium L. is classified as a Schedule (1) drug, and purification must be performed prior to administration [16]. The presence of phytochemical constituents in S. anacardium L. depends on the solvent used for extraction. Acetone extraction yielded the highest amounts of phytochemicals, with the following values (mg/g): total phenols 162.56, flavonoids 82.62, saponins 109.09, and tannins 42.25 [17]. Butein (3,4,2′,4′-tetrahydroxychalcone) [18,19], anacardic acid [16], and amentoflavone [20] are the principal phytocompounds of S. anacardium L. The presence of anacardic acid was 5.62% after extraction using the Siddha purification process [16]. HPTLC quantification reported a total amentoflavone content of 10 g/kg in S. anacardium seed. However, no report is available for butein. The structure, molecular weight, and formula are represented in Figure 2. They have been extensively studied for their roles in oxidative stress, neuroinflammation, and neuroprotection in both in vitro and in vivo settings. The botanical characteristics, pharmacological applications [21] and toxicological effects [22] of S. anacardium L. have been reported earlier. This review discusses the role of S. anacardium L., and its selective phytocompounds in protecting against neuronal oxidative stress, inflammation, and degeneration.
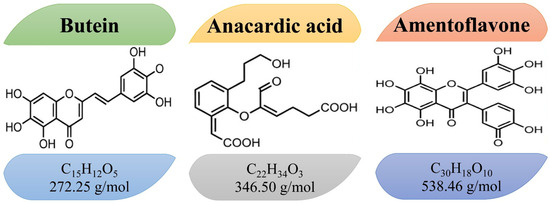
Figure 2.
The structure, molecular weight and formula of the phytocompounds butein, anacardic acid, and amentoflavone.
2. Oxidative Stress and Inflammation Mediated Neuronal Damage
The brain heavily relies on oxidative metabolism; therefore, oxidative stress is regarded as a key factor in neurodegeneration. As shown in Figure 1, several factors such as smoking, radiation, alcohol intake, heavy metals, processed food additives, and others induce oxidative stress in neuron cells. The antioxidant enzymes involved in neuronal degeneration are discussed here.
2.1. Glutathione Peroxidase (GPx)
Glutathione peroxidase (EC 1.11.1.9 and EC 1.11.1.12) is a common name for a family of several isozymes that catalyze the reduction of H2O2 or organic hydroperoxides to water or alcohols through reduced glutathione [23]. GPx enzymes are selenium-dependent and classified as follows: (1) GPx1, found in the cytosol, nucleus, and mitochondria; (2) GPx2, which accumulates in the cytosol and nucleus; (3) GPx3, present in the cytosol; and (4) phospholipid GPx, located in the nucleus, cytosol, mitochondria, bound to membranes [24,25]. GPx is a key component of the neuronal antioxidant defense network, functioning in coordination with GSH, GST, SOD, and catalase to maintain redox homeostasis [26], neutralizes free radical compounds [27], protects cells from oxidative stress. GPx4, a selenoprotein, is a crucial regulator of oxytosis and ferroptosis. Several studies indicate that reduced levels of GPx lead to neurodegenerative disorders [28,29].
2.2. Glutathione (GSH)
GSH is a tripeptide that encompasses cysteine, glutamate, and glycine [30]. GSH is primarily secreted in the liver via de novo and scavenger pathways [31]. GSH redox status plays a significant role in regulating most cellular metabolic processes and maintaining the balance between oxidation and reduction [32,33]. GSH performs an endogenous role in conserving the intracellular antioxidant system, redox equilibrium, cell signalling, gene expression, and cell differentiation [34]. To eliminate ROS or RNS, two GSH molecules are oxidized to produce GSSG, which can then be reduced back into two GSH molecules via GSH reductase to sustain redox homeostasis [35]. During oxidative stress, the oxidized form of GSH inside the cell becomes depleted. Indeed, a decrease in the cellular GSH concentration and an increase in GSSG are considered early stages of apoptotic events [36].
2.3. Glutathione S-Transferase (GST)
Multiple forms of GST (EC 2.5.1.18) appear to be an evolutionary response by cells to chemical toxicity and oxidative stress. GSTs are found in various cellular organelles such as the cytosol, mitochondria, endoplasmic reticulum, nucleus, and plasma membrane [37]. GSTS are associated with phase-II detoxification enzymes that protect cellular molecules from reactive electrophiles and facilitate the conjugation of glutathione to a wide range of endogenous and exogenous electrophilic compounds. GSTs are classified into membrane-bound microsomal and cytosolic family members [38]. They play a role in maintaining GSH levels in different cellular compartments and also detoxify endogenous toxic metabolites, superoxide radicals, and exogenous toxic chemicals [39].
2.4. Superoxide Dismutase (SOD)
SOD is a highly effective antioxidant that mainly contributes to cellular defense against oxidative stress. Its significant properties include a very high catalytic rate of reaction and high stability under physico-chemical stress [40]. SOD is classified into four different types based on its metal centers, including Cu, Zn-, Fe-, Mn-, and Ni [41]. Cu, Zn-SOD (SOD1) is primarily found in the cytosolic and lysosomal fractions, but it is also present in the mitochondrial intermembrane space. Mn-SOD (SOD2) is in the mitochondrial matrix. Both Cu, Zn-SOD and Mn-SOD are abundant in neural tissue [42].
2.5. Catalase (CAT)
Catalase plays an essential role in a versatile response to H2O2 and is strategically positioned as an auxiliary to GPx. It serves as a second-tier defense against ROS. Naturally, peroxisomes contain crystalline inclusions of catalases and exhibit prominent antioxidant functions [43]. Catalases are classified as monofunctional, bifunctional, and pseudo-catalase. Monofunctional catalases are mainly reported with similar molecular features across animals, plants, fungi, and bacteria. They exhibit minor peroxidase activity, targeting small organic substrates. Catalase’s function across a broad range of pH levels (5–10), are resistant to organic solvents since they are made up of glycoproteins, and their activity can be inhibited by 3-amino-1,2,4-triazole [44].
2.6. Neuroinflammation and Its Role in Neuronal Damage
Oxidative stress and inflammation are cognitive players of programmed cell death [45,46]. As shown in Figure 3, oxidative stress reduces the levels of enzymatic antioxidants and increases ROS, which ultimately triggers the expression of inflammatory genes, pro-inflammatory mediators and pro-apoptotic genes.
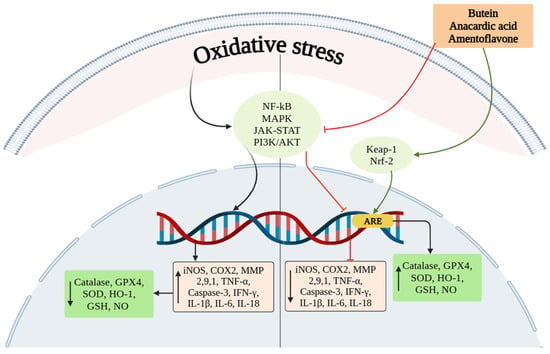
Figure 3.
Oxidative stress activates signaling molecules such as NF-ĸB, MAPK, JAK/STAT, and PI3K/AKT that ultimately upregulates the expression of inflammatory mediators and downregulates enzymatic antioxidants. Meanwhile, treatment with SAE and its phytocomponents induce the expression of Nrf2 and inhibits NF-ĸB expression. Nrf2 expression upregulates the level of enzymatic antioxidants and reduces inflammatory mediators.
Pathological states such as glutamate excitotoxicity, bacterial or viral infections, ischemic or haemorrhagic stroke, or oxidative stress to the CNS or brain cause initial injury and lead to cytokine-mediated inflammation and activation of inflammatory genes. These stimuli activate either canonical or non-canonical NF-κB pathways, which have been implicated in neuroinflammation-related pathogenesis [47]. Inducing the expression of these genes results in the activation of apoptotic proteins and ultimately causes cell death (Figure 4).
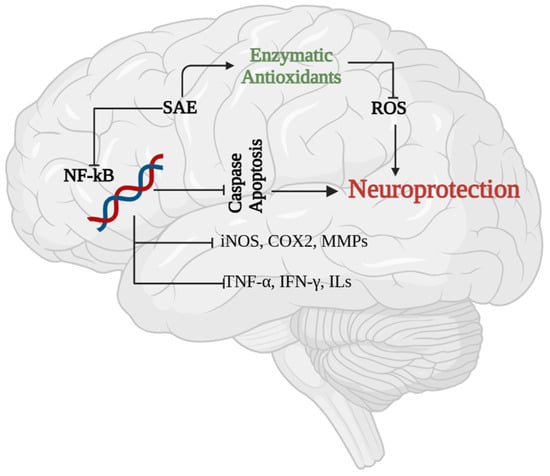
Figure 4.
Neuroprotective effect of SAE. The expression of NF-ĸB is higher in inflamed neuron cells, which regulates apoptosis through activation of caspases. Treatment with SAE and its phytocomponents were shown to inhibit NF-ĸB, caspase expression and pro-inflammatory mediators. Meanwhile SAE treatment was shown to enhance enzymatic antioxidants in neuron cells that reduces the level of ROS and protects neuron cells from apoptosis.
2.7. NF-κB and Neuronal Inflammation
NF-κB expression plays a crucial role in inflammation, immune responses [48], cell cycle, and cell survival [49]. NF-κB is a ‘master immune regulator’ [48] and a member of the Rel family of transcription factors. In mammals, p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2) are five distinct NF-κB family members that share similar amino acid sequences [50]. The term NF-κB refers to dimers composed of any combination of two transcription factors from this family. NF-κB is mostly present in the cytoplasm until it is activated by pro-inflammatory stimuli [48]. IκB degradation induces the release of NF-κB, which is ubiquitously expressed in neurons, glial cells, and cerebral blood vessels, and regulates the transcription of chemokines, cytokines, adhesion molecules, proinflammatory enzymes, and transcription factors within the neuronal environment [47].
3. Effect of S. anacardium L. Crude Extract on Neuronal Oxidative Stress and Damage
3.1. Role of SAE on Antioxidant Genes
The flavonoids purified from S. anacardium L. were shown to increase the expression of genes p38 and Nrf2, as well as enzymes like catalase and SOD, to counteract oxidative stress [51]. SAE-mediated inhibition of NF-ĸB and AP-1 suppressed the LPS-induced expression of pro-inflammatory cytokines (IL-1β, IL-12p40, and NO) [52]. LPS is a classical TLR4 ligand [53] that stimulates MAPK family members of the monocyte lineage, induces the expression of NF-ĸB, AP-1, Sp1, C/EBPβ, Spi-1, and interferon regulatory factors [52], and provokes the induction of inflammatory genes including MMPs [54], iNOS [55], and COX-2 [56].
LPS induced cognitive impairment and neuroinflammation in C57BL/6J mice, confirmed through immunofluorescence, ELISA, and Western blot. LPS increased the expressions of COX-2 and iNOS in brain homogenates [57]. Biflavonoids from S. anacardium L. have been reported to inhibit COX-1 and COX-2 in a dose-dependent manner in vitro [58]. SAE activates the Nrf2/Keap1 signaling axis, leading to transcriptional upregulation of antioxidant enzymes and restoration of cellular redox balance. Oxidative stress indicators, including LDH leakage, increased caspase-3 levels, mitochondrial dysfunction, and DNA damage, were reversed by SAE. The potential of SAE to increase GSH levels by affecting the redox state [59] was demonstrated in vivo using models of mammary carcinoma [27] and DL [60].
3.2. Recovery of Neuronal Function by SAE
SAE reversed ultrastructural changes in the hippocampal neuron cell bodies of rats subjected to long-term immobilization stress, affecting both pyramidal and granule cells. Treatment decreased the number of degenerating cell bodies; this process is linked to increased corticosteroid levels and free radical production. Hippocampal tissues are a target for glucocorticoids, which may lead to neurodegeneration. SAE significantly reduced glucocorticoid levels and was followed by stress treatment [61]. A similar effect was observed in Wistar albino rats with AlCl3-induced dementia [62]. β-amyloid plaques disrupt cell homeostasis and increase calcium ion levels in mitochondria and cytosol, which together lead to excess ROS levels [63]. SAE enhanced learning and reduced toxicity-related memory problems. Additionally, cholinesterase activity and amyloid plaque build-up in the brain decreased after treatment with SAE [62].
SAE has been reported to improve spatial memory by inhibiting acetylcholinesterase, reducing oxidative stress, and preventing glutamate-induced calcium influx [64]. The neuroprotective properties of SAE were demonstrated in NH4Cl-induced hyperammonemic rats [65] and in L-monosodium glutamate-treated in vitro and in vivo models. Glutamate is essential for rapid responses to stimuli and neurological functions such as cognition, memory, movement, and sensation [66]; however, excessive levels of glutamate can cause brain damage by hyperactivating ionotropic glutamate receptors through the excitotoxicity pathway [67].
Glutamate-induced oxidative stress occurs through various mechanisms, leading to glutathione depletion, increased Ca2+ levels, excessive ROS production, and inhibition of cystine uptake [68]. SAE has been shown to protect hippocampal neurons in albino rats [61] and male Wistar rats treated with high doses of L-monosodium glutamate (4 g/kg) [69]. The strong interactions and conformational stability of S. anacardium L. derivatives with AD targets such as AChE [70,71], NMDA, TTBK1, and BACE-1 have been reported in silico.
Experimental evidence strongly supports the role of SAE on activating antioxidant genes and recovering neural function. The various methods of extraction, key phytochemicals, and molecular targets are represented in Table 1.

Table 1.
Overview of Semecarpus anacardium L. extracts/compounds detailing origin, method of extraction, key phytochemicals, experimental concentrations and observed cellular and systemic effects.
Collectively, these findings indicate that SAE exerts neuroprotective effects through integrated modulation of oxidative stress, inflammatory signalling, and apoptosis.
4. The Role of Butein in Neuronal Damage
Butein is a naturally occurring plant-derived metabolite, first extracted from Toxicodendron vernicifluum, formerly known as Rhus verniciflua [72], and its presence has also been reported in Semecarpus anacardium, Dalbergia odorifera, and the flowers of Butea monosperma [73]. The molecular weight, toxicological and pharmacological properties of butein are illustrated in Table 2.
Table 2.
Toxicological and pharmacokinetic profile of butein.
4.1. Butein Activates Nrf2/ARE Pathway
Nrf2 expression is linked to neurological disorders because its deficiency causes mitochondrial failure, oxidative stress, and neuroinflammation [84]. Butein promotes the upregulation of the Nrf2/ARE signaling pathway, which depends on PI3K/AKT activation and increases HO-1 levels to exert anti-neuroinflammatory effects in HT22 and BV2 microglia cells [85]. Butein significantly reduces glutamate-induced cell death and ROS production in HT22 cells by increasing HO-1 levels, a key component of the antioxidant system. The translocation of Nrf2 regulates various antioxidant genes, leading to ARE-mediated induction of phase-II detoxifying enzymes, including HO-1, catalase, glutathione, glutathione-S transferase, glutathione reductase, and glutathione peroxidase [86]. The transcription factor Nrf2 activation counters oxidative stress through its endogenous inhibitor Keap1 and reduces cellular damage [87]. Additionally, inhibition of Nrf2 by siRNA or trigonelline results in increased neuroinflammation in BV2 microglia cells. Butein-induced expression of HO-1, which reduces oxidative stress via Nrf2, was shown in vitro [88,89] and in vivo [90]. Nrf2 binding to ARE can activate downstream antioxidant enzymes, including HO-1, GSH, NQO-1, and GST. These enzymes help lower ROS levels and protect cells and tissues.
Corticosterone is a glucocorticoid stress hormone linked to several neuronal disorders. Butein countered corticosterone-induced oxidative stress-related toxicity in mouse neuroblastoma Neuro2A (N2A) cells. Corticosterone caused apoptosis through mitochondrial dysfunction, caspase-3 activation, and ROS production. Butein significantly lowered ROS levels, LDH leakage, caspase-3 activity, mitochondrial potential loss, and DNA damage [91].
4.2. Butein Suppressess Transcription Factor NF-ĸB
NF-ĸB and the MAPK family (JNK, p38, and ERK) are widely distributed in cells and tissues, playing a crucial role in neuroinflammation, and their signaling is upregulated in SH-SY5Y cells. Phosphorylation of the tau protein promotes neuroinflammation through the ERK pathway, which leads to the activation and nuclear translocation of NF-ĸB [92].
The activity of NF-ĸB was inhibited by pretreatment with butein, which increased the viability of SH-SY5Y cells grown in conditioned medium from BsV2 microglial cells [93]. Activated microglia contain high levels of intracellular ROS that cause oxidative damage and inflammatory conditions [94], where NF-ĸB mediates the secretion of pro-inflammatory cytokines [94], leading to chronic inflammatory reactions [95] and resulting in neuronal damage or death [96].
Butein demonstrated its neuroprotective effects by inhibiting the NF-ĸB signaling pathway in microglial cells [85,93] and in a traumatic spinal cord injury model [18]. Cytotoxic factors released from BV2 microglial cells reduced SH-SY5Y neuronal viability before exposure to Butein. TLR4 mediated BV2 microglial activation and induced secretion of NO, PGE2, iNOS, COX-2, and pro-inflammatory cytokines [97]. This activation leads to neuronal synaptic dysfunction and neuronal death [98]. Studies on HT22 cells showed that butein reduces neuroinflammation by decreasing NO and PGE2 secretion and lowering inducible NOS and COX-2 expression via the NF-ĸB signaling pathway [85].
4.3. Effect of Butein on SCI
SCI is a serious spinal cord complication that causes significant dysfunction in the affected area. The inflammatory response is crucial in SCI development, involving key cell types such as macrophages, endothelial cells, microglia, and astrocytes [99]. The IKK/NF-κB signaling pathway plays a critical role in controlling inflammation and cell death, impacting SCI pathology. Targeting this pathway is a promising approach to improve locomotor recovery, decrease immune cell infiltration, and minimize apoptosis in rats following SCI. Butein reduces the IKK/NF-ĸB pathway, as shown in vivo. Traumatic SCI damages neural structures and leads to neurological deficits, with NF-ĸB signaling involved in secondary SCI damage [18]. Butein inhibited the IKK/NF-ĸB pathway and decreased apoptotic protein expression in spinal cord tissue. Apoptosis is a key factor in secondary SCI damage [100]. Neuronal and oligodendrocyte apoptosis triggers caspase-3 expression, causing axonal degeneration and loss of neural function [101,102]. Butein mitigated the activation of the IKK/NF-ĸB pathway and reduced inflammatory cell infiltration and caspase-3 activation in the spinal cords of Sprague-Dawley rats with SCI within 24 h [18]. Inflammation significantly contributes to SCI, with NF-ĸB being the main transcriptional regulator of inflammatory genes [103]. The IKK/NF-ĸB cascade plays a central role in regulating inflammation and apoptosis [18].
4.4. Role of Butein on MMP Expression
Butein reduced expression of genes including Bcl-2 [104], c-Myc [105], and COX-2 [106]. Bcl-2 enhances the production of inflammatory mediators, making control of Bcl-2 expression a key strategy for managing inflammatory and allergic reactions. Bcl-2 inhibitors have been reported to prevent airway inflammation [107], human tubulointerstitial inflammation [108], and experimental allergic rhinitis [109].
Butein significantly reduced IL-1β-induced inflammatory reactions in human and mouse osteoarthritis models. It suppressed the expression of COX-2, iNOS, TNF-α, IL-6, MMP-1, MMP-3, and MMP-13 [110]. In the CNS, MMPs are shown to degrade basal laminae components, leading to BBB disruption and contributing to neuroinflammation. In response to cellular stress, brain cells produce both constitutive and inducible MMPs [111]. Transcriptome analyses [112] and patient data [113] demonstrate the link between MMP-9 expression and inflammatory diseases. Therefore, natural and synthetic MMP inhibitors are emerging as treatments for inflammatory conditions [114]. NF-ĸB-mediated suppression of MMP-9 was shown in vitro with Butein [105]. SAE exemplified the normalization of MMP-1, MMP-2, MMP-3, TIMP-1, and TIMP-2 levels in a mammary carcinoma model [115]. The presence of flavonoids in the extract may enhance membrane stability and prevent lysosomal hydrolase secretion [116,117]. Lysosomal activity plays a critical role in MMP-9 secretion and the inflammatory response [118].
The dual role of butein in activating antioxidative enzymes and reducing inflammation is supported by various experimental studies. In summary, Figure 5 illustrates the butein mediated upregulation of Nrf2, which enhances the expression of antioxidative enzymes. Meanwhile, butein downregulates NF-ĸB expression, leading to a reduction in pro-inflammatory mediators. Thus, butein exerts dual regulatory control over oxidative stress and neuroinflammation through coordinated modulation of antioxidant–inflammatory signaling interplay. Table 3 summarizes the molecular targets and mechanism of action of butein.
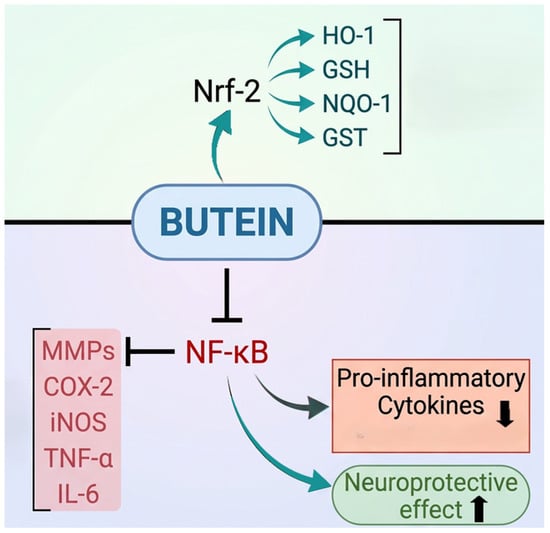
Figure 5.
Butein increases Nrf2 expression, which boosts antioxidant enzyme levels that counteract oxidative stress. At the same time, butein decreases NF-κB expression, leading to lower levels of pro-inflammatory cytokines and related factors.
Table 3.
Molecular targets and mechanisms of butein in neuronal inflammation.
5. Anacardic Acid and Neuroinflammation
The presence of phenolic, carboxylic, and a 15-carbon alkyl side chain functional group makes it desirable for biological applications. Anacardic acid serves as a potential starting material for synthesizing various biologically active compounds [119]. The toxicological and pharmacological properties of anacardic acid are illustrated in Table 4.
Table 4.
Illustrates the therapeutic outcomes, toxicological, and pharmacokinetic profile of phytocompound anacardic acid.
5.1. Role of Anacardic Acid on Enzymatic Antioxidants
Anacardic acid modulates redox homeostasis by enhancing antioxidant enzyme expression while concurrently suppressing oxidative enzyme activity [126]. Anacardic acid was also found to elevate GSH levels in the prefrontal cortex and hippocampus of mice following acute intraperitoneal doses of 25 and 50 mg/Kg. This indicates that the effect was consistent across different brain regions, doses, and methods of administration [127]. Anacardic acid increased overall SOD gene expression in both mitochondria and the cell cytoplasm. Treatment with anacardic acid increased the expression of GPx4 [128] that regulates lipid peroxidation and inflammatory cytokines. The GPx4−/−AT mouse model demonstrated the protective role of GPx4 against systemic low-grade inflammation [129].
Moreover, GPx4 plays a crucial role in iron-dependent cell death [116,130]. TBI-mediated inhibition of GPx4 expression results in ROS accumulation, redox imbalance, and ferroptosis induction [131,132] TBI is a severe cause of neuronal disability that leads to iron-dependent lipid peroxidation and oxidative cell death [133]. It is characterized by the pathophysiological processes of edema, inflammation, ferroptosis, and programmed cell death. Anacardic acid mitigates ferroptosis by upregulating GPx4 expression, reducing lipid peroxidation, and limiting iron-dependent oxidative damage. It lessens ferroptosis severity by reducing inflammation and oxidative stress and by modifying the expression of key ferroptosis proteins. Furthermore, anacardic acid decreases iron deposition in tissues as part of its anti-ferroptosis effects and enhances BBB permeability [128].
5.2. Protective Effect of Anacardic Acid on PD
Anacardic acid exerts a protective effect against PD [127,134]. Oral administration of anacardic acid demonstrates preventive antioxidant activity in the rat nigrostriatal system and cerebral cortex in a pesticide rotenone-induced experimental model of PD. Anacardic acid completely blunted rotenone-provoked lipoperoxidation [127,134] by stimulating t-SOD [134]. Additionally, it increased NO levels and reduced the redox balance of GSH/GSSG in the substantia nigra and striatum [127]. There was a significant increase in the gene expression of both SOD-1 and SOD-2 in the striatum, with 2490- and 190-fold increases, respectively [134]. SOD is predominantly expressed in glial cells and throughout the CNS. Clinical and genetic evidence shows a direct correlation between SOD gene mutations and neurodegenerative diseases [135].
Furthermore, the protective effect of anacardic acid was demonstrated in a pesticide rotenone-induced experimental model of PD in Wistar rats. Sufficient amounts of anacardic acid crossed the BBB, reached the CNS, and exerted beneficial effects. An increase in SOD gene expression and t-SOD activity confirmed the neuroprotective effect of anacardic acid [134]. The results support the findings of Augusto et al. on PD induced in Swiss mice [127].
Neuro histological and neuroimaging studies have shown ongoing and end-stage neuroinflammatory processes in PD. Samples of peripheral blood and cerebrospinal fluid from PD patients reveal variations in inflammation markers and immune cell populations that can worsen neuroinflammation and contribute to neurodegeneration [136]. Oxidative stress causes cellular damage, triggers NF-ĸB expression, and activates inflammatory processes that lead to cytokine secretion in neurodegenerative diseases such as PD [137,138].
5.3. Regulation of Anacardic Acid on Inflammatory Mediators
Anacardic acid demonstrates anti-inflammatory effects by inhibiting anti-oxidative enzymes, pro-inflammatory mediators, and NF-ĸB. It notably lowers TNF-α- induced mRNA levels of NF-κB, a nuclear transcription factor associated with inflammation. Suppressing NF-κB diminishes free oxygen radicals and reduces oxidative stress [139]. In a carrageenan-induced peritonitis model, anacardic acid lowered total leukocyte and neutrophil migration and reduced MPO activity. In neutrophils, MPO is crucial for activating p38 MAPK and NF-κB pathways. MPO also stimulates the production of cytokines such as IL-6 and IL-8, as well as ROS [140]. Anacardic acid has been shown to reduce pro-IL-1β levels in the striatum [127] and inhibit the gene expression and activity of MMP-2 and MMP-9 [141]. MMP-9 supports glial activation and neurodegeneration in both monkey and mouse models of PD induced by MPTP or rotenone [142,143]. Anacardic acids were also shown to decrease MMP-9 and increase TIMP-1 protein levels in vivo [127] Phenolic compounds were shown to downregulate inflammatory markers in various brain regions [141].
5.4. Anacardic Acid vs. Apoptotic Molecules
The role of apoptotic molecules in inflammation depends on their substrates. proIL-1β and proIL-18 are key in inflammation and serve as substrates for caspase-1 [144]. The participation of caspases in apoptosis, pyroptosis, and necroptosis is a shared characteristic. Caspase-driven proteolysis causes either gain-of-function or loss-of-function effects on their substrates, leading to inflammation [145]. Anacardic acid was shown to reduce caspase-3 levels while increasing the expression of Bcl-2 [146]. Caspases, activated by TNF-α, initiated programmed cell death by destroying critical components of the cellular infrastructure that mediate cell damage [147]. Bcl-2 family members can be either pro-apoptotic or anti-apoptotic, and their balance depends on the release of cytochrome C [148]. However, caspases can also influence the homeostasis of pro-apoptotic or anti-apoptotic signals from the Bcl-2 family [149].
5.5. Anacardic Acid vs. Voltage-Gated Ion Channels
Intracellular Ca2+ homeostasis regulates neurodegenerative processes [150], where SAE treatment normalized intracellular Ca2+ levels in PC-12 cells [69]. Reduced levels of ROS, calcium influx, toxicity, and increased cell viability were observed in PC-12 cells treated with bhilawanol and anacardic acid. Calcium entry across the cell membrane is ROS-dependent [151], and higher calcium entry was significantly decreased by the treatment. Bhilawanol and anacardic acid are lipid-soluble compounds found in S. anacardium L. which potentially inhibit AChE in a dose- and time-dependent manner. In addition, a glutamate receptor antagonist and AChE inhibitors are FDA-approved drugs for AD [146].
Furthermore, the neuroprotective activity of anacardic acid has been demonstrated in an experimental epilepsy model. An imbalance in the function of oxygen- or nitrogen-derived reactive species affects brain activity, leading to mitochondrial dysfunction, DNA damage, changes in neural signalling, and hindrance of neurogenesis [152] Oxidative stress-mediated disruption of calcium signalling, mitochondrial impairment, and cellular damage result in neuronal cell death, seizures, and systemic toxicity [153]. Anacardic acid reduced epileptic seizures in a dose-dependent manner, potentially through inhibition of Na+ voltage-dependent channels [125] The mechanism of action of anacardic acid and its molecular targets are listed in Table 5.
Table 5.
Represents the therapeutic targets, molecular mechanism, and the concentrations reported for anacardic acid (↓ decrease; ↑ increase).
This coordinated regulation of ferroptosis, oxidative stress, and inflammation highlights anacardic acid as a multifunctional neuroprotective agent.
6. Amentoflavone
Amentoflavone belongs to the class of biflavonoids and polyflavonoids known as 3′, 8″-biapigenin [154]. The toxicological and pharmacological properties of amentoflavone illustrated in Table 6.
Table 6.
Illustrates the therapeutic outcomes, toxicological, and pharmacokinetic profile of phytocompound amentoflavone.
6.1. Amentoflavone vs. Antioxidants
Amentoflavone reduces oxidative stress by enhancing Nrf2-mediated antioxidant responses while simultaneously suppressing TLR4/NF-κB-driven inflammation. It alleviates oxidative stress and neuroinflammation-induced cerebral ischemia/reperfusion in rats via the TLR4/NF-κB signaling pathway. Pro-inflammatory cytokines are involved in the body’s inflammatory response to traumatic brain injury. Treatment with amentoflavone significantly decreases serum levels of TNF-α, IL-1β, and IL-6 compared to the control group [157]. Low levels of catalase, SOD, and GPx limit the brain’s ability to eliminate superoxide anion and hydrogen peroxide, making brain tissue more vulnerable to ROS and oxidative stress [167]. Amentoflavone significantly raised brain antioxidant markers GSH and CAT and lowered MDA levels [157].
Amentoflavone activates AMPK-dependent Nrf2 signaling, promoting transcription of antioxidant enzymes and protecting against Aβ-induced neurotoxicity. In transgenic AD mice, the Nrf2 pathway was compromised, alongside increased brain Aβ levels. Treatment with amentoflavone significantly boosted Nrf2 expression and its translocation, which in turn increased HO-1 and NQO-1 levels, thereby reducing oxidative stress in the brain. Suppressing Nrf2 expression in neuronal cells weakened amentoflavone’s protective effects, indicating that Nrf2 plays a critical role in its neuroprotective activity [168].
Its neuroprotective role was demonstrated in transgenic ADAT/Nrf2-KO mice, where accumulation of Aβ and p-tauS404 was observed in its absence [169]. Additionally, amentoflavone inhibited ferroptosis-mediated inflammation via the SLC7A11/GPx4 axis in homocysteine-induced neuronal dysfunction. Homocysteine-exposed HT22 cells showed increased mRNA levels of ferroptosis-related genes and iron accumulation [170].
6.2. Amentoflavone Influences the Expression of NF-ĸB
The role of amentoflavone in fighting stroke was shown with cerebral ischemia/reperfusion (IR) injury in unilateral common carotid artery occlusion (CCAO). Amentoflavone increased GSH and CAT activities and reduced neuroinflammation by negatively regulating the TLR4/NF-ĸB signaling pathway, providing neuroprotective benefits [157]. Proinflammatory cytokines are known mediators of brain damage after HI injury. Yang et al., noted that elevated systemic and cerebral IL-6 levels are part of traumatic brain injury and inflammation [171].
In addition to inhibiting the TLR4/MyD88/NF-ĸB cascade, amentoflavone activated the Nrf2/HO-1 pathway in LPS-induced BV2 microglia cells and reversed neuroinflammation [172]. TLR4-mediated inflammation is a key factor that contributes to neurodegeneration in PD [173]. Prior research supports the role of amentoflavone in blocking the TLR4/MyD88/NF-κB signaling pathway [174].
Administering amentoflavone to rats with ischemic injury reduced serum cytokines (TNF-α, IL-1β, and IL-6) compared to the control. A lower level of the TBK1 pathway is linked to neuroinflammation, which plays a key role in selective autophagy and inflammatory IFN signalling. Similarly, NF-ĸB is important in the secretion of iNOS synthetase and inflammation—brain cells such as astrocytes, microglia, and oligodendrocytes exhibit activated NF-ĸB complexes. Amentoflavone treatment significantly decreased NF-ĸB expression and improved TBK1 and IFN-β function [157].
The strong neuroprotective effect of amentoflavone is shown by improving neurological function, increasing motor coordination, and boosting locomotor activity. Amentoflavone reduced ischemia/reperfusion-induced brain injury through the HMGB1-mediated TLR4/NF-ĸB signalling pathway in a unilateral common carotid artery occlusion (CCAO) rat model. IR control rats showed hemorrhage with pyknotic nuclei in neurons of the cerebrum. In contrast, brain sections from treated animals displayed mild congestion of cerebral blood vessels, along with some vacuolar degeneration in neurons and perivascular edema [157].
Amentoflavone inhibited the activation and nuclear translocation of the NF-ĸB subunit p65. As a result, amentoflavone suppressed inflammation, apoptosis, and prevented the excessive discharge of hippocampal neurons, thereby avoiding pilocarpine-induced epilepsy [175]. Flavonoids have been reported to interact with MAPK signalling pathways that regulate various cellular functions [176]. Amentoflavone increased the expression of PI3K and Akt, as well as the Bcl-2/Bax ratio, and protected against the progressive degeneration of dopaminergic neurons [177] in vitro, amentoflavone treatment reduced nuclear condensation and cell viability loss induced by 1-methyl-4-phenylpyridinium.
6.3. Amentoflavone Controls HI Injury
Similar research supports the role of amentoflavone in protecting against hypoxic-ischemic damage in rat brains. Administering amentoflavone systemically 3.5 h after HI injury offers strong neuroprotection in neonatal cases. Its dual function of reducing cytotoxic damage and providing anti-inflammatory effects enhances outcomes in HI injury [166]. Amentoflavone reduces microglial inflammation after HI injury. It blocks the upstream cell death cascades that cause both necrotic and apoptotic neuronal death following HI injury. The presence of the active form (p18) of caspase 3 indicates that amentoflavone inhibits caspase 3 activation [166]. Brain proinflammatory cytokines can mediate brain damage after HI injury. For example, microglia, astrocytes, and neurons secrete early response cytokines such as IL-1β and TNF-α [178].
Amentoflavone inhibits the induction of proinflammatory mediators, including iNOS, COX-2, IL-1β, and TNF-α, in microglial BV-2 cells. Amentoflavone protected hippocampal neurons in epilepsy mice by reducing inflammation and preventing cell death. It also decreased the gene expression of inflammatory mediators like IL-1β and iNOS in the substantia nigra pars compacta of MPTP-induced mice. Additionally, amentoflavone significantly reduced OX-42 immunoreactivity, a biomarker for activated microglia and LPS-induced worsening of HI brain damage, supporting the neuroprotective effects of amentoflavone in neonatal HI injury [166]. Hampered caspase-3 and p21 function and increased Bcl-2/Bax expression were observed with amentoflavone treatment in SY5Y cells [177]. The role of amentoflavone on various neurological conditions, its molecular target, and reported concentrations were listed in Table 7.
Table 7.
Represents the disease, molecular targets, and concentrations of amentoflavone (↑ increase; ↓ decrease).
Overall, amentoflavone exerts neuroprotective effects through coordinated modulation of oxidative stress, inflammatory signaling, and ferroptosis pathways.
7. Mechanism of Action of S. anacardium L. and Its Phytocompounds
Studies have demonstrated that SAE activates the Nrf2/Keap-1 signaling pathway, thereby enhancing the expression of endogenous antioxidant enzymes under conditions of oxidative stress and inflammation (Figure 6). In addition, S. anacardium extract (SAE) and its bioactive compounds have been reported to suppress NF-κB signaling, resulting in the downregulation of key inflammatory mediators, including MMPs, iNOS, and COX-2. Furthermore, several intracellular signaling cascades such as AMPK, PI3K, AKT, mTOR, MAPK, PPAR-γ, ERK1/2, and JAK/STAT are modulated in response to NF-κB activation status and play critical roles in the progression of neuroinflammation. However, direct evidence elucidating the interaction between S. anacardium extracts or their phytoconstituents and these molecular pathways remains scarce and warrants further investigation. Phytochemical classes including alkaloids, terpenoids, flavonoids, saponins, glycosides, and steroids substantially contribute to the therapeutic potential of medicinal plants. Notably, the seeds of S. anacardium extracts are rich in phenolic compounds and flavonoids, which are primarily responsible for their strong antioxidant capacity [16]. Polyphenols have been widely reported to attenuate oxidative stress through activation of Nrf2 signaling [179,180], enhancement of antioxidant defense systems [181], and inhibition of NF-κB–mediated inflammatory pathways [182]; Comparable mechanistic effects have also been observed for specific compounds such as butein, anacardic acid, and amentoflavone. As a crude extract, SAE is likely to exert pleiotropic effects by targeting multiple signaling pathways simultaneously [183]; however, such multi-target actions have not yet been conclusively validated through experimental studies. In addition, growing evidence suggests that synergistic interactions among dietary phytochemicals may further enhance their efficacy in mitigating oxidative stress and promoting neuroprotection [184]. Variability in extraction methods and solvent systems significantly influences the phytochemical composition of SAE, thereby altering biological activity. Standardization of key bioactive compounds such as butein, anacardic acid, and amentoflavone remains essential for reproducibility and clinical translation.
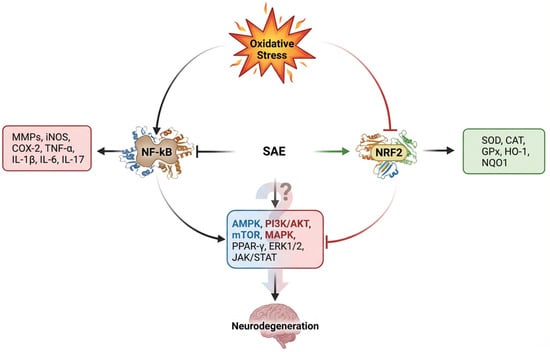
Figure 6.
Mechanistic overview of SAE-mediated modulation of oxidative stress and inflammatory pathways in neurons. Extensive evidence supports the oxidative stress mediated upregulation of NF-ĸB and inflammation where the levels of Nrf2 reduced with oxidative stress. SAE and its phytocomponents were shown to inhibit NF-ĸB expression and activation of Nrf2. Higher expression of Nrf2 elevates the level of enzymatic antioxidants which protect the cells from neurodegeneration. Consequently, reduced level of pro-inflammatory mediators was observed with NF-ĸB inhibition. However, the role of SAE and its phytocomponents on the regulation of transcription factors, such as AMPK, PI3K/AKT, mTOR, MAPK, PPAR-γ, ERK1/2, and JAK/STAT needs to be explored.
8. Future Perspective
This review highlights the neuroprotective potential of SAE and its key phytocomponents-butein, anacardic acid, and amentoflavone-in modulating oxidative stress and neuroinflammation, which are central drivers of neurodegenerative diseases. Collectively, these compounds exert dual regulatory effects by activating Nrf2-mediated antioxidant pathways while concurrently suppressing NF-κB-driven inflammatory signaling. Despite robust preclinical evidence, several limitations hinder translational advancement. Variability in extract composition, lack of standardized formulations, insufficient pharmacokinetic and ADMET profiling, and limited in vivo studies remain critical challenges. Furthermore, most studies rely on acute or simplified disease models that do not fully capture the complexity of human neurodegenerative disorders. Future research should focus on standardized extract characterization, mechanistic validation using multi-omics approaches, and evaluation in clinically relevant models. Integration of advanced delivery systems to improve bioavailability, along with well-designed clinical studies, will be essential to translate these promising phytocompounds into therapeutic interventions. Collectively, these findings position SAE and its phytocompounds as a promising multi-target therapeutic candidate, bridging traditional medicine and modern neuropharmacology.
Author Contributions
Conceptualization and original draft, S.R.K.P.; Co-writing, figure, and tables, S.H. and R.S.; Review, T.W.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful to Sankarganesh Devaraj, Associate, Department of Biotechnology, School of Bio Sciences and Technology, Vellore Institute of Technology (VIT) Vellore–632014 Tamil Nadu, India, for critically evaluating the manuscript and providing suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AChE | Acetylcholinesterase Enzyme |
| AD | Alzheimer’s Disease |
| AKT | Protein Kinase B |
| ALS | Amyotrophic Lateral Sclerosis |
| AMPK | AMP-Activated Protein Kinase |
| AP-1 | Activator Protein-1 |
| ARE | Antioxidant Responsive Element |
| BACE-1 | β-Secretase 1 |
| Bax | Bcl-2-Associated Protein X |
| BBB | Blood Brain Barrier |
| BChE | Butyrylcholinesterase |
| BCL | B-Cell Lymphoma |
| BV2 | Murine Microglial Cell Line |
| C/EBPβ | Ccaat Enhancer Binding Protein Beta |
| CAT | Catalase |
| cMyc | Cellular Myc Oncogene |
| CNS | Central Nervous System |
| CO | Carbon Monoxide |
| COX | Cyclooxygenase |
| COX-2 | Cyclooxygenase-2 |
| DL | Dalton’s Lymphoma |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ERK | Extracellular Signal-Regulated Kinase |
| FTH1 | Ferritin Heavy Chain 1 |
| GPX | Glutathione Peroxidase |
| GPX4 | Glutathione Peroxidase 4 |
| GSH | Glutathione |
| GSSG | Oxidized Gluthatione |
| GST | Glutathione S-Transferase |
| GSK-3β | Glycogen synthase kinase-3 beta |
| HD | Huntington’s Disease |
| HI | Hypoxic-Ischemic Brain Injury |
| HMGB-1 | High Mobility Group Box 1 |
| HO-1 | Heme Oxygenase 1 |
| IFN | Interferon |
| IKK | Inhibitor of Nuclear Factor-Κb (Iκb) Kinase |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| JNK | Jun N-Terminal Kinase |
| Keap1 | Kelch-Like ECH-Associated Protein 1 |
| LDH | Lactate Dehydrogenase |
| LPO | Lipoperoxidation |
| MAPK | Mitogen-Activated Protein Kinase |
| MDA | Malondialdehyde |
| MMPs | Matrix Metalloproteinases |
| MND | Motor Neuron Diseases |
| MyD88 | Myeloid Differentiation Primary Response Protein 88 |
| NF-ĸB | Nuclear Factor-Kappa B |
| NMDA | N-Methyl D-Aspartate |
| NO | Nitric Oxide |
| NQO1 | Quinone Oxidoreductase 1 |
| Nrf2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| PD | Parkinson’s Disease |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphatidylinositol 3-Kinase |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 |
| PUFA | Polyunsaturated Fatty Acids |
| ROS | Reactive Oxygen Species |
| RSV | Respiratory Syncytial Virus |
| SAE | Semecarpus anacardium L., Extract |
| SCA | Spinocerebellar Ataxia |
| SCI | Spinal Cord Injury |
| SH-SY5Y | SK-N-SH Neuroblastoma Cell Line |
| SLC7A11 | Solute Carrier Family 7-Member 11 |
| SMA | Spinal Muscular Atrophy |
| SOD | Superoxide Dismutase |
| SP-1 | Specificity Protein |
| SLC7A11 | Solute Carrier Family 7 Member 11 |
| TBI | Traumatic Brain Injury |
| TBK1 | Tank-Binding-Kinase1 |
| TFR1 | Transferrin Receptor 1 |
| TH | tyrosine hydroxylase |
| TIMP | Tissue Inhibitor of Metalloproteinase |
| TLR | Toll-Like Receptor |
| TNF | Tumor Necrosis Factor |
| t-SOD | Total Superoxide Dismutase |
| TTBK1 | Tau-Tubulin Kinase 1 |
References
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Mukherjee, A.; Becerra Calixto, A.D.; Chavez, M.; Delgado, J.P.; Soto, C. Mitochondrial transplant to replenish damaged mitochondria: A novel therapeutic strategy for neurodegenerative diseases? Prog. Mol. Biol. Transl. Sci. 2021, 177, 49–63. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.E.S.; de Vasconcelos, A.S.; da Costa Vilhena, T.; da Silva, T.L.; da Silva Barbosa, A.; Gomes, A.R.Q.; Dolabela, M.F.; Percário, S. Oxidative Stress in Alzheimer’s Disease: Should We Keep Trying Antioxidant Therapies? Cell. Mol. Neurobiol. 2015, 35, 595–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 1224. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuča, K.; Musílek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Das, K.; Yendigeri, S.; Patil, B.; Bagoji, I.; Reddy, R.; Bagali, S.; Biradar, M.; Saha, S. Subchronic hypoxia pretreatment on brain pathophysiology in unilateral common carotid artery occluded albino rats. Indian J. Pharmacol. 2018, 50, 185–191. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Tornatore, L.; Thotakura, A.K.; Bennett, J.; Moretti, M.; Franzoso, G. The nuclear factor kappa B signaling pathway: Integrating metabolism with inflammation. Trends Cell Biol. 2012, 22, 557–566. [Google Scholar] [CrossRef]
- Kumar, A.; Negi, G.; Sharma, S.S. Suppression of NF-κB and NF-κB regulated oxidative stress and neuroinflammation by BAY 11-7082 (IκB phosphorylation inhibitor) in experimental diabetic neuropathy. Biochimie 2012, 94, 1158–1165. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Park. Relat. Disord. 2012, 18, S210–S212. [Google Scholar] [CrossRef]
- Varin, M.; Bentea, E.; Michotte, Y.; Sarre, S. Oxidative stress in genetic mouse models of Parkinsons disease. Oxidative Med. Cell. Longev. 2012, 2012, 624925. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.; Viswan Lilly, R.; Velmurugan, A.; Sundhara Moorthy, K.R.; Sudarsanam, S.R.; Parameswaran, S.; Kadarkarai, K. Quantification of anacardic acid, the toxic component in raw and purified samples of Semecarpus anacardium L. by Siddha purification processes. J. Complement. Integr. Med. 2022, 19, 947–953. [Google Scholar] [CrossRef]
- Srinivasan, A.; Suresh, B.D.; Senthilkumar, N.; Murugesan, S. Physicochemical properties and phytochemical constituents of Semecarpus anacardium L. seed oil. Adv. Appl. Sci. Res. 2016, 7, 151–154. [Google Scholar]
- Lu, M.; Wang, S.; Han, X.; Lv, D. Butein inhibits NF-κB activation and reduces infiltration of inflammatory cells and apoptosis after spinal cord injury in rats. Neurosci. Lett. 2013, 542, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Sandur, S.K.; Sung, B.; Sethi, G.; Kunnumakkara, A.B.; Aggarwal, B.B. Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-κB and NF-κB-regulated gene expression through direct inhibition of IκBα kinase β on cysteine 179 residue. J. Biol. Chem. 2007, 282, 17340–17350. [Google Scholar] [CrossRef]
- Gil, R. Anacardoside from the seeds of Semecarpus anacardium. Phytochemistry 1995, 39, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Pal, D. Antioxidant Potentials and Pharmacological Activities of Marking Nut (Semecarpus anacardium L.f.). In Nuts and Seeds in Health and Disease Prevention; Academic Press: Cambridge, MA, USA, 2011. [Google Scholar] [CrossRef]
- Mishra, A.K.; S.L, N.; Jain, A.; Jagtap, C.Y.; Dane, G.; Paroha, S.; Sahoo, P.K. Effectiveness of Semecarpus anacardium Linn. fruits in cancer and inflammatory diseases: A mini review. Fitoterapia 2024, 175, 105978. [Google Scholar] [CrossRef]
- Margis, R.; Dunand, C.; Teixeria, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Pickard, K.; Nicol, F.; Beckett, G.J.; Duthie, G.G.; Arthur, J.R. Effects of organic and inorganic selenium supplementation on selenoenzyme activity in blood lymphoctyes, granulocytes, platelets and erythrocytes. Clin. Sci. 2000, 98, 593–599. [Google Scholar] [CrossRef]
- Herbette, S.; Roeckel-Drevet, P.; Drevet, J.R. Seleno-independent glutathione peroxidases: More than simple antioxidant scavengers. FEBS J. 2007, 274, 2163–2180. [Google Scholar] [CrossRef]
- de Carvalho e Martins, C.M.; da Silva Santos Oliveira, A.S.; da Silva, L.A.A.; Primo, M.G.S.; de Carvalho Lira, V.B. Biological Indicators of Oxidative Stress [Malondialdehyde, Catalase, Glutathione Peroxidase, and Superoxide Dismutase] and Their Application in Nutrition. In Biomarkers in Nutrition; Springer: Cham, Switzerland, 2022. [Google Scholar] [CrossRef]
- Mathivadhani, P.; Shanthi, P.; Sachdanandam, P. Effect of Semecarpus anacardium Linn. nut milk extract on glutathione and its associated enzymes in experimentally induced mammary carcinoma. J. Med. Food 2006, 9, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Dar, N.J.; John, U.; Bano, N.; Khan, S.; Bhat, S.A. Oxytosis/Ferroptosis in Neurodegeneration: The Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4). Mol. Neurobiol. 2024, 61, 1507–1526. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Casu, M.; Butler, N.; Breda, C.; Campesan, S.; Clapp, J.; Green, E.W.; Dhulkhed, D.; Kyriacou, C.P.; Giorgini, F. Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat. Genet. 2013, 45, 1249–1254. [Google Scholar] [CrossRef]
- Hopkins, F.G. On glutathione: A reinvestigation. J. Biol. Chem. 1929, 84, 269–320. [Google Scholar] [CrossRef]
- Block, G.S.; Coates, R.J.; Eley, J.W.; Greenberg, R.S. Dietary glutathione intake in humans and the relationship between intake and plasma total glutathione level. Nutr. Cancer 1994, 21, 33–46. [Google Scholar] [CrossRef]
- Jefferies, H.; Coster, J.; Khalil, A.; Bot, J.; McCauley, R.D.; Hall, J.C. Glutathione. ANZ J. Surg. 2003, 73, 517–522. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. The suffer containing amino acids: An overview. J. Nutr. 2006, 136, 1636S–1640S. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995, 251, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef]
- Raza, H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: Implications in oxidative stress, toxicity and disease. FEBS J. 2011, 278, 4243–4259. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anticancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Allocati, N.; Federici, L.; Masulli, M.; Di Ilio, C. Glutathione transferases in bacteria. FEBS J. 2009, 276, 58–75. [Google Scholar] [CrossRef]
- Bafana, A.; Dutt, S.; Kumar, A.; Kumar, S.; Ahuja, P.S. The basic and applied aspects of superoxide dismutase. J. Mol. Catal. B Enzym. 2011, 68, 129–138. [Google Scholar] [CrossRef]
- Smith, M.W.; Doolittle, R.F. A comparison of evolutionary rates of the two major kinds of superoxide dismutase. J. Mol. Evol. 1992, 34, 175–184. [Google Scholar] [CrossRef]
- Lalkovičová, M.; Danielisová, V. Neuroprotection and antioxidants. Neural Regen. Res. 2016, 11, 865–874. [Google Scholar] [CrossRef]
- Sepasi Tehrani, H.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018, 140, 5–12. [Google Scholar] [CrossRef]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Protein Cell 2010, 1, 888–897. [Google Scholar] [CrossRef]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Rev. Neurosci. 2004, 5, S18–S25. [Google Scholar] [CrossRef]
- Hald, A.; Lotharius, J. Oxidative stress and inflammation in Parkinson’s disease: Is there a causal link? Exp. Neurol. 2005, 193, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-κB signaling pathways in neurological inflammation: A mini review. Front. Mol. Neurosci. 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.E.; Walker, A.K.; Weickert, C.S. Neuroinflammation in schizophrenia: The role of nuclear factor kappa B. Transl. Psychiatry 2021, 11, 528. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Meffert, M.K. Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006, 13, 852–860. [Google Scholar] [CrossRef]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Kumar, A.D.N.; Bevara, G.B.; Kaja, L.K.; Badana, A.K.; Malla, R.R. Protective effect of 3-O-methyl quercetin and kaempferol from Semecarpus anacardium against H2O2 induced cytotoxicity in lung and liver cells. BMC Complement. Altern. Med. 2016, 16, 376. [Google Scholar] [CrossRef]
- Singh, D.; Aggarwal, A.; Mathias, A.; Naik, S. Immunomodulatory activity of Semecarpus anacardium extract in mononuclear cells of normal individuals and rheumatoid arthritis patients. J. Ethnopharmacol. 2006, 108, 398–406. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Yang, C.C.; Hsiao, L.D.; Yang, C.M. Galangin inhibits LPS-induced MMP-9 expression via suppressing protein kinase-dependent AP-1 and FoxO1 activation in rat brain astrocytes. J. Inflamm. Res. 2020, 13, 945–960. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, Q.; Fan, H.; Zhao, H.; Yang, Y. Myocardin-Related Transcription Factor A Mediates LPS-Induced iNOS Transactivation. Inflammation 2020, 43, 1351–1361. [Google Scholar] [CrossRef]
- Tsukayama, I.; Mega, T.; Hojo, N.; Toda, K.; Kawakami, Y.; Takahashi, Y.; Suzuki-Yamamoto, T. Diosgenin suppresses COX-2 and mPGES-1 via GR and improves LPS-induced liver injury in mouse. Prostaglandins Other Lipid Mediat. 2021, 156, 106580. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef]
- Selvam, C.; Jachak, S.M. A cyclooxygenase (COX) inhibitory biflavonoid from the seeds of Semecarpus anacardium. J. Ethnopharmacol. 2004, 95, 209–212. [Google Scholar] [CrossRef]
- Sanz, M.J.; Ferrandiz, M.L.; Cejudo, M.; Terencio, M.C.; Gil, B.; Bustos, G.; Ubeda, A.; Gunasegaran, R.; Alcaraz, M.J. Influence of a series of natural flavonoids on free radical generating systems and oxidative stress. Xenobiotica 1994, 24, 689–699. [Google Scholar] [CrossRef]
- Verma, N.; Vinayak, M. Semecarpus anacardium nut extract promotes the antioxidant defence system and inhibits anaerobic metabolism during development of lymphoma. Biosci. Rep. 2009, 29, 151–164. [Google Scholar] [CrossRef]
- Shukla, S.D.; Jain, S.; Sharma, K.; Bhatnagar, M. Stress induced neuron degeneration and protective effects of Semecarpus anacardium Linn. and Withania somnifera Dunn. in hippocampus of albino rats: An ultrastructural study. Indian J. Exp. Biol. 2000, 38, 1007–1013. [Google Scholar]
- Gaurav, S.; Nitin, K.; Hansraj, S.; Mamta, S.; Nutan, K. In-vivo shielding effects of Semecarpus anacardium extract in presenile dementia. Int. J. Pharm. Res. 2020, 12, 862–868. [Google Scholar] [CrossRef]
- Shigematsu, K.; McGeer, P.L. Accumulation of amyloid precursor protein in damaged neuronal processes and microglia following intracerebral administration of aluminum salts. Brain Res. 1992, 593, 117–123. [Google Scholar] [CrossRef]
- Mughairbi FAl Khan, F.; Ahmed, S.; Nawaz, R. Semecarpus anacardium decreases oxidative stress & improves memory through inhibiting acetylcholinesterase and reducing glutamate induced calcium influx. IBRO Neurosci. Rep. 2023, 15, S353. [Google Scholar] [CrossRef]
- Vijayakumar, N.; Subramanian, P. Neuroprotective effect of Semecarpus anacardium against hyperammonemia in rats. J. Pharm. Res. 2010, 3, 1564–1568. [Google Scholar]
- Steckler, T.; Sahgal, A.; Aggleton, J.P.; Drinkenburg, W.H.I.M. Recognition memory in rats—III. Neurochemical substrates. Prog. Neurobiol. 1998, 54, 333–348. [Google Scholar] [CrossRef]
- Chen, Z.; Ljunggren, H.G.; Bogdanovic, N.; Nennesmo, I.; Winblad, B.; Zhu, J. Excitotoxic neurodegeneration induced by intranasal administration of kainic acid in C57BL/6 mice. Brain Res. 2002, 931, 135–145. [Google Scholar] [CrossRef]
- Rössler, O.G.; Bauer, I.; Chung, H.Y.; Thiel, G. Glutamate-induced cell death of immortalized murine hippocampal neurons: Neuroprotective activity of heme oxygenase-1, heat shock protein 70, and sodium selenite. Neurosci. Lett. 2004, 362, 253–257. [Google Scholar] [CrossRef]
- Al Mughairbi, F.; Khan, F.; Ilyas, S.; Shad, Y.; Choudhary, M.I. From old remedy to modern therapy: Neuroprotective effects of Semecarpus anacardium on the l-Monosodium Glutamate treated rats and neuronal cells. In Proceedings of the 4th International Conference on Educational Neuroscience, Abu Dhabi, United Arab Emirates, 10–11 March 2019; Volume 34. [Google Scholar] [CrossRef]
- Adhami, H.R.; Linder, T.; Kaehlig, H.; Schuster, D.; Zehl, M.; Krenn, L. Catechol alkenyls from Semecarpus anacardium: Acetylcholinesterase inhibition and binding mode predictions. J. Ethnopharmacol. 2012, 139, 142–148. [Google Scholar] [CrossRef]
- Vijayalakshmi, M.; Sundarapandian, V.; Siva Bharathi, V.; Pavadai, P.; Sundar, K.; Kunjiappan, S.; Pandian, S.R.K. Graph Theoretical Network Analysis and Pharmacoinformatics-Based Investigation of Bioactive Compounds from Semecarpus anacardium Linn. for Alzheimer’s Disease. J. Comput. Biophys. Chem. 2025, 24, 287–305. [Google Scholar] [CrossRef]
- Kim, J.H.; Jung, C.H.; Jang, B.H.; Go, H.Y.; Park, J.H.; Choi, Y.K.; Hong SIl Shin, Y.C.; Ko, S.G. Selective cytotoxic effects on human cancer cell lines of phenolic-rich ethyl-acetate fraction from Rhus verniciflua stokes. Am. J. Chin. Med. 2009, 37, 609–620. [Google Scholar] [CrossRef]
- Padmavathi, G.; Rathnakaram, S.R.; Monisha, J.; Bordoloi, D.; Roy, N.K.; Kunnumakkara, A.B. Potential of butein, a tetrahydroxychalcone to obliterate cancer. Phytomedicine 2015, 22, 1163–1171. [Google Scholar] [CrossRef]
- Padmavathi, G.; Roy, N.K.; Bordoloi, D.; Arfuso, F.; Mishra, S.; Sethi, G.; Bishayee, A.; Kunnumakkara, A.B. Butein in health and disease: A comprehensive review. Phytomedicine 2017, 25, 118–127. [Google Scholar] [CrossRef]
- Chen, Y.-N.; Huang, T.-F.; Chang, C.-H.; Hsu, C.-C.; Lin, K.-T.; Wang, S.-W.; Peng, H.-C.; Chung, C.-H. Antirestenosis Effect of Butein in the Neointima Formation Progression. J. Agric. Food Chem. 2012, 60, 6832–6838. [Google Scholar] [CrossRef]
- Lim, S.S.; Jung, S.H.; Ji, J.; Shin, K.H.; Keum, S.R. Synthesis of flavonoids and their effects on aldose reductase and sorbitol accumulation in streptozotocin-induced diabetic rat tissues. J. Pharm. Pharmacol. 2001, 53, 653–668. [Google Scholar] [CrossRef]
- Kushwaha, V.; Singh, K.; Mishra, M.K. Investigation of the neuroprotective effect of Butein against scopolamine-induced Alzheimer’s like symptoms in rats. J. Pharm. Negat. Results 2022, 13, 408–416. [Google Scholar] [CrossRef]
- Mohammed, R.A.; Al-Shawi, N.N. Butein mitigates 5-FU-triggered hepatotoxicity via antioxidant, anti-inflammatory, and anti-apoptotic pathways. Toxicol. Rep. 2025, 15, 102120. [Google Scholar] [CrossRef]
- Ugan, R.A.; Un, H. The Protective Roles of Butein on Indomethacin Induced Gastric Ulcer in Mice. Eurasian J. Med. 2020, 52, 265–270. [Google Scholar] [CrossRef]
- Gao, L.; Cui, S.; Huang, Z.; Cui, H.; Awad Alahmadi, T.; Manikandan, V. Antinociceptive and anti-inflammatory activities of butein in different nociceptive and inflammatory mice models. Saudi J. Biol. Sci. 2021, 28, 7090–7097. [Google Scholar] [CrossRef]
- Golmei, P.; Kasna, S.; Kumar, S. Unveiling the therapeutic potential of butein: A comprehensive review. Health Sci. Rev. 2024, 13, 100197. [Google Scholar] [CrossRef]
- Kim, N.A.; Oh, H.K.; Lee, J.C.; Choi, Y.H.; Jeong, S.H. Comparison of solubility enhancement by solid dispersion and micronized butein and its correlation with in vivo study. J. Pharm. Investig. 2021, 51, 53–60. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A.Y. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9443165 (accessed on 26 December 2025). [CrossRef]
- Maldonado, P.P.; Guevara, C.; Olesen, M.A.; Orellana, J.A.; Quintanilla, R.A.; Ortiz, F.C. Neurodegeneration in Multiple Sclerosis: The Role of Nrf2-Dependent Pathways. Antioxidants 2022, 11, 1146. [Google Scholar] [CrossRef]
- Lee, D.S.; Jeong, G.S. Butein provides neuroprotective and anti-neuroinflammatory effects through Nrf2/ARE-dependent haem oxygenase 1 expression by activating the PI3K/Akt pathway. Br. J. Pharmacol. 2016, 173, 2894–2909. [Google Scholar] [CrossRef]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta-Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Tungalag, T.; Park, K.W.; Yang, D.K. Butein Ameliorates Oxidative Stress in H9c2 Cardiomyoblasts through Activation of the NRF2 Signaling Pathway. Antioxidants 2022, 11, 1430. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sung, J.; Kim, Y.; Jeong, H.S.; Lee, J. Inhibitory Effects of Butein on Adipogenesis through Upregulation of the Nrf2/HO-1 Pathway in 3T3-L1 Adipocytes. Prev. Nutr. Food Sci. 2017, 22, 306. [Google Scholar] [CrossRef]
- Wang, Z.; Ka, S.O.; Lee, Y.; Park, B.H.; Bae, E.J. Butein induction of HO-1 by p38 MAPK/Nrf2 pathway in adipocytes attenuates high-fat diet induced adipose hypertrophy in mice. Eur. J. Pharmacol. 2017, 799, 201–210. [Google Scholar] [CrossRef]
- Ohmoto, M.; Shibuya, Y.; Taniguchi, S.; Nakade, T.; Nomura, M.; Ikeda-Matsuo, Y.; Daikoku, T. Protective effects of butein on corticosterone-induced cytotoxicity in Neuro2A cells. IBRO Rep. 2020, 8, 82–90. [Google Scholar] [CrossRef]
- Khan, M.I.; Khan, M.Z.; Shin, J.H.; Shin, T.S.; Lee, Y.B.; Kim, M.Y.; Kim, J.D. Neuroprotective effects of green tea seed isolated saponin due to the amelioration of tauopathy and alleviation of neuroinflammation: A therapeutic approach to Alzheimer’s disease. Molecules 2022, 27, 2079. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, Y.; Zhang, Y.; Liu, F.; Rose, G.M.; He, X.; Yi, X.; Ren, R.; Li, Y.; Zhang, Y.; et al. Butein attenuates the cytotoxic effects of LPS-stimulated microglia on the SH-SY5Y neuronal cell line. Eur. J. Pharmacol. 2020, 868, 172858. [Google Scholar] [CrossRef]
- Cai, B.; Seong, K.J.; Bae, S.W.; Chun, C.; Kim, W.J.; Jung, J.Y. A synthetic diosgenin primary amine derivative attenuates LPS-stimulated inflammation via inhibition of NF-κB and JNK MAPK signaling in microglial BV2 cells. Int. Immunopharmacol. 2018, 61, 204–214. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Schlichter, L.C. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 2008, 28, 2221–2230. [Google Scholar] [CrossRef]
- Dai, X.J.; Li, N.; Yu, L.; Chen, Z.Y.; Hua, R.; Qin, X.; Zhang, Y.M. Activation of BV2 microglia by lipopolysaccharide triggers an inflammatory reaction in PC12 cell apoptosis through a toll-like receptor 4-dependent pathway. Cell Stress Chaperones 2015, 20, 321–331. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Veerhuis, R.; Rozemuller, J.M.; Eikelenboom, P. Soothing the Inflamed Brain: Effect of Non-Steroidal Anti-Inflammatory Drugs on Alzheimers Disease Pathology. CNS Neurol. Disord.-Drug Targets 2011, 10, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Botchway, B.O.; Zhang, S.; Zhou, J.; Liu, X. Inhibition of NF-κB signaling pathway by resveratrol improves spinal cord injury. Front. Neurosci. 2018, 12, 690. [Google Scholar] [CrossRef]
- Keane, R.W.; Kraydieh, S.; Lotocki, G.; Bethea, J.R.; Krajewski, S.; Reed, J.C.; Dietrich, W.D. Apoptotic and anti-apoptotic mechanisms following spinal cord injury. J. Neuropathol. Exp. Neurol. 2001, 60, 422–429. [Google Scholar] [CrossRef]
- Hagg, T.; Oudega, M. Degenerative and spontaneous regenerative processes after spinal cord injury. J. Neurotrauma 2006, 23, 263–280. [Google Scholar] [CrossRef]
- Jiang, S.; Bendjelloul, F.; Ballerini, P.; D’Alimonte, I.; Nargi, E.; Jiang, C.; Huang, X.; Rathbone, M.P. Guanosine reduces apoptosis and inflammation associated with restoration of function in rats with acute spinal cord injury. Purinergic Signal. 2007, 3, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Niederberger, E.; Geisslinger, G. The IKK-NF-κB pathway: A source for novel molecular drug targets in pain therapy? FASEB J. 2008, 22, 3432–3442. [Google Scholar] [CrossRef]
- Kim, N.Y.; Pae, H.O.; Oh, G.S.; Kang, T.H.; Kim, Y.C.; Rhew, H.Y.; Chung, H.T. Butein, a plant polyphenol, induces apoptosis concomitant with increased caspase-3 activity, decreased Bcl-2 expression and increased Bax expression in HL-60 cells. Pharmacol. Toxicol. 2001, 88, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Choi, Y.H.; Moon, S.K.; Kim, W.J.; Kim, G.Y. Butein suppresses the expression of nuclear factor-kappa B-mediated matrix metalloproteinase-9 and vascular endothelial growth factor in prostate cancer cells. Toxicol. Vitr. 2010, 24, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Lau, G.T.Y.; Huang, H.; Lin, S.M.; Leung, L.K. Butein downregulates phorbol 12-myristate 13-acetate-induced COX-2 transcriptional activity in cancerous and non-cancerous breast cells. Eur. J. Pharmacol. 2010, 648, 24–30. [Google Scholar] [CrossRef]
- Tian, B.P.; Xia, L.X.; Bao, Z.Q.; Zhang, H.; Xu, Z.W.; Mao, Y.Y.; Cao, C.; Che, L.Q.; Liu, J.K.; Li, W.; et al. Bcl-2 inhibitors reduce steroid-insensitive airway inflammation. J. Allergy Clin. Immunol. 2017, 140, 418–430. [Google Scholar] [CrossRef]
- Ko, K.; Wang, J.; Perper, S.; Jiang, Y.; Yanez, D.; Kaverina, N.; Ai, J.; Liarski, V.M.; Chang, A.; Peng, Y.; et al. Bcl-2 as a Therapeutic Target in Human Tubulointerstitial Inflammation. Arthritis Rheumatol. 2016, 68, 2740–2751. [Google Scholar] [CrossRef]
- Kim, H.Y.; Jeong, H.J.; Kim, H.M. Anti-allergic and anti-inflammatory effects of the Bcl-2 inhibitor ABT-737 on experimental allergic rhinitis models. Eur. J. Pharmacol. 2018, 833, 34–43. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, H.; Jin, Y.; Wang, Q.; Chen, L.; Feng, Z.; Chen, H.; Wu, Y. Butein inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes and slows the progression of osteoarthritis in mice. Int. Immunopharmacol. 2017, 42, 115–123. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix metalloproteinases in neuroinflammation. GLIA 2002, 39, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.; Hanash, S. Expression of matrix metalloproteinase 9 (MMP-9/gelatinase B) in adenocarcinomas strongly correlated with expression of immune response genes. Silico Biol. 2003, 3, 301–311. [Google Scholar]
- Naik, S.P.; Mahesh, P.A.; Jayaraj, B.S.; Madhunapantula, S.R.V.; Jahromi, S.R.; Yadav, M.K. Evaluation of inflammatory markers interleukin-6 (IL-6) and matrix metalloproteinase-9 (MMP-9) in asthma. J. Asthma 2017, 54, 584–593. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Sang, Q.X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef]
- Mathivadhani, P.; Shanthi, P.; Sachdanandam, P. Effect of Semecarpus anacardium nut extract on ECM and proteases in mammary carcinoma rats. Vasc. Pharmacol. 2007, 46, 419–426. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wei, D.G.; Li, R.S. Capsaicin induces ferroptosis of NSCLC by regulating SLC7A11/GPX4 signaling in vitro. Sci. Rep. 2022, 12, 11996. [Google Scholar] [CrossRef]
- Verstraeten, S.V.; Keen, C.L.; Schmitz, H.H.; Fraga, C.G.; Oteiza, P.I. Flavan-3-ols and procyanidins protect liposomes against lipid oxidation and disruption of the bilayer structure. Free. Radic. Biol. Med. 2003, 34, 84–92. [Google Scholar] [CrossRef]
- Bramwell, K.K.C.; Mock, K.; Ma, Y.; Weis, J.H.; Teuscher, C.; Weis, J.J. β-Glucuronidase, a Regulator of Lyme Arthritis Severity, Modulates Lysosomal Trafficking and MMP-9 Secretion in Response to Inflammatory Stimuli. J. Immunol. 2015, 195, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Hamad, F.B.; Mubofu, E.B. Potential biological applications of bio-based anacardic acids and their derivatives. Int. J. Mol. Sci. 2015, 16, 8569. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.L.N.; Annoni, R.; Silva, P.R.P.; Borelli, P.; Fock, R.A.; Trevisan, M.T.S.; Mauad, T. Acute, subacute toxicity and mutagenic effects of anacardic acids from cashew (Anacardium occidentale Linn.) in mice. J. Ethnopharmacol. 2011, 135, 730–736. [Google Scholar] [CrossRef]
- Sriram, S.; Kumar, M.S.; Shourie, G.K.; Palukurthi, A.; Kadam, S.; Srikanth, T.M. Ninety-day toxicity and genotoxic effects of synthetically derived fully saturated forms of anacardic acid in mice. Regul. Toxicol. Pharmacol. 2024, 147, 105538. [Google Scholar] [CrossRef]
- Chung, S.; Shin, E.J.; Choi, H.; Park, J.H.; Hwang, J. Anacardic acid mitigates liver fat accumulation and impaired glucose tolerance in mice fed a high-fat and high-sucrose diet. Food Sci. Nutr. 2020, 8, 796–804. [Google Scholar] [CrossRef]
- Gnanaprakasam, J.N.R.; López-Bañuelos, L.; Vega, L. Anacardic 6-pentadecyl salicylic acid induces apoptosis in breast cancer tumor cells, immunostimulation in the host and decreases blood toxic effects of taxol in an animal model. Toxicol. Appl. Pharmacol. 2021, 410, 115359. [Google Scholar] [CrossRef] [PubMed]
- Júnior, A.L.G.; Tchekalarova, J.D.; da Conceição Machado, K.; Silva, S.W.C.; Paz, M.F.C.J.; Nogueira, T.R.; de Matos Monteiro Lira, B.S.; Zihad, S.M.N.K.; Islam, M.T.; Ali, E.S.; et al. Antidepressant-like effect of anacardic acid in mice via the L-arginine–nitric oxide–serotonergic system. Phytother. Res. 2019, 33, 2126–2138. [Google Scholar] [CrossRef]
- Luiz Gomes, A.; Dimitrova Tchekalarova, J.; Atanasova, M.; da Conceição Machado, K.; de Sousa Rios, M.A.; Paz, M.F.C.J.; Găman, M.-A.; Găman, A.M.; Yele, S.; Shill, M.C.; et al. Anticonvulsant effect of anacardic acid in murine models: Putative role of GABAergic and antioxidant mechanisms. Biomed. Pharmacother. 2018, 106, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, P.; Sethy, K. Antioxidant profile of anacardic acid derivatives and their effects on oxidative stress-induced diseases: An update. Russ. J. Bioorg. Chem. 2025, 51, 2247–2260. [Google Scholar] [CrossRef]
- Augusto, R.L.; Mendonça, I.P.; de Albuquerque Rego, G.N.; Pereira, D.D.; da Penha Gonçalves, L.V.; dos Santos, M.L.; de Souza, R.F.; Moreno, G.M.M.; Cardoso, P.R.G.; de Souza Andrade, D.; et al. Purified anacardic acids exert multiple neuroprotective effects in pesticide model of Parkinson’s disease: In vivo and in silico analysis. IUBMB Life 2020, 72, 1765–1779. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Z.; Guo, J.; Ma, Y.; Li, J.; Ji, H.; Chen, Z.; Zheng, J. Anacardic acid improves neurological deficits in traumatic brain injury by anti-ferroptosis and anti-inflammation. Exp. Neurol. 2023, 370, 114568. [Google Scholar] [CrossRef] [PubMed]
- Schwärzler, J.; Mayr, L.; Radlinger, B.; Grabherr, F.; Philipp, M.; Texler, B.; Grander, C.; Ritsch, A.; Hunjadi, M.; Enrich, B.; et al. Adipocyte GPX4 protects against inflammation, hepatic insulin resistance and metabolic dysregulation. Int. J. Obes. 2022, 46, 951–959. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Fang, J.; Yuan, Q.; Du, Z.; Zhang, Q.; Yang, L.; Wang, M.; Yang, W.; Yuan, C.; Yu, J.; Wu, G.; et al. Overexpression of GPX4 attenuates cognitive dysfunction through inhibiting hippocampus ferroptosis and neuroinflammation after traumatic brain injury. Free. Radic. Biol. Med. 2023, 204, 68–81. [Google Scholar] [CrossRef]
- Gao, J.; Li, Y.; Song, R. SIRT2 inhibition exacerbates p53-mediated ferroptosis in mice following experimental traumatic brain injury. NeuroReport 2021, 32, 1001–1008. [Google Scholar] [CrossRef]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr. Neuropharmacol. 2017, 16, 1224–1238. [Google Scholar] [CrossRef]
- Medeiros-Linard, C.F.B.; Andrade-da-Costa BLda, S.; Augusto, R.L.; Sereniki, A.; Trevisan, M.T.S.; Perreira Rde, C.R.; de Souza, F.T.C.; Braz, G.R.F.; Lagranha, C.J.; de Souza, I.A.; et al. Anacardic Acids from Cashew Nuts Prevent Behavioral Changes and Oxidative Stress Induced by Rotenone in a Rat Model of Parkinson’s Disease. Neurotox. Res. 2018, 34, 250–262. [Google Scholar] [CrossRef]
- Chidambaram, S.B.; Anand, N.; Varma, S.R.; Ramamurthy, S.; Vichitra, C.; Sharma, A.; Mahalakshmi, A.M.; Essa, M.M. Superoxide dismutase and neurological disorders. IBRO Neurosci. Rep. 2024, 16, 373–394. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-κB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.M. ROS and brain diseases: The good, the bad, and the ugly. Oxid. Med. Cell. Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef]
- Önal, B.; Özen, D.; Demir, B.; Gezen Ak, D.; Dursun, E.; Demir, C.; Akkan, A.G.; Özyazgan, S. The anti-inflammatory effects of anacardic acid on a TNF-α-induced human saphenous vein endothelial cell culture model. Curr. Pharm. Biotechnol. 2020, 21, 710–719. [Google Scholar] [CrossRef]
- Gomes Junior, A.L.; Islam, M.T.; Nicolau, L.A.D.; de Souza, L.K.M.; Araújo, T.D.S.L.; Lopes de Oliveira, G.A.; de Melo Nogueira, K.; da Silva Lopes, L.; Medeiros, J.V.R.; Mubarak, M.S.; et al. Anti-inflammatory, antinociceptive, and antioxidant properties of ana-cardic acid in experimental models. ACS Omega 2020, 5, 19506–19515. [Google Scholar] [CrossRef]
- Nambiar, J.; Bose, C.; Venugopal, M.; Banerji, A.; Patel, T.B.; Kumar, G.B.; Nair, B.G. Anacardic acid inhibits gelatinases through the regulation of Spry2, MMP-14, EMMPRIN and RECK. Exp. Cell Res. 2016, 349, 139–151. [Google Scholar] [CrossRef]
- Annese, V.; Herrero, M.T.; Di Pentima, M.; Gomez, A.; Lombardi, L.; Ros, C.M.; De Pablos, V.; Fernandez-Villalba, E.; De Stefano, M.E. Metalloproteinase-9 contributes to inflammatory glia activation and nigro-striatal pathway degeneration in both mouse and monkey models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Brain Struct. Funct. 2015, 220, 703–727. [Google Scholar] [CrossRef]
- Singh, D.; Srivastava, S.K.; Chaudhuri, T.K.; Upadhyay, G. Multifaceted role of matrix metalloproteinases (MMPs). Front. Mol. Biosci. 2015, 2, 19. [Google Scholar] [CrossRef]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef]
- Bibo-Verdugo, B.; Salvesen, G.S. Caspase mechanisms in the regulation of inflammation. Mol. Asp. Med. 2022, 88, 101085. [Google Scholar] [CrossRef]
- Al Mughairbi, F.; Nawaz, R.; Khan, F.; Hassan, A.; Mahmood, N.; Ahmed, H.T.; Alshamali, A.; Ahmed, S.; Bashir, A. Neuroprotective effects of Bhilawanol and Anacardic acid during glutamate-induced neurotoxicity. Saudi Pharm. J. 2021, 29, 1043–1049. [Google Scholar] [CrossRef]
- Friedlander, R.M. Apoptosis and Caspases in Neurodegenerative Diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Ijomone, O.M.; Aluko, O.M.; Okoh, C.O.A.; Martins, A.C.; Aschner, M. Role for calcium signaling in manganese neurotoxicity. J. Trace Elem. Med. Biol. 2019, 56, 146–155. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Rosa, J.M.; Dafre, A.L.; Rodrigues, A.L.S. Antidepressant-like responses in the forced swimming test elicited by glutathione and redox modulation. Behav. Brain Res. 2013, 253, 165–172. [Google Scholar] [CrossRef]
- Martinc, B.; Grabnar, I.; Vovk, T. The Role of Reactive Species in Epileptogenesis and Influence of Antiepileptic Drug Therapy on Oxidative Stress. Curr. Neuropharmacol. 2012, 10, 328–343. [Google Scholar] [CrossRef]
- Xiong, X.; Tang, N.; Lai, X.; Zhang, J.; Wen, W.; Li, X.; Li, A.; Wu, Y.; Liu, Z. Insights into Amentoflavone: A Natural Multifunctional Biflavonoid. Front. Pharmacol. 2021, 12, 768708. [Google Scholar] [CrossRef]
- Hou, T.; Yang, M.; Yan, K.; Fan, X.; Ci, X.; Peng, L. Amentoflavone Ameliorates Carrageenan-Induced Pleurisy and Lung Injury by Inhibiting the NF-κB/STAT3 Pathways via Nrf2 Activation. Front. Pharmacol. 2022, 13, 763608. [Google Scholar] [CrossRef]
- Ijaz, M.U.; Ghafoor, N.; Hayat, M.F.; Almutairi, B.O.; Atique, U. Amentoflavone mediated hepatoprotection to counteract paraquat instigated hepatotoxicity via modulating Nrf2/keap1 pathway: A biochemical, inflammatory, apoptotic and histopathological study. Pestic. Biochem. Physiol. 2024, 198, 105715. [Google Scholar] [CrossRef]
- Saeedan, A.S.; Abdel-Rahman, R.F.; Soliman, G.A.; Ogaly, H.A.; Abdel-Kader, M.S. Amentoflavone attenuates oxidative stress and neuroinflammation induced by cerebral ischemia/reperfusion in rats by targeting HMGB1-mediated TLR4/NF-κB signaling pathway. Saudi Pharm. J. 2023, 31, 101798. [Google Scholar] [CrossRef]
- Su, C.; Yang, C.; Gong, M.; Ke, Y.; Yuan, P.; Wang, X.; Li, M.; Zheng, X.; Feng, W. Antidiabetic Activity and Potential Mechanism of Amentoflavone in Diabetic Mice. Molecules 2019, 24, 2184. [Google Scholar] [CrossRef]
- Ishola, I.O.; Chatterjee, M.; Tota, S.; Tadigopulla, N.; Adeyemi, O.O.; Palit, G.; Shukla, R. Antidepressant and anxiolytic effects of amentoflavone isolated from Cnestis ferruginea in mice. Pharmacol. Biochem. Behav. 2012, 103, 322–331. [Google Scholar] [CrossRef]
- Kim, G.L.; Jang, E.H.; Lee, D.-E.; Bang, C.; Kang, H.; Kim, S.; Yoon, S.Y.; Lee, D.H.; Na, J.H.; Lee, S.; et al. Amentoflavone, active compound of Selaginella tamariscina, inhibits in vitro and in vivo TGF-β-induced metastasis of human cancer cells. Arch. Biochem. Biophys. 2020, 687, 108384. [Google Scholar] [CrossRef]
- Lee, K.-C.; Chen, W.-T.; Liu, Y.-C.; Lin, S.-S.; Hsu, F.-T. Amentoflavone Inhibits Hepatocellular Carcinoma Progression Through Blockage of ERK/NF-ĸB Activation. In Vivo 2018, 32, 1097–1103. [Google Scholar] [CrossRef]
- Alherz, F.A.; El-Masry, T.A.; Negm, W.A.; El-Kadem, A.H. Potential cardioprotective effects of Amentoflavone in doxorubicin-induced cardiotoxicity in mice. Biomed. Pharmacother. 2022, 154, 113643. [Google Scholar] [CrossRef]
- Fang, G.; Li, X.; Yang, F.; Huang, T.; Qiu, C.; Peng, K.; Wang, Z.; Yang, Y.; Lan, C. Amentoflavone mitigates doxorubicin-induced cardiotoxicity by suppressing cardiomyocyte pyroptosis and inflammation through inhibition of the STING/NLRP3 signalling pathway. Phytomedicine 2023, 117, 154922. [Google Scholar] [CrossRef]
- Han, B.H.; Cofell, B.; Everhart, E.; Humpal, C.; Kang, S.-S.; Lee, S.K.; Kim-Han, J.S. Amentoflavone Promotes Cellular Uptake and Degradation of Amyloid-Beta in Neuronal Cells. Int. J. Mol. Sci. 2022, 23, 5885. [Google Scholar] [CrossRef]
- Rong, S.; Wan, D.; Fan, Y.; Liu, S.; Sun, K.; Huo, J.; Zhang, P.; Li, X.; Xie, X.; Wang, F.; et al. Amentoflavone Affects Epileptogenesis and Exerts Neuroprotective Effects by Inhibiting NLRP3 Inflammasome. Front. Pharmacol. 2019, 10, 856. [Google Scholar] [CrossRef]
- Shin, D.H.; Bae, Y.C.; Kim-Han, J.S.; Lee, J.H.; Choi, I.Y.; Son, K.H.; Kang, S.S.; Kim, W.K.; Han, B.H. Polyphenol amentoflavone affords neuroprotection against neonatal hypoxic-ischemic brain damage via multiple mechanisms. J. Neurochem. 2006, 96, 561–572. [Google Scholar] [CrossRef]
- Althurwi, H.N.; Abdel-Rahman, R.F.; Soliman, G.A.; Ogaly, H.A.; Alkholifi, F.K.; Abd-Elsalam, R.M.; Alqasoumi, S.I.; Abdel-Kader, M.S. Protective Effect of Beta-Carotene against Myeloperoxidase- Mediated Oxidative Stress and Inflammation in Rat Ischemic Brain Injury. Antioxidants 2022, 11, 2344. [Google Scholar] [CrossRef]
- Chen, C.; Li, B.; Cheng, G.; Yang, X.; Zhao, N.; Shi, R. Amentoflavone Ameliorates Aβ1–42-Induced Memory Deficits and Oxidative Stress in Cellular and Rat Model. Neurochem. Res. 2018, 43, 857–868. [Google Scholar] [CrossRef]
- Ren, P.; Chen, J.; Li, B.; Zhang, M.; Yang, B.; Guo, X.; Chen, Z.; Cheng, H.; Wang, P.; Wang, S.; et al. Nrf2 Ablation Promotes Alzheimer’s Disease-Like Pathology in APP/PS1 Transgenic Mice: The Role of Neuroinflammation and Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 3050971. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, B.; Jin, X. Amentoflavone attenuates homocysteine-induced neuronal ferroptosis-mediated inflammatory response: Involvement of the SLC7A11/GPX4 axis activation. Brain Res. Bull. 2024, 215, 111005. [Google Scholar] [CrossRef]
- Yang, S.H.; Gangidine, M.; Pritts, T.A.; Goodman, M.D.; Lentsch, A.B. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock 2013, 40, 471–475. [Google Scholar] [CrossRef]
- Rong, S.; Yang, C.; Wang, F.; Wu, Y.; Sun, K.; Sun, T.; Wu, Z. Amentoflavone Exerts Anti-Neuroinflammatory Effects by Inhibiting TLR4/MyD88/NF- κ B and Activating Nrf2/HO-1 Pathway in Lipopolysaccharide-Induced BV2 Microglia. Mediat. Inflamm. 2022, 2022, 5184721. [Google Scholar] [CrossRef]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2019, 68, 316844. [Google Scholar] [CrossRef]
- Sakthivel, K.M.; Guruvayoorappan, C. Amentoflavone inhibits iNOS, COX-2 expression and modulates cytokine profile, NF-κB signal transduction pathways in rats with ulcerative colitis. Int. Immunopharmacol. 2013, 17, 907–916. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, T.; Niu, J.G.; He, Z.Q.; Liu, Y.; Wang, F. Amentoflavone protects hippocampal neurons: Anti-inflammatory, antioxidative, and antiapoptotic effects. Neural Regen. Res. 2015, 10, 1125–1133. [Google Scholar] [CrossRef]
- Lee, C.W.; Na, Y.; Park, N.H.; Kim, H.S.; Ahn, S.M.; Kim, J.W.; Kim, H.K.; Jang, Y.P. Amentoflavone inhibits UVB-induced matrix metalloproteinase-1 expression through the modulation of AP-1 components in normal human fibroblasts. Appl. Biochem. Biotechnol. 2012, 166, 1137–1147. [Google Scholar] [CrossRef]
- Cao, Q.; Qin, L.; Huang, F.; Wang, X.; Yang, L.; Shi, H.; Wu, H.; Zhang, B.; Chen, Z.; Wu, X. Amentoflavone protects dopaminergic neurons in MPTP-induced Parkinson’s disease model mice through PI3K/Akt and ERK signaling pathways. Toxicol. Appl. Pharmacol. 2017, 319, 80–90. [Google Scholar] [CrossRef]
- Calvert, J.W.; Zhang, J.H. Pathophysiology of an hypoxic-ischemic insult during the perinatal period. Neurol. Res. 2005, 27, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Stranieri, C.; Guzzo, F.; Gambini, S.; Cominacini, L.; Fratta Pasini, A.M. Intracellular polyphenol wine metabolites oppose oxidative stress and upregulate Nrf2/ARE pathway. Antioxidants 2022, 11, 2055. [Google Scholar] [CrossRef] [PubMed]
- Lonati, E.; Carrozzini, T.; Bruni, I.; Mena, P.; Botto, L.; Cazzaniga, E.; Del Rio, D.; Labra, M.; Palestini, P.; Bulbarelli, A. Coffee-derived phenolic compounds activate Nrf2 antioxidant pathway in an in vitro I/R injury model: A nutritional approach preventing age-related damages. Molecules 2022, 27, 1049. [Google Scholar] [CrossRef] [PubMed]
- Eghbaliferiz, S.; Iranshahi, M. Prooxidant activity of polyphenols, flavonoids, anthocyanins and carotenoids: Updated review of mechanisms and catalyzing metals. Phytother. Res. 2016, 30, 1379–1391. [Google Scholar] [CrossRef]
- Karunaweera, N.; Raju, R.; Gyengesi, E.; Münch, G. Plant polyphenols as inhibitors of NF-κB-induced cytokine production—A potential anti-inflammatory treatment for Alzheimer’s disease? Front. Mol. Neurosci. 2015, 8, 24. [Google Scholar] [CrossRef]
- Villegas-Aguilar, M.D.C.; Fernández-Ochoa, Á.; Cádiz-Gurrea, M.D.L.L.; Pimentel-Moral, S.; Lozano-Sánchez, J.; Arráez-Román, D.; Segura-Carretero, A. Pleiotropic biological effects of dietary phenolic compounds and their metabolites on energy metabolism, inflammation and aging. Molecules 2020, 25, 596. [Google Scholar] [CrossRef]
- Yu, J.; Xiao, X.; Chen, B.; Deng, Z.; Chen, X.; Fan, Y.; Li, H. Synergistic and antagonistic activity of selected dietary phytochemicals against oxidative stress-induced injury in cardiac H9c2 cells via the Nrf2 signaling pathway. Foods 2024, 13, 2440. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.