Abstract
Airborne particulate matter with a diameter of <10 μm (PM10) can damage the corneal epithelium by inducing oxidative stress, disrupting the NRF2 antioxidant pathway, and triggering epithelial barrier dysfunction and inflammation. However, the role of mitochondria in mediating PM10-induced damage remains unexplored. This study investigated the impact of PM10 on mitochondrial homeostasis in both immortalized human corneal epithelial cells (HCE-2) and the mouse corneal epithelium, as well as the protective effects of SKQ1. For in vivo assessment, female C57BL/6 mice were exposed to either control air or PM10 (±SKQ1) in a whole-body exposure chamber for 2 weeks (3 h/day, 5 days/week, with weekends off). In vitro, HCE-2 cells were exposed to 100 μg/mL PM10 (±SKQ1) for 24 h, and mitochondrial function and morphology were evaluated. In vitro, PM10 significantly impaired mitochondrial function by reducing basal, maximal, and ATP-linked respiration; reserve capacity; and coupling efficiency compared to the control and SKQ1 groups. PM10 also downregulated mitofusin1 (MFN1) and optic atrophy1 (OPA1) and upregulated dynamin-related protein1 (DRP1) and mitochondrial fission protein1 (FIS1) in HCE-2 cells. In addition, PM10 exposure significantly decreased the mitochondrial membrane potential; mitochondrial DNA copy number; and cytochrome c oxidase subunit 4 isoform 1 (COX4i1), mitochondrial transcription factor A (TFAM), and peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) levels. SKQ1 pre-treatment significantly attenuated these effects. In vivo, PM10 exposure significantly decreased the levels of MFN1, TFAM, COX4i1, and superoxide dismutase (SOD2), whereas SKQ1 treatment significantly reversed these effects. Overall, these findings demonstrate that PM10 exposure induces mitochondrial fragmentation, disrupts mitochondrial biogenesis and quality control, and reduces mitochondrial respiration, resulting in mitochondrial dysfunction. SKQ1 effectively reversed these changes, suggesting its potential as a therapeutic strategy to protect corneal epithelial cells from PM10-induced mitochondrial damage.
1. Introduction
Exposure to particulate matter less than 10 µm in aerodynamic diameter (PM10) is associated with increased morbidity and mortality [1,2,3]. Among its many adverse effects, PM10 exposure has been linked to significant ocular damage, including increased outpatient visits for eye discomfort [4], worsened tear film stability in patients with dry eye disease [5], and an elevated risk of Sjögren’s syndrome [6] and keratitis [7]. Our recent work has shown that PM10 exposure reduces the level of nuclear erythroid 2-related factor 2 (NRF2) and its transcriptional activity, leading to impaired antioxidant defenses and inflammation both in vitro [8,9] and in vivo [8]. NRF2 is a key regulator of antioxidant defenses [10], activated in response to oxidative stress, and its regulation occurs at multiple levels, including the transcriptional [11], post-transcriptional, translational [12], and protein stability levels [13]. Under basal conditions, NRF2 is bound to Kelch-like ECH-associated protein 1 (Keap1), a substrate adaptor of the Cullin-3-based E3 ubiquitin ligase in the cytosol, and remains inactive [10,14]. Upon activation, it translocates to the nucleus, where it binds to specific sites called antioxidant response elements (AREs) on DNA to regulate the expression of cell-protective genes [10,14]. NRF2 is a key regulator of mitochondrial structure, function, metabolism, and bioenergetics through its control of redox balance, antioxidant defense, and quality control pathways [15]. Mitochondria are key organelles essential for multiple facets of cellular function, including apoptosis [16] and energy production [16]. ATP production by mitochondria via oxidative phosphorylation (OxPhos) is the most efficient cellular pathway for generating energy [17]. During OxPhos, electrons are transported via the electron transport chain (ETC), creating a proton gradient that powers ATP synthase to generate ATP [17,18]. NRF2 reduces mitochondrial ROS by inducing genes involved in glutathione and NADPH production, thereby preserving ETC integrity and ATP production [15,19]. Structurally, NRF2 supports mitochondrial dynamics, regulates the balance between fission and fusion, and promotes mitophagy to remove damaged organelles [15]. It also influences biogenesis via crosstalk with transcription factors, such as PGC-1α and NRF1, ensuring adequate mitochondrial mass in response to stress [20,21]. Additionally, NRF2 stabilizes the mitochondrial membrane potential and prevents the opening of the permeability transition pore, thereby reducing susceptibility to calcium overload and apoptosis [15]. The cryoprotective role of NRF2 makes it an ideal candidate for therapeutic targeting. Therefore, compounds that activate NRF2 can be used as a therapeutic strategy to improve overall mitochondrial health and function [22]. One such compound is SKQ1 (10-(6-plastoquinonyl) decyltriphenylphosphonium), a mitochondrial antioxidant that accumulates in the inner mitochondrial membrane [23,24,25]. The administration of SKQ1 significantly increased the mRNA levels of NRF2 and NRF2-controlled genes encoding antioxidant enzymes in a rat model of hyperoxia [26]. Recently, we verified that SKQ1 is protective and restores NRF2 levels in primary human corneal epithelial cells (HCECs) and transformed human corneal epithelial cells (HCE-2) exposed to PM10 [8,9], as well as in the mouse cornea exposed to PM10 using a whole-body exposure chamber [8]. SKQ1 is also protective against oxidative stress-mediated damage in various animal models of disease [27,28,29,30]. Clinically, an ophthalmic formulation of SKQ1 (Visomitin) has been effective in preventing the pathology of anesthetic-induced dry eye syndrome after surgery or prolonged general anesthesia, demonstrating human relevance [31,32].
Therefore, our current study aimed to better understand the effects of PM10 on mitochondrial health and function in vitro in HCE-2 cells and in vivo in the mouse cornea and whether SKQ1 is protective.
2. Materials and Methods
2.1. Mice
Eight-week-old female C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). They were housed in compliance with the National Institutes of Health guidelines and treated humanely and in accordance with both the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research) and the Institutional Animal Care and Use Committee of Wayne State University (IACUC 24-03-6665). The animal study protocol was approved by the Institutional Review Board (IACUC) of Wayne State University (IACUC 24-03-6665 and date of approval 30 May 2024–29 May 2027.
2.2. Whole-Body Exposure to PM10
For PM10 exposure, a whole-body exposure chamber (CH Technologies, Westwood, NJ, USA) was used, as previously described [8,33]. Briefly, mice were either exposed to control air or 0.5–1 mg/m3 PM10, for 3 h a day for 5 days per week for 2 weeks [33]. This range was chosen to reflect exposure levels similar to those reported in countries with high pollution levels [34,35,36].
2.3. SKQ1 Treatment
SKQ1 (catalog# 934826-68-3, BOC Sciences, Shirley, NY, USA) was used at a previously published dose of 7.5 µM to treat mice exposed to control air and PM10 [8,37]. Eyes were topically treated with 5 µL SKQ1 (7.5 µM) or PBS, three times on the day before the first chamber exposure and then once each day before exposure for two weeks, as previously described [8].
2.4. Separating Corneal Epithelium from Stroma
The corneal epithelium was separated from the stroma by modifying a previously published protocol [38]. Briefly, the corneas (right and left) of each mouse were dissected and incubated in 0.02 M EDTA-PBS buffer (pH 7.2) at 37 °C for 1 h, and the epithelium was separated from the stroma. For all in vivo experiments, four groups were tested: control air + PBS (control), control air + SKQ1 (SKQ1), PM10 + PBS (PM10), and PM10 + SKQ1.
2.5. Tissue Culture and Treatments
HCE-2 ([50.B1], catalog# CRL-11135, ATCC, Gaithersburg, MA, USA) cells were grown and cultured as previously reported [9]. PM10 (SRM 2787) was purchased from the National Institute of Standards and Technology (NIST, Gaithersburg, MD, USA). PM10 was resuspended in sterile phosphate-buffered saline (PBS) to prepare a 10 mg/mL stock solution and subsequently diluted to a final concentration of 100 µg/mL using culture medium. The PM10 dosage was chosen after performing a dose curve [9,39] and was used in all our previous studies [8,9,39,40]. More details regarding PM10 can be found online at https://tsapps.nist.gov/srmext/certificates/2787.pdf (accessed on 31 October 2025).
SKQ1 (catalog# 934826-68-3, BOC Sciences, Shirley, NY, USA) was dissolved in 50% ethanol to obtain a 162 mM stock solution, which was subsequently diluted to 50 nM using culture medium. The dosage of SKQ1 used in this study was chosen based on a dose curve and a previously published report [29] and was successfully used in our previous publications [8,9,39,40]. To test the protective effects of SKQ1, cells were pre-treated with 50 nM SKQ1 for 1 h and then exposed to 100 µg/mL PM10, as described previously [9]. An SKQ1 control was included, in which cells were treated with only 50 nM SKQ1 for 24 h. Untreated cells were used as the control. For all experiments, the cells were divided into four groups: control (untreated), SKQ1 (SKQ1-treated), PM10, and PM10 + SKQ1.
2.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
RNA extraction from HCE-2 cells (n = 3/group) and the mouse corneal epithelium (n = 5 per group) was performed using a previously described protocol [9]. Briefly, cDNA templates were generated from all samples. They were then diluted 20-fold with DEPC-treated water. SYBR Green PCR Master Mix (Bio-Rad Laboratories, Hercules, CA, USA) was used for qRT-PCR. For HCE-2 cells, the mRNA levels of MFN-1, MFN2; OPA1; FIS1; DRP1; PGC-1α; TFAM; and COX4i1 were tested using real-time RT-PCR (CFX Connect real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA). In the mouse corneal epithelium, the levels of NRF2, SOD2, MFN1, MFN2, DRP1, PGC-1α, TFAM, and COX4i1 were analyzed. The primer sequences are listed in Table 1. The fold differences in gene expression were calculated relative to 18S rRNA (in vitro) and β-actin (in vivo) using 2−ΔΔCT and expressed as relative mRNA concentration ± SEM.

Table 1.
Primer sequences used for RT-qPCR (human and mouse).
2.7. Protein Analysis
The Protein Simple Abby system (Biotechne, Minneapolis, MN, USA) was used to detect the levels of SOD2, MFN1, TFAM, COX4i1, and PGC-1α in HCE-2 cells (n = 3/group) and the mouse corneal epithelium (n = 5/group/treatment). Briefly, control and SKQ1-, PM10-, and PM10 ± SKQ1-exposed cells were harvested after 24 h and lysed in RIPA buffer with protease and phosphatase inhibitors (Santa Cruz Biotech, Dallas, TX, USA). The resulting supernatants were collected after centrifugation at 12,500 RPM to remove any debris. Protein levels in the supernatant were determined using the Bradford Assay (BioRad, Hercules, CA, USA), as described previously [9]. Samples were diluted with PBS to a concentration of 0.25 μg/μL, according to the manufacturer’s guidelines. Samples were subsequently prepared for analysis on the Abby Simple Western system following the Protein Simple kit and the manufacturer’s guidelines. The primary antibodies SOD2, MFN1, TFAM, COX4i1, and PGC-1α (Proteintech, Rosemont, IL, USA) were used at a 1:50 dilution ratio. The secondary anti-rabbit HRP-conjugated antibody supplied by the manufacturer was used as a detection probe. Subsequently, the samples were re-probed using Abby’s RePlex feature with HRP-conjugated β-tubulin (Proteintech Labs, Rosemont, IL, USA) at a 1:200 dilution. Densiometric readings were obtained using Protein Simple Abby software, Compass for SW’ Version 7.0.0 (Biotechne, Minneapolis, MN, USA), and normalized against β-tubulin. Molecular weights were pre-determined through antibody validation conducted before the experiment. The bands are composites of multiple exposures and are displayed in an HDR format. This fully automated process ensured that the band intensity was neither overexposed nor underexposed. Data were acquired via raw images from the ‘Compass or SW’ program. The entire Abby system is automated, and once the input information is entered, manual changes cannot be made. The program presets all calculations.
2.8. Immunofluorescence Staining
For COX4i1 staining, HCE-2 cells were grown on 12 mm coverslips coated with fibronectin, collagen, and albumin (FNC, catalog# NC2000952, Athena ES, Baltimore, MD, USA) in 60 mm Petri dishes; pre-treated with SKQ1; and exposed to PM10, as described in the Tissue Culture and Treatments Section. Immunofluorescence was performed as previously described [9]. Cells from the control and SKQ1-, PM10-, and PM10 + SKQ1-exposed groups (n = 3/group) were fixed in 1% paraformaldehyde (PFA) for 10 min at room temperature, permeabilized using 0.2% Triton X-100 for 5 min, blocked with blocking buffer (PBS containing 1% bovine serum albumin (BSA) for 1 h, and incubated with anti-COX4i1 antibody (1:200) diluted in blocking buffer for 1 h. Coverslips were then washed three times, incubated with goat anti-rabbit IgG coupled to CoraLite Plus 488 (1:1500 in blocking buffer) for 1 h, washed, and mounted onto slides using an antifade mounting medium (Vectashield) containing DAPI (Vector Laboratories Inc., Burlingame, CA, USA). A Leica TCS SP8 microscope (Deerfield, IL, USA) was used to image the cells, and all images were processed in a similar manner using Adobe Photoshop version, 7.0.1. All experiments were performed in triplicate.
2.9. JC-1 Staining Using Flow Cytometry
HCE-2 cells were grown in 100 mm cell culture dishes and exposed to PM10 ± SKQ1, as described above in the Tissue Culture and Treatments Section. The assay was performed following the manufacturer’s instructions. Briefly, the cells were dissociated using TrypLE (ThermoFisher Scientific, Waltham, MA, USA) and transferred to FACS tubes for further analysis. The cells were centrifuged at 300× g for 5 min at room temperature. The cells were then resuspended in 100 µL of assay medium. Cells treated with 10 µM carbonyl cyanide m-chlorophenyl hydrazone (CCCP) for 1 h were used as positive controls. A total of 100 µL of JC-1 staining solution was added to each sample individually and incubated for 15 min. JC-1 aggregates were detected in the PE channel, and monomers were detected in the FITC channel. The experiments were performed in triplicate to ensure reproducibility.
2.10. JC-1 Live Staining
HCE-2 cells were grown on 12 mm coverslips coated FNC (, Athena ES, Baltimore, MD, USA) in a 12-well plate for 24 h; pre-treated with SKQ1; and exposed to PM10, as described in the Tissue Culture and Treatments Section. Cells treated with 10 µM CCCP for 1 h were used as the positive control. Live staining was performed using the JC-1 mitochondrial membrane potential assay kit (Cayman Chemical, Ann Arbor, MI, USA) following the manufacturer’s protocol. Briefly, the cells were incubated with 1 µM JC-1 in cell culture media at 37 °C for 15 min in 5% CO2. The cells were washed three times and imaged using a Zeiss Axiophot 200M Apotome microscope (White Plains, NY, USA). All images were processed in a similar manner using Adobe Photoshop, version 7.0.1.
2.11. Seahorse Assay
The Seahorse Extracellular Flux Analyzer XFe96 (Agilent, Santa Clara, CA, USA) was used to measure the O2 consumption rate in a 96-well format according to the manufacturer’s guidelines. The machine detects changes in O2 content in a very small volume of 7 μL above the plated cells using a fluorescence biosensor. Measurements were taken from intact cells at short, repeated intervals, making them noninvasive and physiologically relevant. HCE-2 cells were seeded at 40,000 cells/well in an XFe96 well cell culture microplate previously coated with FNC and exposed to PM10 ± SKQ1, as described above in the Tissue Culture and Treatments Section, and incubated for 24 h at 37 °C in 5% CO2. A sensor cartridge was placed on the utility plate, and the sensors were submerged in the calibrant in a 37 °C non-CO2 incubator overnight. Prior to the assay, the culture medium was removed and replaced with fresh medium, and the cells were placed in a CO2-free incubator for 1 h at 37 °C. Stock solutions of oligomycin, FCCP, antimycin A, and rotenone were diluted according to the manufacturer’s instructions and dispensed into Ports A–C of the sensor cartridge. The XF96 analyzer was used to examine basal and maximal respiration, coupling efficiency, ATP production and spare respiration capacity.
2.12. Statistical Analysis
Statistical analysis was performed using a one-way ANOVA followed by Bonferroni’s multiple comparison test (GraphPad Prism version 8). The data generated from qRT-PCR, Abby, flow cytometry, and Seahorse were tested for significance set at p < 0.05. Tissue culture studies included three biological replicates per group; these studies were repeated once more, while mouse experiments initially used five mice per group; these mouse studies were also repeated once more, resulting in a total of n = 10 mice per group. Although a formal power analysis was not performed, the sample sizes were consistent with the ranges typically recommended by power analysis. Randomization and blinding procedures were applied where feasible, particularly in mouse experiments, to minimize bias. The results are presented as the mean ± SEM derived from combined replicates across independent experiments.
3. Results
3.1. PM10 Altered Mitochondrial Function, Which Was Reversed by SKQ1 Pre-Treatment
To evaluate the effects of PM10 on mitochondrial function, we measured mitochondrial respiration by assessing the oxygen consumption rate (OCR) in HCE-2 cells. The data show that exposure to PM10 significantly impaired the mitochondrial oxygen consumption rate compared to both the control and SKQ1 groups (Figure 1A). Specifically, after 24 h of PM10 exposure, there was a significant reduction in several key parameters of mitochondrial function. Basal respiration (Figure 1B), which reflects the energy demand of the cell under normal conditions, showed a significant decrease after PM10 exposure compared to the control (p < 0.001) and SKQ1 groups (p < 0.05). The maximum respiration (Figure 1C), which indicates the maximal capacity of mitochondria to consume oxygen, showed a significant decline after PM10 exposure compared to the control (p < 0.001) and SKQ1 (p < 0.001) groups. Spare capacity (Figure 1D), which refers to the ability of cells to produce additional ATP through respiration energy demands, showed a significant decrease after PM10 exposure compared to the control (p < 0.001) and SKQ1 groups (p < 0.001). ATP production (Figure 1E), indicating the ability of mitochondria to generate energy, was also significantly reduced after PM10 exposure compared to that in the control (p < 0.001) and SKQ1 groups (p < 0.001). Finally, coupling efficiency (Figure 1F), which measures how effectively oxygen consumption is linked to ATP synthesis, also showed a significant decrease after PM10 exposure compared to the control (p < 0.05) and SKQ1 (p < 0.05) groups. SKQ1 pre-treatment significantly restored the oxygen consumption rate (Figure 1A), basal respiration (Figure 1B, p < 0.01), maximum respiration (Figure 1C, p < 0.05), spare capacity (Figure 1D, p < 0.01), ATP production (Figure 1E, p < 0.001), and coupling efficiency (Figure 1F, p < 0.05), demonstrating its protective role in preserving mitochondrial function under PM10-induced stress.
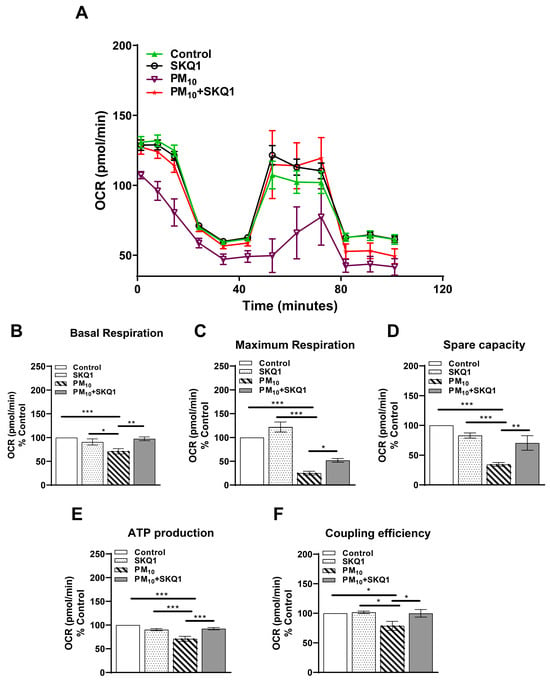
Figure 1.
Mitochondrial function in HCE-2 cells following exposure to 100 µg/mL PM10 with or without 50 nM SKQ1 was evaluated using a Seahorse bioanalyzer after 24 h. A representative profile of the oxygen consumption rate (OCR) shows a reduced OCR after PM10 exposure compared to the control and SKQ1 groups, which was improved by SKQ1 (A). PM10 significantly reduced basal respiration (B), maximum respiration (C), spare capacity (D), ATP production (E), and coupling efficiency (F). SKQ1 pre-treatment significantly reversed these effects. Significant changes are denoted by p-values indicated by symbols: * p < 0.05; ** p < 0.01; *** p < 0.001. n = 3 biological replicates for each group.
3.2. PM10 Reduced the Mitochondrial Membrane Potential, Which Was Reversed by SKQ1 Pre-Treatment
Following PM10 exposure (100 µg/mL), the mitochondrial membrane potential was measured using JC-1 live staining and flow cytometry, as shown in Figure 2 and Figure 3. JC-1 staining showed that cells exposed to PM10 predominantly exhibited JC-1 monomers, which emitted green fluorescence (indicated by arrows), and showed a marked absence of JC-1 aggregates, which emitted red fluorescence. This shift from red to green fluorescence indicates a significant reduction in the mitochondrial membrane potential after PM10 exposure compared to the control and SKQ1 groups (Figure 2). Flow cytometry scatter plots in Figure 3A also confirm these findings by demonstrating the loss of JC-1 aggregates in PM10-exposed cells. A quantitative analysis of the flow data (Figure 3B) further confirmed that PM10 significantly increased the number of depolarized mitochondria (p < 0.001), along with a significant reduction in the number of mitochondria maintaining a normal mitochondrial potential (p < 0.001), relative to the control and SKQ1 groups. SKQ1 pre-treatment significantly reversed these effects, highlighting the protective role of SKQ1 in the mitochondrial membrane’s integrity under PM10-mediated stress.
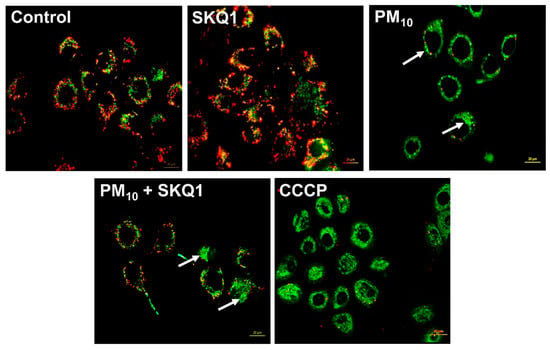
Figure 2.
JC-1 staining in HCE-2 cells following exposure to 100 µg/mL PM10 in the absence or presence of 50 nM SKQ1. Green fluorescence (monomers) reflects a reduced mitochondrial membrane potential, whereas red fluorescence indicates healthy mitochondria. PM10-exposed cells had a lower mitochondrial membrane potential than the control or SKQ1 alone after 24 h. Pre-treatment with SKQ1 before PM10 exposure partially restored the mitochondrial membrane potential. CCCP was used as a positive control. Arrows indicate depolarized mitochondria. n = 3 biological replicates were used for each group in this study.
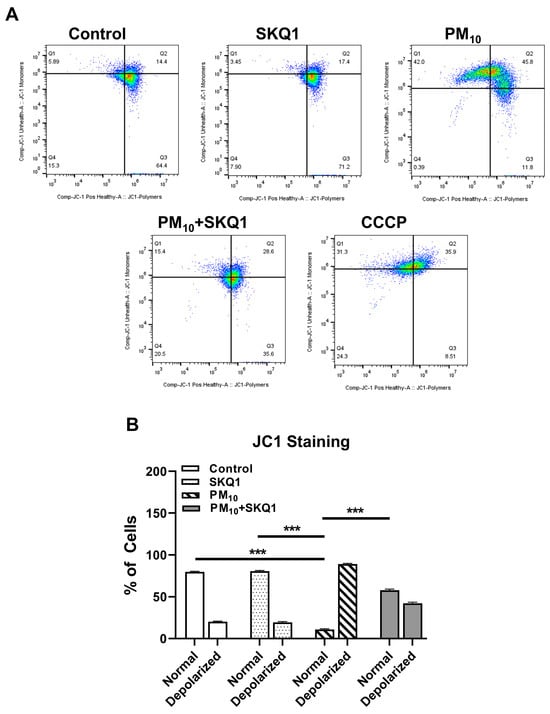
Figure 3.
The mitochondrial membrane potential in HCE-2 cells was assessed by flow cytometry using JC-1 dye following exposure to 100 µg/mL PM10 with or without 50 nM SKQ1. Scatter plots (A) and graphical quantification (B) show that PM10-exposed cells have more depolarized mitochondria (lower mitochondrial membrane potential) than the control or SKQ1 alone (normal mitochondria) after 24 h. Pre-treatment with SKQ1 before PM10 exposure significantly reversed these changes. CCCP was used as a positive control. *** p < 0.001; n = 3 biological replicates were used for each group in this study.
3.3. SKQ1 Reversed PM10-Induced Effects on Mitochondrial Biogenesis, Quality Control, Structure, and Health
PM10 exposure caused a significant alteration in the expression of key regulators involved in mitochondrial structure, biogenesis, and quality control, as shown in Figure 4, Figure 5 and Figure 6. At the mRNA level (Figure 4), PM10 significantly decreased the transcript levels of fusion-related genes MFN1 (4A, p < 0.001, p < 0.001), MFN2 (4B, p < 0.01, p < 0.05), and OPA1 (4C, p < 0.001, p < 0.001) compared to the control and SKQ1 groups, which maintained mitochondrial network integrity through fusion. Additionally, PM10 exposure also reduced the expression of genes involved in mitochondrial biogenesis and function, such as PGC-1α (4F, p < 0.05, p < 0.01) and TFAM (4G, p < 0.001, p < 0.001). However, COX4i1 (4H, p < 0.05) levels were only significantly reduced by PM10 compared to the control group. These decreases indicate impaired mitochondrial replication, transcription, and respiratory capacity. Conversely, PM10 significantly increased the mRNA levels of fission-related genes such as FIS1 (4D, p < 0.001, p < 0.001) and DRP1 (4E, p < 0.05, p < 0.001) relative to the control and SKQ1 groups. Pre-treatment with SKQ1 significantly increased the transcript levels of MFN1 (4A, p < 0.05), MFN2 (4B, p < 0.001), OPA1 (4C, p < 0.01), PGC-1α (4F, p < 0.05), TFAM (4G, p < 0.01), and COX4i1 (4H, p < 0.05) but significantly lowered the levels of FIS1 (4D, p < 0.001) and DRP1 (4E, p < 0.001). Compared to the SKQ1 group, FIS1 expression (4D, p < 0.01) was significantly increased, while DRP1 expression (4E, p < 0.01) was significantly decreased.
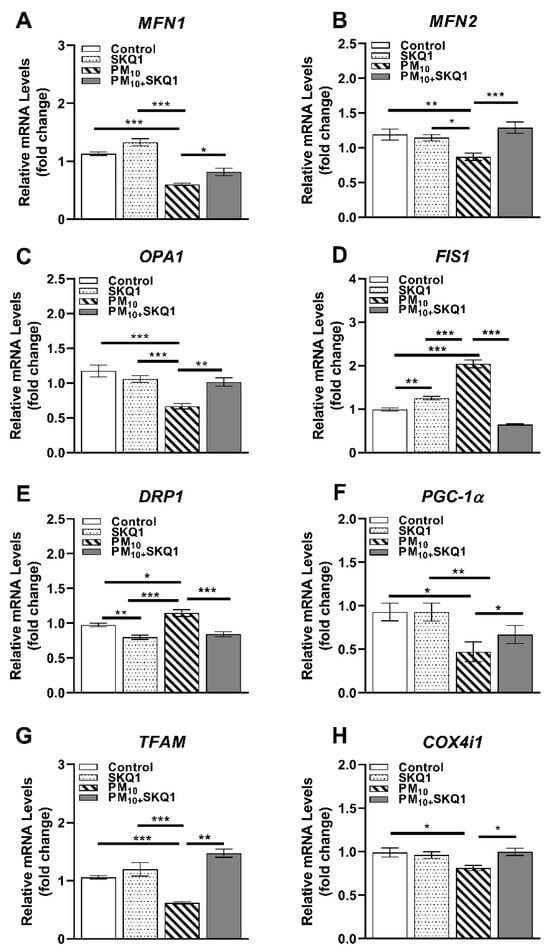
Figure 4.
qRT-PCR in HCE-2 cells exposed to 100 µg/mL PM10 in the presence or absence of 50 nM SKQ1. Compared to the control and SKQ1 groups, PM10 exposure significantly downregulated MFN1 (A), MFN2 (B), OPA1 (C), PGC-1α (F), TFAM (G), and COX4i1 (H) but upregulated FIS1 (D) and DRP1 (E) after 24 h. SKQ1 pre-treatment reversed these effects. * p < 0.05, ** p < 0.01, *** p < 0.001, n = 3 biological replicates were used for each group in this study.
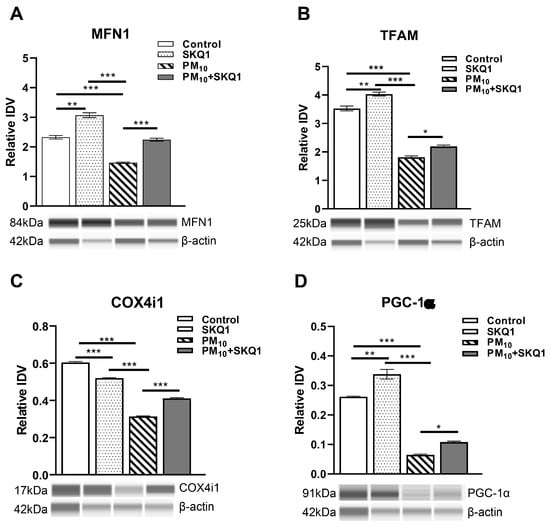
Figure 5.
Protein analysis in HCE-2 cells exposed to 100 µg/mL PM10 in the presence or absence of 50 nM SKQ1 using the Protein Simple Abby system. PM10 exposure significantly downregulated MFN1 (A), TFAM (B), COX4i1 (C), and PGC-1α (D) compared to the control and SKQ1 alone after 24 h. Pre-treatment with SKQ1 before PM10 exposure reversed these effects. * p < 0.05, ** p < 0.01, *** p < 0.001. n = 3 biological replicates were used for each group in this study.
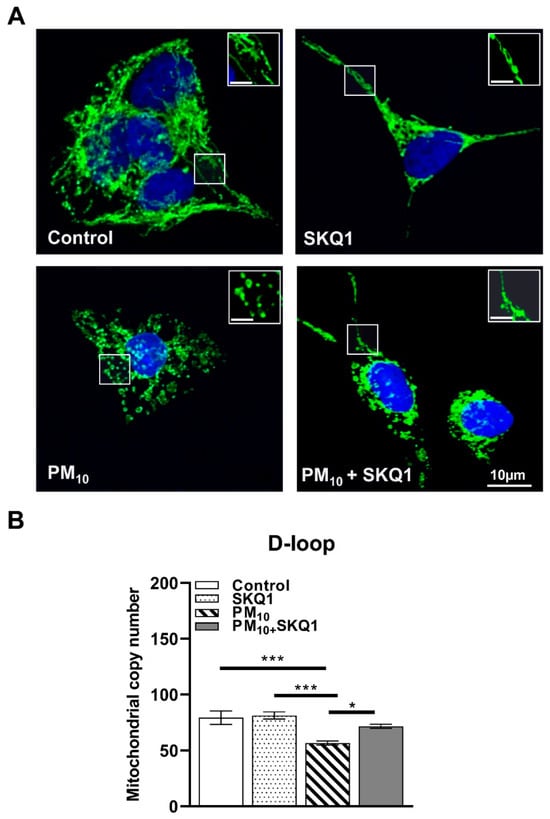
Figure 6.
Representative images of mitochondrial morphology (A) and DNA copy number (B) after exposure of HCE-2 cells to 100 µg/mL PM10 ± 50 nM SKQ1. PM10 exposure induced mitochondrial fragmentation and reduced mtDNA copy number compared to control and SKQ1 alone after 24 h. SKQ1 pre-treatment partially restored normal elongated morphology and increased reduction in DNA copy number. n = 3 biological replicates were used for each group in this study. The white frames indicate enlarged areas to clearly illustrate the differences in morphology. * p < 0.05, *** p < 0.001.
At the protein level (Figure 5), PM10 exposure similarly caused a significant reduction in MFN1 (5A, p < 0.001), TFAM (5B, p < 0.001), COX4i1 (5C, p < 0.001) and PGC-1α (5D, p < 0.001) compared to the control and SKQ1 groups. Pre-treatment with SKQ1 significantly increased the levels of MFN1 (5A, p < 0.001), TFAM (5B, p < 0.05), COX4i1 (5C, p < 0.001) and PGC-1α (5D, p < 0.05). Compared to the control group, the SKQ1 group showed significantly higher levels of MFN1 (5A, p < 0.001), TFAM (5B, p < 0.001) and PGC-1α (5D, p < 0.001). In contrast, COX4i1 levels (5C, p < 0.001) were significantly reduced in the SKQ1 group compared to the control.
Immunofluorescence analysis (Figure 6A) showed that PM10 exposure caused fragmented mitochondria, characterized by small punctate mitochondria instead of the elongated, interconnected network observed in the control and SKQ1 groups. SKQ1 pre-treatment partially restored the elongated mitochondrial structure, suggesting improved mitochondrial dynamics and health. Furthermore, PM10-exposed cells showed a significant reduction in the relative mitochondrial DNA (mtDNA) copy number (Figure 6B) measured specifically within the D-loop region, compared to the control (p < 0.001) and SKQ1 (p < 0.001) groups, suggesting compromised mitochondrial replication. SKQ1 pre-treatment significantly reversed this effect (Figure 6B, p < 0.05). These changes collectively suggest that PM10 disrupts the balance between mitochondrial fusion and fission, decreases mitochondrial biogenesis, and impairs mitochondrial respiratory components, leading to fragmented and dysfunctional mitochondria. SKQ1 effectively mitigated these effects, indicating the recovery of mitochondrial structure and function.
3.4. The Effects of PM10 and SKQ1 on the Mouse Corneal Epithelium
The mRNA and protein levels of the regulators of mitochondrial structure, biogenesis, and quality control are shown in Figure 7 and Figure 8. Exposure to PM10 caused a significant decrease compared to the control and SKQ1 groups in the mRNA expression of several critical genes: NRF2 (7A, p < 0.001, p < 0.001), SOD2 (7B, p < 0.05, p < 0.001), MFN1 (7C, p < 0.001, p < 0.001), PGC-1α (7F, p < 0.001, p < 0.001), TFAM (7G, p < 0.001, p < 0.001), and COX4i1 (7H, p < 0.001, p < 0.001). MFN2 levels significantly declined compared to SKQ1 (7D, p < 0.001) but not compared to the control group. These genes are essential for antioxidant defense, mitochondrial biogenesis, fusion, and electron transport. In contrast, PM10 significantly upregulated the mRNA levels of DRP1 (7E, p < 0.001, p < 0.001), a key mediator of mitochondrial fission, indicating enhanced mitochondrial fragmentation. Importantly, SKQ1 pre-treatment effectively mitigated (p < 0.001) PM10-mediated changes in mRNA expression. In addition, compared to the control group, the SKQ1 group showed significantly higher expression levels of MFN1 (7C, p < 0.001) and MFN2 (7D, p < 0.001).
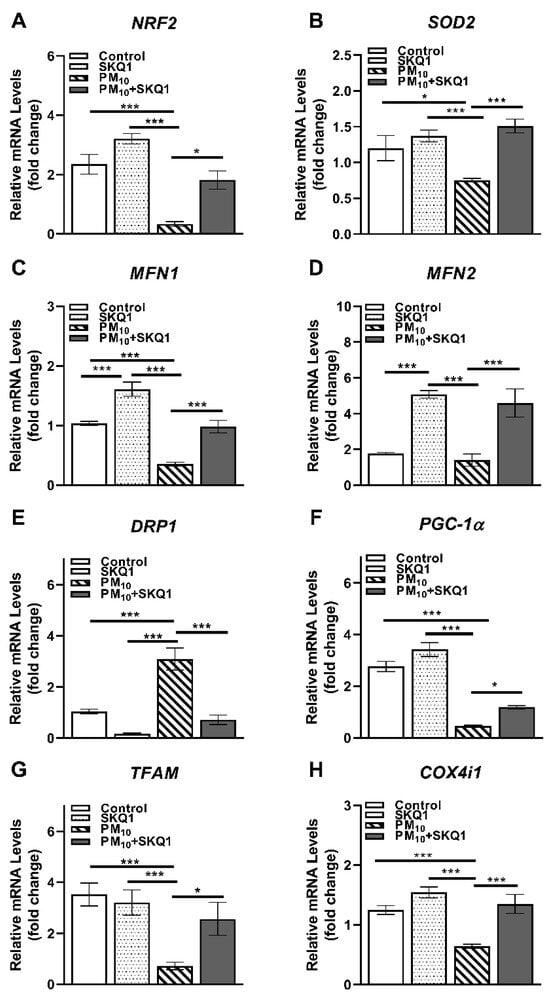
Figure 7.
qRT-PCR in mouse corneal epithelium after 2 weeks of exposure to either control air or PM10 (0.5–1 mg/m3) ± 7.5 µM SKQ1. Data showed that compared to control and SKQ1 groups, PM10 exposure significantly downregulated NRF2 (A), SOD2 (B), MFN1 (C), MFN2 (D), PGC-1α (F), TFAM (G) and COX4i1 (H) but upregulated DRP1 (E). SKQ1 pre-treatment reduced PM10-mediated effects. * p < 0.05, *** p < 0.001, n = 5 biological replicates for each group.
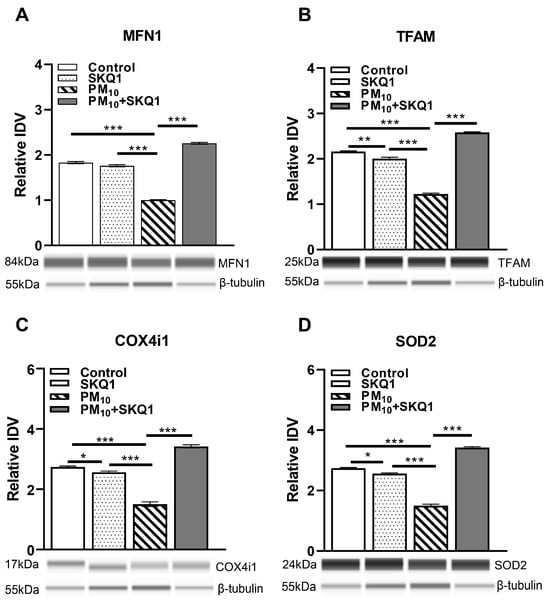
Figure 8.
Protein analysis of mouse corneal epithelial sheets after 2 weeks of exposure to either control air or PM10 (0.5–1 mg/m3) ± 7.5 µM SKQ1 using Protein Simple Abby system. Data indicated that PM10 exposure significantly downregulated MFN1 (A), TFAM (B), COX4i1 (C), and SOD2 (D) compared to control and SKQ1 alone. Pre-treatment with 7.5 µM SKQ1 reduced PM10-mediated effects. * p < 0.05, ** p < 0.01, *** p < 0.001. n = 5 biological replicates per group.
At the protein level (Figure 8), PM10 exposure significantly reduced the expression of MFN1 (8A, p < 0.001, p < 0.001), TFAM (8B, p < 0.001, p < 0.001), COX4i1 (8C, p < 0.001, p < 0.001), and SOD2 (8D, p < 0.001, p < 0.001) compared to the control and SKQ1 groups. SKQ1 pre-treatment significantly counteracted these reductions for MFN1 (5A, p < 0.001), TFAM (5B, p < 0.001), COX4i1 (5C, p < 0.001) and SOD2 (5D, p < 0.001). In addition, compared to the control group, the SKQ1 group showed significantly lower expression levels of TFAM (8B, p < 0.01), COX4i1 (8C, p < 0.05) and SOD2 (7D, p < 0.05).
Collectively, these results demonstrate that PM10 exposure disrupts mitochondrial homeostasis by reducing biogenesis and fusion markers while upregulating fission markers. SKQ1 pre-treatment is protective and reverses these detrimental changes, thereby preserving mitochondrial integrity and function.
4. Discussion
Continuous exposure to the external environment makes the ocular surface highly vulnerable to airborne pollutants, making it an ideal model for studying pollution-mediated changes [41]. Particulate matter (PM) is a major airborne contaminant composed of heterogeneous particles ranging from 0.1 to 10 µm that exert detrimental effects on ocular tissues [42,43]. Based on the aerodynamic diameter, PM is categorized into PM10 (coarse particles ≤ 10 µm), PM2.5 (fine, ≤2.5 µm), and PM0.1 (ultrafine, <0.1 µm) [44]. Epidemiological and clinical studies have linked PM exposure to tear film instability [45], dry eye disease [46], oxidative stress [41] and inflammation [41]. In vitro studies using corneal epithelial cells have shown that PM exposure impairs several cellular processes, including wound healing [47], apoptosis [48,49], autophagy [50], mitochondrial integrity [49], and senescence [9,48]. While the impact of PM2.5 on ocular surface toxicity is well documented, relatively few studies have investigated the impact of PM10. Previous work from our laboratory established that PM10 exposure disrupts redox homeostasis via the dysregulation of the NRF2 pathway both in vitro and in vivo and that the mitochondria-targeted antioxidant SKQ1 confers significant protection against PM10 [8,9]. However, the specific effects of PM10 on mitochondrial dynamics in corneal epithelial cells remain unclear.
Thus, this study evaluated the impact of PM10 on mitochondrial function and structural dynamics in HCE-2 cells (in vitro) and the mouse corneal epithelium (in vivo). Key functional biomarkers, including ROS production, mitochondrial membrane potential, and bioenergetic capacity, reflect oxidative stress and metabolism [51,52,53]. Airborne pollutants, such as PM2.5 and PM10, are potent inducers of oxidative stress [54], and ROS production is mainly attributed to their heavy metal constituents (cadmium, chromium, copper, iron, and nickel) [55]. Multiple studies have reported that exposure to PM leads to increased mitochondrial ROS production [56,57,58,59]. In our study, we similarly observed elevated mitochondrial superoxide levels in HCE-2 cells using the MitoSOX assay, which detects mitochondrial superoxide, a type of ROS [8]. Mitochondria serve as the main source of intracellular ROS and as prime targets of PM-induced oxidative injury [60,61,62]. Ultrafine particles, PM2.5, and PM10 have been reported to accumulate within the mitochondrial matrix [63], causing structural damage, including crista disruption and mitochondrial swelling, in tissues such as the lungs, brain, and olfactory epithelium [58,63,64]. These ultrastructural abnormalities are associated with a decline in respiratory capacity [64]. Consistent with these findings in human alveolar basal epithelial cells (A549) [64] and mouse microglial cells (BV2) [65] exposed to PM2.5 and other fine particulates, our oxygen consumption rate analyses demonstrated that PM10 significantly reduced basal respiration, ATP production, maximum respiratory capacity, and coupling efficiency in HCE-2 cells. Similar mitochondrial respiratory impairments and ATP depletion have been documented in human nasal olfactory mucosal cells [58] and mitochondria isolated from PM10-exposed rat lung tissues [66].
Elevated ROS levels can cause the depolarization of the mitochondrial membrane [67]. In this line, we detected the loss of mitochondrial membrane potential in HCE-2 cells treated with PM10, corroborating previous reports on lung mitochondria treated with PM10 [66]. Mitochondrial morphology, including fission, fusion, and network organization, critically affects mitochondrial function [53]. We found that PM10 caused the fragmentation of the normally elongated mitochondrial network in HCE-2 cells, a pattern also described in A549 cells after PM2.5 exposure [64]. This observation led us to evaluate the key regulators of mitochondrial dynamics in these cell types. PM10 significantly downregulated the fusion proteins MFN1, MFN2, and OPA1 and upregulated the fission mediators DRP1 and FIS1, both in vitro and in vivo. This molecular profile parallels the findings in human vascular endothelial cells (EA.hy926) [68], human bronchial epithelial cells (16HBE) [69], human bronchial epithelial cells (BEAS-2B) [70], and A549 cells [64] exposed to PM2.5, where mitochondrial fragmentation is accompanied by suppressed fusion proteins and increased fission.
Mitochondrial size and abundance are correlated with mtDNA copy number, all of which fluctuate according to metabolic demand and extrinsic cellular stressors [71]. The mtDNA copy number serves as a biomarker of mitochondrial integrity [72] and typically decreases under stress. Our data show that PM10 significantly reduced the mtDNA copy number in HCE-2 cells, consistent with the reductions reported in pulmonary endothelial cells [73] and A549 cells [64] exposed to PM2.5. TFAM, the primary mtDNA packaging protein, is essential for maintaining mtDNA stability [74]. Mitochondrial ROS can suppress TFAM expression and impair biogenesis [74,75]. Our previous data showed that PM10 significantly increased mitochondrial ROS production and lipid peroxidation (Malondialdehyde levels) and reduced ATP levels [8], and our current data show decreased PGC-1α, TFAM, and COX4i1 levels, both in vitro and in vivo, after PM10 exposure, indicating impaired biogenesis and the disruption of mitochondrial quality control mechanisms. NRF2 regulates the expression of PGC-1α [20], which governs mitochondrial biogenesis [76] and antioxidant defenses, such as SOD2, whose levels were also reduced after PM10 exposure. This simultaneous reduction compromises both mitochondrial antioxidant capacity and biogenic regulation, providing a mechanistic explanation for the mitochondrial defects observed following PM10 exposure.
The PM10 dosage used for in vivo experiments in this study ranged from 0.5 to 1 mg/m3. This exposure regimen was selected to mimic the in vivo PM10 levels reported in regions such as China, India, and parts of the Middle East [34,35,36]. Similarly, the in vitro PM10 dosage was established through dose–response curves [9,39] and was consistently applied in our previous studies [8,9,39], where it effectively induced oxidative stress, barrier dysfunction, and cellular senescence.
The pharmacological modulation of mitochondrial dynamics has been shown to mitigate PM-induced oxidative stress [76], and several natural and synthetic compounds have been investigated for their potential to enhance mitochondrial function [77,78,79]. In this context, we evaluated the mitochondria-targeted antioxidant SKQ1 and demonstrated its efficacy in protecting mitochondrial health from PM10-induced toxicity. SKQ1 dosages of 7.5 µM for in vivo experiments and 50 nM for in vitro experiments were determined based on dose–response experiments and are supported by prior published data [8,9,33,39]. The topical application of 7.5 µM SKQ1 demonstrates significant clinical relevance, as it follows the same treatment regimen established in a rabbit model study, which successfully prevented anesthesia-induced dry eye disease [37]. Moreover, ophthalmic SKQ1 solution (Visomitin) has completed phase 3 clinical trials, confirming its efficacy in alleviating the symptoms of dry eye disease, thereby supporting its potential therapeutic use in clinical settings [80]. Collectively, our data indicate that PM10-induced ROS are central mediators of mitochondrial dysfunction. The ability of SKQ1 to reverse PM10-driven impairments in mitochondrial morphology, respiration, membrane potential, mtDNA stability, and biogenesis strongly supports the notion that oxidative stress is the primary upstream event. The restoration of mitochondrial integrity and function by SKQ1 further demonstrated that the neutralization of mitochondrial ROS can mitigate PM10-induced damage.
5. Limitations
This study presents valuable and robust data on the effects of PM10 on mitochondrial homeostasis on the ocular surface and the protective effects of SKQ1. However, certain limitations of this study should be acknowledged, such as the use of a single immortalized human corneal epithelial cell line and mouse model, as well as a single dose and time point with a relatively small sample size. These factors may influence the extent to which the findings represent the full complexity of the human ocular response to PM10 and its broader applicability. The two-week acute exposure model provides important insights but may not fully capture chronic or varying real-world conditions. The protective effects of SKQ1 were tested as a prophylactic agent, at specific doses (50 nM in vitro and 7.5 µM in vivo) and assessed at definite time points, with long-term safety and potential off-target effects to be explored in future studies. Additionally, while immunofluorescence offers valuable information, it has limited resolution and ultrastructural details compared to electron microscopy.
6. Conclusions
In conclusion, PM10 exposure disrupts multiple aspects of mitochondrial health in corneal epithelial cells, including respiratory function, membrane potential, structural dynamics, mtDNA maintenance, and mitochondrial biogenesis. The protective effects of SKQ1 highlight its therapeutic potential for targeting mitochondrial oxidative stress, thereby preserving ocular surface homeostasis in environments affected by high levels of air pollution.
Author Contributions
Conceptualization, M.S. and L.D.H.; methodology, M.S. and L.D.H.; software, M.S. and R.W.; formal analysis, M.S., R.W., F.S.M. and S.A.M.; investigation, M.S., R.W., F.S.M. and S.A.M.; resources, L.D.H.; data curation, M.S.; writing—original draft preparation, M.S. and L.D.H.; writing, review and editing, M.S., A.S.I. and L.D.H.; visualization, M.S., A.S.I. and S.A.M.; supervision, L.D.H.; project administration, L.D.H.; funding acquisition, L.D.H. All authors have read and agreed to the published version of the manuscript.
Funding
R01EY035231 (LDH), R01EY034964 (ASI), and P30EY04068 (LDH), and Research to Prevent Blindness.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (IACUC) of Wayne State University (IACUC 24-03-6665 and date of approval 30 May 2024 (30 May 2024–29 May 2027).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
We would like to thank Mohamed Shawky for all the help with the seahorse experiments.
Conflicts of Interest
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
- Yoon, S.; Han, S.; Jeon, K.J.; Kwon, S. Effects of collected road dusts on cell viability, inflammatory response, and oxidative stress in cultured human corneal epithelial cells. Toxicol. Lett. 2018, 284, 152–160. [Google Scholar] [CrossRef]
- Maesano, C.N.; Morel, G.; Matynia, A.; Ratsombath, N.; Bonnety, J.; Legros, G.; Da Costa, P.; Prud’homme, J.; Annesi-Maesano, I. Impacts on human mortality due to reductions in PM10 concentrations through different traffic scenarios in Paris, France. Sci. Total Environ. 2020, 698, 134257. [Google Scholar] [CrossRef]
- Yi, O.; Hong, Y.-C.; Kim, H. Seasonal effect of PM10 concentrations on mortality and morbidity in Seoul, Korea: A temperature-matched case-crossover analysis. Environ. Res. 2010, 110, 89–95. [Google Scholar] [CrossRef]
- Chang, C.-J.; Yang, H.-H.; Chang, C.-A.; Tsai, H.-Y. Relationship between Air Pollution and Outpatient Visits for Nonspecific Conjunctivitis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 429–433. [Google Scholar]
- Kim, Y.; Choi, Y.H.; Kim, M.K.; Paik, H.J.; Kim, D.H. Different adverse effects of air pollutants on dry eye disease: Ozone, PM(2.5), and PM(10). Environ. Pollut. 2020, 265, 115039. [Google Scholar] [CrossRef]
- Zhang, T.P.; Dou, J.; Wang, L.; Wang, S.; Wang, P.; Zhou, X.H.; Yang, C.M.; Li, X.M. Exposure to particulate pollutant increases the risk of hospitalizations for Sjögren’s syndrome. Front. Immunol. 2022, 13, 1059981. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, J.W.; Kim, E.J.; Lee, M.Y.; Nam, C.W.; Chung, I.S. Spatial analysis between particulate matter and emergency room visits for conjunctivitis and keratitis. Ann. Occup. Environ. Med. 2018, 30, 41. [Google Scholar] [CrossRef] [PubMed]
- Somayajulu, M.; McClellan, S.A.; Wright, R.; Pitchaikannu, A.; Croniger, B.; Zhang, K.; Hazlett, L.D. Airborne Exposure of the Cornea to PM(10) Induces Oxidative Stress and Disrupts Nrf2 Mediated Anti-Oxidant Defenses. Int. J. Mol. Sci. 2023, 24, 3911. [Google Scholar] [CrossRef] [PubMed]
- Somayajulu, M.; Muhammed, F.S.; Wright, R.; McClellan, S.A.; Hazlett, L.D. Mechanisms of PM(10) Disruption of the Nrf2 Pathway in Cornea. Int. J. Mol. Sci. 2024, 25, 3754. [Google Scholar] [CrossRef]
- Suzuki, T.; Takahashi, J.; Yamamoto, M. Molecular Basis of the KEAP1-NRF2 Signaling Pathway. Mol. Cells 2023, 46, 133–141. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Y.; Cao, J.; Wu, C.; Tang, L.; Bian, W.; Chen, Y.; Yu, L.; Wu, Y.; Li, S.; et al. Targeting epigenetic and post-translational modifications of NRF2: Key regulatory factors in disease treatment. Cell Death Discov. 2025, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Nrf2 as a regulator of mitochondrial function: Energy metabolism and beyond. Free Radic. Biol. Med. 2022, 189, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It is all about energy. Front. Physiol. 2023, 14, 1114231. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; The Mechanism of Oxidative Phosphorylation; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Deng, X.; Lin, N.; Fu, J.; Xu, L.; Luo, H.; Jin, Y.; Liu, Y.; Sun, L.; Su, J. The Nrf2/PGC1α Pathway Regulates Antioxidant and Proteasomal Activity to Alter Cisplatin Sensitivity in Ovarian Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 4830418. [Google Scholar] [CrossRef]
- Qi, X.M.; Zhang, W.Z.; Zuo, Y.Q.; Qiao, Y.B.; Zhang, Y.L.; Ren, J.H.; Li, Q.S. Nrf2/NRF1 signaling activation and crosstalk amplify mitochondrial biogenesis in the treatment of triptolide-induced cardiotoxicity using calycosin. Cell Biol. Toxicol. 2024, 41, 2. [Google Scholar] [CrossRef]
- Bhat, A.A.; Moglad, E.; Goyal, A.; Afzal, M.; Thapa, R.; Almalki, W.H.; Kazmi, I.; Alzarea, S.I.; Ali, H.; Gaur, A.; et al. Nrf2 pathways in neuroprotection: Alleviating mitochondrial dysfunction and cognitive impairment in aging. Life Sci. 2024, 357, 123056. [Google Scholar] [CrossRef]
- Skulachev, M.V.; Antonenko, Y.N.; Anisimov, V.N.; Chernyak, B.V.; Cherepanov, D.A.; Chistyakov, V.A.; Egorov, M.V.; Kolosova, N.G.; Korshunova, G.A.; Lyamzaev, K.G.; et al. Mitochondrial-targeted plastoquinone derivatives. Effect on senescence and acute age-related pathologies. Curr. Drug Targets 2011, 12, 800–826. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Antonenko, Y.N.; Cherepanov, D.A.; Chernyak, B.V.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; Korshunova, G.A.; Lyamzaev, K.G.; Pletjushkina, O.Y.; et al. Prevention of cardiolipin oxidation and fatty acid cycling as two antioxidant mechanisms of cationic derivatives of plastoquinone (SkQs). Biochim. Biophys. Acta 2010, 1797, 878–889. [Google Scholar] [CrossRef]
- Chernyak, B.V.; Antonenko, Y.N.; Domnina, L.V.; Ivanova, O.Y.; Lyamzaev, K.G.; Pustovidko, A.V.; Rokitskaya, T.I.; Severina, I.I.; Simonyan, R.A.; Trendeleva, T.A.; et al. Novel penetrating cations for targeting mitochondria. Curr. Pharm. Des. 2013, 19, 2795–2806. [Google Scholar] [CrossRef] [PubMed]
- Vnukov, V.V.; Gutsenko, O.I.; Milutina, N.P.; Kornienko, I.V.; Ananyan, A.A.; Danilenko, A.O.; Panina, S.B.; Plotnikov, A.A.; Makarenko, M.S. Influence of SkQ1 on Expression of Nrf2 Gene, ARE-Controlled Genes of Antioxidant Enzymes and Their Activity in Rat Blood Leukocytes under Oxidative Stress. Biochemistry 2015, 80, 1598–1605. [Google Scholar] [CrossRef]
- Pavshintsev, V.V.; Podshivalova, L.S.; Frolova, O.Y.; Belopolskaya, M.V.; Averina, O.A.; Kushnir, E.A.; Marmiy, N.V.; Lovat, M.L. Effects of Mitochondrial Antioxidant SkQ1 on Biochemical and Behavioral Parameters in a Parkinsonism Model in Mice. Biochemistry 2017, 82, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Austad, S.N.; Ballinger, S.; Buford, T.W.; Carter, C.S.; Smith, D.L., Jr.; Darley-Usmar, V.; Zhang, J. Targeting whole body metabolism and mitochondrial bioenergetics in the drug development for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 511–531. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Troger, A.; Spahiu, V.; Perekhvatova, N.; Skulachev, M.; Petrov, A.; Chernyak, B.; Asbell, P. The Role of SKQ1 (Visomitin) in Inflammation and Wound Healing of the Ocular Surface. Ophthalmol. Ther. 2019, 8, 63–73. [Google Scholar] [CrossRef]
- Kryl’skii, E.D.; Popova, T.N.; Zhaglin, D.A.; Razuvaev, G.A.; Oleynik, S.A. SkQ1 Improves Immune Status and Normalizes Activity of NADPH-Generating and Antioxidant Enzymes in Rats with Adjuvant-Induced Rheumatoid Arthritis. Biochemistry 2023, 88, 1092–1104. [Google Scholar] [CrossRef]
- Brzheskiy, V.V.; Efimova, E.L.; Vorontsova, T.N.; Alekseev, V.N.; Gusarevich, O.G.; Shaidurova, K.N.; Ryabtseva, A.A.; Andryukhina, O.M.; Kamenskikh, T.G.; Sumarokova, E.S.; et al. Results of a Multicenter, Randomized, Double-Masked, Placebo-Controlled Clinical Study of the Efficacy and Safety of Visomitin Eye Drops in Patients with Dry Eye Syndrome. Adv. Ther. 2015, 32, 1263–1279. [Google Scholar] [CrossRef]
- Gusev, E.A.; Chemodanov, D.V.; Sungurov, V.A.; Neverovsky, E.A.; Grebenchikov, O.A.; Likhvantsev, V.V. Mitochondria-targeted antioxidants in the prevention of the cornea erosion when performing surgery under general anesthesia. Anesteziol. Reanimatol. 2017, 61, 224–227. [Google Scholar]
- McClellan, S.A.; Wright, R.; Muhammed, F.; Hazlett, L.D. Impact of Airborne Exposure to PM(10) Increases Susceptibility to P. aeruginosa Infection. Int. J. Environ. Res. Public Health 2024, 21, 722. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, H.; Katra, I.; Koutrakis, P.; Friger, M.D. Contribution of dust storms to PM10 levels in an urban arid environment. J. Air Waste Manag. Assoc. 2014, 64, 89–94. [Google Scholar] [CrossRef]
- Why Is Beijing’s Air Quality So Bad Again? The Economist. 15 March 2021. Available online: https://www.economist.com/the-economist-explains/2021/03/15/why-is-beijings-air-quality-so-bad-again (accessed on 30 October 2025).
- Desk, W.W. Delhi Air Quality Dips Beyond ‘Severe’ Due to Dust Storm. India News News. 01:02 IST 13 June 2018. Available online: https://www.wionews.com/india-news/delhi-air-quality-dips-beyond-severe-due-to-dust-storm-144178 (accessed on 30 October 2025).
- Zernii, E.Y.; Gancharova, O.S.; Baksheeva, V.E.; Golovastova, M.O.; Kabanova, E.I.; Savchenko, M.S.; Tiulina, V.V.; Sotnikova, L.F.; Zamyatnin, A.A., Jr.; Philippov, P.P.; et al. Mitochondria-Targeted Antioxidant SkQ1 Prevents Anesthesia-Induced Dry Eye Syndrome. Oxid. Med. Cell. Longev. 2017, 2017, 9281519. [Google Scholar] [CrossRef]
- Hazlett, L.D.; McClellan, S.A.; Rudner, X.L.; Barrett, R.P. The Role of Langerhans Cells in Pseudomonas aeruginosa Infection. Investig. Ophthalmol. Vis. Sci. 2002, 43, 189–197. [Google Scholar]
- Somayajulu, M.; McClellan, S.A.; Muhammed, F.; Wright, R.; Hazlett, L.D. PM(10) and Pseudomonas aeruginosa: Effects on corneal epithelium. Front. Cell. Infect. Microbiol. 2023, 13, 1240903. [Google Scholar] [CrossRef] [PubMed]
- Somayajulu, M.; Wright, R.; Muhammed, F.; McClellan, S.A.; Ibrahim, A.; Hazlett, L.D. PM(10) dysregulates epithelial barrier function in human corneal epithelial cells that is restored by antioxidant SKQ1. Toxicol. Appl. Pharmacol. 2024, 492, 117122. [Google Scholar] [CrossRef] [PubMed]
- Upaphong, P.; Thonusin, C.; Wanichthanaolan, O.; Chattipakorn, N.; Chattipakorn, S.C. Consequences of exposure to particulate matter on the ocular surface: Mechanistic insights from cellular mechanisms to epidemiological findings. Environ. Pollut. 2024, 345, 123488. [Google Scholar] [CrossRef]
- US Environmental Protection Agency (EPA). Particulate Matter (PM) Pollution; US Environmental Protection Agency (EPA): Washington, DC, USA, 2020.
- Dijkhoff, I.M.; Drasler, B.; Karakocak, B.B.; Petri-Fink, A.; Valacchi, G.; Eeman, M.; Rothen-Rutishauser, B. Impact of airborne particulate matter on skin: A systematic review from epidemiology to in vitro studies. Part. Fibre Toxicol. 2020, 17, 35. [Google Scholar] [CrossRef]
- Gokul, T.; Kumar, K.R.; Prema, P.; Arun, A.; Balaji, P.; Faggio, C. Particulate pollution and its toxicity to fish: An overview. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2023, 270, 109646. [Google Scholar] [CrossRef]
- Yang, Q.; Li, K.; Li, D.; Zhang, Y.; Liu, X.; Wu, K. Effects of fine particulate matter on the ocular surface: An in vitro and in vivo study. Biomed. Pharmacother. 2019, 117, 109177. [Google Scholar] [CrossRef]
- Xia, M.; Yang, Y.; Sun, J.; Huang, R.; Huang, Y.; Zhang, M.; Yao, X. Time-series analysis of the association between air pollution exposure and outpatient visits for dry eye disease: A case study in Zhengzhou, China. Front. Public Health 2024, 12, 1352057. [Google Scholar] [CrossRef]
- Cui, Y.H.; Hu, Z.X.; Gao, Z.X.; Song, X.L.; Feng, Q.Y.; Yang, G.; Li, Z.J.; Pan, H.W. Airborne particulate matter impairs corneal epithelial cells migration via disturbing FAK/RhoA signaling pathway and cytoskeleton organization. Nanotoxicology 2018, 12, 312–324. [Google Scholar] [CrossRef]
- Li, X.; Kang, B.; Eom, Y.; Zhong, J.; Lee, H.K.; Kim, H.M.; Song, J.S. SIRT1 Protects Against Particulate Matter-Induced Oxidative Stress in Human Corneal and Conjunctival Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2022, 63, 19. [Google Scholar] [CrossRef]
- Park, E.J.; Chae, J.B.; Lyu, J.; Yoon, C.; Kim, S.; Yeom, C.; Kim, Y.; Chang, J. Ambient fine particulate matters induce cell death and inflammatory response by influencing mitochondria function in human corneal epithelial cells. Environ. Res. 2017, 159, 595–605. [Google Scholar] [CrossRef]
- Fu, Q.; Lyu, D.; Zhang, L.; Qin, Z.; Tang, Q.; Yin, H.; Lou, X.; Chen, Z.; Yao, K. Airborne particulate matter (PM2.5) triggers autophagy in human corneal epithelial cell line. Environ. Pollut. 2017, 227, 314–322. [Google Scholar] [CrossRef]
- Pucadyil, T.J.; Chipuk, J.E.; Liu, Y.; O’Neill, L.; Chen, Q. The multifaceted roles of mitochondria. Mol. Cell 2023, 83, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, T.B. The Functional Impact of Mitochondrial Structure Across Subcellular Scales. Front. Physiol. 2020, 11, 541040. [Google Scholar] [CrossRef] [PubMed]
- Rickard, B.P.; Overchuk, M.; Chappell, V.A.; Kemal Ruhi, M.; Sinawang, P.D.; Nguyen Hoang, T.T.; Akin, D.; Demirci, U.; Franco, W.; Fenton, S.E.; et al. Methods to Evaluate Changes in Mitochondrial Structure and Function in Cancer. Cancers 2023, 15, 2564. [Google Scholar] [CrossRef]
- Lim, E.Y.; Kim, G.D. Particulate Matter-Induced Emerging Health Effects Associated with Oxidative Stress and Inflammation. Antioxidants 2024, 13, 1256. [Google Scholar] [CrossRef] [PubMed]
- Matei, E.; Râpă, M.; Mateș, I.M.; Popescu, A.F.; Bădiceanu, A.; Balint, A.I.; Covaliu-Mierlă, C.I. Heavy Metals in Particulate Matter-Trends and Impacts on Environment. Molecules 2025, 30, 1455. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Engen, P.A.; Soberanes, S.; Urich, D.; Forsyth, C.B.; Nigdelioglu, R.; Chiarella, S.E.; Radigan, K.A.; Gonzalez, A.; Jakate, S.; et al. Particulate matter air pollution causes oxidant-mediated increase in gut permeability in mice. Part. Fibre Toxicol. 2011, 8, 19. [Google Scholar] [CrossRef]
- Wittkopp, S.; Staimer, N.; Tjoa, T.; Gillen, D.; Daher, N.; Shafer, M.; Schauer, J.J.; Sioutas, C.; Delfino, R.J. Mitochondrial genetic background modifies the relationship between traffic-related air pollution exposure and systemic biomarkers of inflammation. PLoS ONE 2013, 8, e64444. [Google Scholar] [CrossRef]
- Chew, S.; Lampinen, R.; Saveleva, L.; Korhonen, P.; Mikhailov, N.; Grubman, A.; Polo, J.M.; Wilson, T.; Komppula, M.; Rönkkö, T.; et al. Urban air particulate matter induces mitochondrial dysfunction in human olfactory mucosal cells. Part. Fibre Toxicol. 2020, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhu, Z.Z.; Zhang, X.; Nordio, F.; Bonzini, M.; Schwartz, J.; Hoxha, M.; Dioni, L.; Marinelli, B.; Pegoraro, V.; et al. Airborne particulate matter and mitochondrial damage: A cross-sectional study. Environ. Health 2010, 9, 48. [Google Scholar] [CrossRef]
- Soberanes, S.; Misharin, A.V.; Jairaman, A.; Morales-Nebreda, L.; McQuattie-Pimentel, A.C.; Cho, T.; Hamanaka, R.B.; Meliton, A.Y.; Reyfman, P.A.; Walter, J.M.; et al. Metformin Targets Mitochondrial Electron Transport to Reduce Air-Pollution-Induced Thrombosis. Cell Metab. 2019, 29, 335–347.e5. [Google Scholar] [CrossRef]
- An, Z.; Liu, G.; Shen, L.; Qi, Y.; Hu, Q.; Song, J.; Li, J.; Du, J.; Bai, Y.; Wu, W. Mitochondrial dysfunction induced by ambient fine particulate matter and potential mechanisms. Environ. Res. 2024, 262, 119930. [Google Scholar] [CrossRef] [PubMed]
- Soberanes, S.; Urich, D.; Baker, C.M.; Burgess, Z.; Chiarella, S.E.; Bell, E.L.; Ghio, A.J.; De Vizcaya-Ruiz, A.; Liu, J.; Ridge, K.M.; et al. Mitochondrial Complex III-generated Oxidants Activate ASK1 and JNK to Induce Alveolar Epithelial Cell Death following Exposure to Particulate Matter Air Pollution. J. Biol. Chem. 2009, 284, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Kondratyeva, E.V.; Vitkina, T.I. Effect of Atmospheric Particulate Matter on the Functional State of Mitochondria. Russ. Open Med. J. 2023, 12, 106. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Gao, Y.; Kang, J.; Wang, W.; Yong, Y.L.; Qu, X.; Dang, X.; Shang, D.; Shao, Y.; et al. Fine particulate matter exposure disturbs autophagy, redox balance and mitochondrial homeostasis via JNK activation to inhibit proliferation and promote EMT in human alveolar epithelial A549 cells. Ecotoxicol. Environ. Saf. 2023, 262, 115134. [Google Scholar] [CrossRef]
- Morris, R.H.; Counsell, S.J.; McGonnell, I.M.; Thornton, C. Exposure to urban particulate matter (UPM) impairs mitochondrial dynamics in BV2 cells, triggering a mitochondrial biogenesis response. J. Physiol. 2024, 602, 2737–2750. [Google Scholar] [CrossRef]
- Delgado-Buenrostro, N.L.; Freyre-Fonseca, V.; Cuéllar, C.M.; Sánchez-Pérez, Y.; Gutierrez-Cirlos, E.B.; Cabellos-Avelar, T.; Orozco-Ibarra, M.; Pedraza-Chaverri, J.; Chirino, Y.I. Decrease in respiratory function and electron transport chain induced by airborne particulate matter (PM10) exposure in lung mitochondria. Toxicol. Pathol. 2013, 41, 628–638. [Google Scholar] [CrossRef]
- Lebiedzinska-Arciszewska, M.; Suski, J.; Bonora, M.; Pakula, B.; Pinton, P.; Duszynski, J.; Jakubek-Olszewska, P.; Wieckowski, M.R. The Relation Between Mitochondrial Membrane Potential and Reactive Oxygen Species Formation. Methods Mol. Biol. 2025, 2878, 133–162. [Google Scholar]
- Wang, Y.; Xiong, L.; Yao, Y.; Ma, Y.; Liu, Q.; Pang, Y.; Tang, M. The involvement of DRP1-mediated caspase-1 activation in inflammatory response by urban particulate matter in EA.hy926 human vascular endothelial cells. Environ. Pollut. 2021, 287, 117369. [Google Scholar]
- Jin, X.; Su, R.; Li, R.; Song, L.; Chen, M.; Cheng, L.; Li, Z. Amelioration of particulate matter-induced oxidative damage by vitamin c and quercetin in human bronchial epithelial cells. Chemosphere 2016, 144, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, X.; Li, X.; Lv, S.; Ma, R.; Qi, Y.; Abulikemu, A.; Duan, H.; Guo, C.; Li, Y.; et al. PM(2.5) triggered apoptosis in lung epithelial cells through the mitochondrial apoptotic way mediated by a ROS-DRP1-mitochondrial fission axis. J. Hazard. Mater. 2020, 397, 122608. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Mitochondrial role in life and death of the cell. J. Biomed. Sci. 2000, 7, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, G.; Yang, Z. Mitochondrial superoxide mediates PM2.5-induced cytotoxicity in human pulmonary lymphatic endothelial cells. Environ. Pollut. 2020, 263, 114423. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar]
- Chevtzoff, C.; Yoboue, E.D.; Galinier, A.; Casteilla, L.; Daignan-Fornier, B.; Rigoulet, M.; Devin, A. Reactive oxygen species-mediated regulation of mitochondrial biogenesis in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 1733–1742. [Google Scholar] [CrossRef]
- Chen, L.; Qin, Y.; Liu, B.; Gao, M.; Li, A.; Li, X.; Gong, G. PGC-1α-Mediated Mitochondrial Quality Control: Molecular Mechanisms and Implications for Heart Failure. Front. Cell Dev. Biol. 2022, 10, 871357. [Google Scholar] [CrossRef]
- Gibellini, L.; Bianchini, E.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Natural Compounds Modulating Mitochondrial Functions. Evid. Based Complement. Alternat. Med. 2015, 2015, 527209. [Google Scholar]
- Meng, L.; Wu, G. Recent advances in small molecules for improving mitochondrial disorders. RSC Adv. 2023, 13, 20476–20485. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Friedhoff, L.T.; Petrov, A.; Ousler, G.W.; Watson, M.; Xue, Q.; Ngiam, M. Safety and Efficacy of First-in-Class mtROS scavenger SkQ1 for the Treatment of Dry Eye Disease: A Phase 3 Clinical Trial. Investig. Ophthalmol. Vis. Sci. 2019, 60, 6750. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.