Abstract
Inflammatory bowel disease (IBD), encompassing Crohn’s disease and ulcerative colitis, represents a chronic inflammatory condition of the gastrointestinal tract characterized by immune dysregulation, oxidative stress, and impaired epithelial barrier function. Among the complex molecular networks involved, the transcription factor nuclear factor erythroid 2–related factor 2 (Nrf2) has emerged as a central regulator of redox balance, anti-inflammatory signaling, and mucosal homeostasis. This review provides a comprehensive overview of the pathogenic role of oxidative stress in IBD, detailing the sources and consequences of reactive oxygen species (ROS) accumulation, and the mechanisms by which Nrf2 activation counters these effects. We discuss the dual regulation of Nrf2 through Keap1-dependent and Keap1-independent pathways, its influence on epithelial integrity, immune cell polarization, microbiota composition, and its paradoxical role in IBD-associated tumorigenesis and fibrosis. Furthermore, we highlight emerging therapeutic strategies aimed at modulating Nrf2 activity via pharmacologic agents, dietary phytochemicals, and probiotics. Taken together, these insights position Nrf2 as a pivotal therapeutic target in IBD, offering new avenues for restoring epithelial resilience, mitigating chronic inflammation, and improving clinical outcomes.
1. Introduction
Inflammatory bowel disease represents a chronic relapsing inflammatory disorder of the gastrointestinal tract driven by complex interactions among genetics, immunity, microbiota, and environmental influences. Despite significant advances in understanding immune and microbial mechanisms, the contribution of oxidative stress and its regulatory network, particularly the Nrf2/Keap1 signaling axis, remains incompletely defined. The existing literature highlights oxidative stress as a key amplifier of mucosal injury and immune dysregulation, yet the precise molecular links between redox imbalance, epithelial barrier failure, immune modulation, and intestinal fibrosis or tumorigenesis are insufficiently integrated. This review aims to synthesize current experimental and clinical evidence on the Nrf2-centered redox network in IBD, outlining its crosstalk with inflammatory and metabolic pathways, its physiological and pathological roles in intestinal homeostasis, and its translational relevance. To achieve this, comprehensive literature analysis was performed using recent peer-reviewed publications, with emphasis on mechanistic, preclinical, and therapeutic data that collectively illustrate the evolving understanding of Nrf2 as a promising target in IBD management.
2. IBD at a Glance: Phenotypes, Diagnosis, and Global Burden
Inflammatory bowel disease comprises two related but distinct entities: Crohn’s disease (CD) and ulcerative colitis (UC), that present with chronic, relapsing inflammation of the gastrointestinal tract [1]. The hallmark that separates them is the depth and distribution of tissue injury. CD produces transmural inflammation that can affect any segment from mouth to anus, with a predilection for the terminal ileum [2]. UC is limited to the colon and primarily involves the mucosa in a continuous pattern starting from the rectum [3]. These anatomic and histologic differences underlie divergent clinical courses and complications [4].
Diagnosis rests on the integration of clinical assessment with endoscopy, imaging, and histopathology [4]. Endoscopy and biopsies define lesion distribution and depth, while cross-sectional imaging captures transmural damage and skip lesions typical of CD [5,6,7]. Systemic inflammation can extend beyond the gut and manifest as extraintestinal features, emphasizing that IBD is a multisystem disease [8]. In practice, the mechanistic link is straightforward: sustained mucosal injury in UC drives bloody diarrhea and urgency, whereas transmural penetration in CD fosters strictures, fistulas, and abscesses [9].
IBD can occur at any age, but both disorders most often debut in late adolescence or early adulthood [10]. UC is frequently diagnosed several years later than CD on average [11]. This age pattern has clinical implications: early-life onset coincides with critical periods for growth, education, and family planning, magnifying disease burden even when symptoms wax and wane [12].
Epidemiological studies reveal that the rates are high and continue to rise across the globe [13]. While Western nations still account for the highest overall prevalence, affecting roughly 0.3% of the population, the most rapid growth is now being observed in newly industrialized regions such as East Asia [14]. The burden remains significant in countries with high and medium–high socio-demographic indices and is steadily increasing in those with low and medium indices [15]. This trend reflects broader global changes in environmental conditions, lifestyle patterns, and access to healthcare [16,17].
Outcomes mirror underlying pathology. CD carries higher all-cause mortality than the general population and often necessitates surgery; over a lifetime, most patients require at least one intestinal resection, and a subset will need a permanent stoma [18,19]. UC, while confined to the colon, still leads 10–30% of patients to colectomy [20]. Chronic, uncontrolled inflammation in both conditions increases the risk of intestinal cancer, consistent with the inflammation–dysplasia–carcinoma sequence [21,22]. These trajectories underscore a central principle: the depth and extent of inflammation shape complications, surgical needs, and long-term malignancy risk, guiding surveillance and therapeutic strategy [23,24].
Etiology and Treatment Landscape of IBD: Interplay of Genetics, Immunity, Microbiota, and Redox Control
Inflammatory bowel disease arises from a multifactorial process in which genetic susceptibility interacts with immune dysregulation, environmental exposures, and intestinal dysbiosis; the precise initiating cause remains undefined [17,25]. Genome-wide studies implicate over a hundred risk loci that converge on pathways governing innate and adaptive immunity, epithelial barrier function, microbial sensing, and cellular stress responses, including oxidative and electrophilic stress [17,26]. Variants affecting redox control (e.g., regulators of antioxidant defenses and electrophile response) can lower the threshold for tissue injury, intensify cytokine signaling, and weaken mucosal repair, thereby amplifying inflammation [27,28].
The gut microbiota serves as both trigger and amplifier of disease activity [29]. Microbial antigens and metabolites influence the host immune system by engaging pattern recognition receptors and metabolic checkpoints [30]. At the same time, environmental factors such as diet, smoking, medications, and early-life exposures like breastfeeding alter the composition and activity of the gut microbiota [31,32]. These influences can modify gene expression via epigenetic mechanisms (DNA methylation, histone marks, and microRNAs), linking external cues to persistent changes in epithelial and immune cell behavior [33,34]. The result is a self-reinforcing loop in which barrier defects permit microbial translocation, innate and adaptive responses escalate, and redox imbalance (excess reactive oxygen species with inadequate antioxidant compensation) sustains tissue damage [27,35].
Within this network, oxidative stress is not merely a by-product but a driver of pathophysiology [35]. Reactive oxygen species produced by activated immune cells and stressed epithelial cells oxidize lipids and proteins, weaken tight junctions, and amplify pro-inflammatory signaling pathways, including nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) [36,37]. In contrast, protective cellular mechanisms, especially those regulated by electrophile and antioxidant sensors, support the detoxification of harmful compounds, maintain the balance of glutathione, and help bring inflammation to an end [38,39]. When genetic variation or environmental pressure blunts these defenses, inflammation persists and healing is delayed. The multifactorial pathogenesis of inflammatory bowel disease, illustrating the interplay between genetic, immune, microbial, and environmental determinants, is schematically summarized in Figure 1.
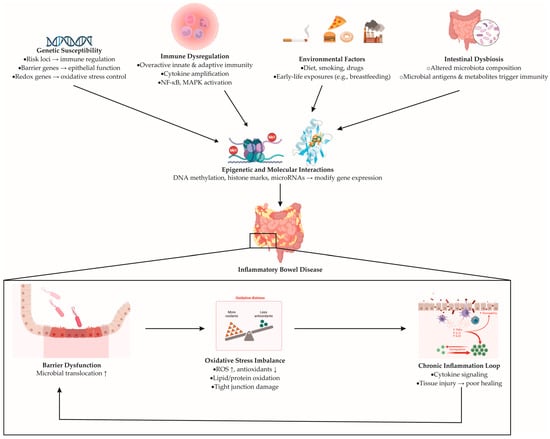
Figure 1.
Proposed mechanisms underlying the pathogenesis of inflammatory bowel disease. Genetic susceptibility (risk variants affecting immune regulation, epithelial barrier genes, and redox-related genes), immune dysregulation (overactivation of innate and adaptive immunity), environmental factors (diet, smoking, medications, early-life exposures), and intestinal dysbiosis (altered gut microbiota composition and microbial metabolites) converge to induce epigenetic and molecular alterations (DNA methylation, histone modifications, and microRNA regulation). These processes drive persistent immune activation and epithelial dysfunction, ultimately leading to IBD development. The downstream cycle includes barrier disruption, oxidative stress imbalance, and chronic inflammation. Barrier injury promotes microbial translocation; oxidative stress—characterized by increased ROS (reactive oxygen species) and reduced antioxidants—amplifies cytokine signaling via NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) and MAPK (mitogen-activated protein kinase) pathways, while inadequate antioxidant defenses impair tissue repair.
Current therapy targets immune activation and mucosal injury but remains imperfect [40]. Standard regimens include aminosalicylates, corticosteroids, immunosuppressants, and biologics aimed at key cytokines or trafficking pathways [41]. Although effective for induction and maintenance in many patients, these agents are limited by primary non-response, loss of response, adverse effects, and treatment-limiting toxicity [42]. These constraints underscore the need for strategies that also restore barrier function and rebalance redox and epithelial stress pathways—complementing immunomodulation to achieve durable remission.
3. Oxidative Stress: Sources, Molecular Targets, and Antioxidant Control
Oxidative stress is a disruption of cellular redox balance that occurs when the generation of reactive oxygen species (ROS) exceeds the capacity of antioxidant defenses to neutralize them [43]. ROS comprise radical and non-radical oxygen-containing species produced continuously in aerobic cells, with mitochondria as a major source during oxidative phosphorylation [43]. At physiological levels, ROS function as second messengers that tune pathways governing proliferation, differentiation, apoptosis, host defense, and inflammation [44]. When ROS accumulate, their high reactivity drives indiscriminate chemistry with nearby macromolecules, damaging membrane lipids, proteins, and nucleic acids and disturbing membrane fluidity and cell volume regulation [45].
The principal pro-oxidant species include superoxide (O2•−), hydrogen peroxide (H2O2), and the hydroxyl radical (•OH) [45]. Other important contributors include singlet oxygen, hypochlorous acid (HOCl), ozone (O3), chloramines, and lipid peroxyl and hydroperoxide species [46]. Many of these reactive molecules are produced through enzymatic reactions in immune cells, such as those involving nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and myeloperoxidase, or through metal-catalyzed redox cycling [47]. A moderate level of ROS supports the destruction of microbiota, wound healing, and tissue repair [48,49,50]. However, when ROS production becomes excessive, it overwhelms the body’s detoxification systems, promotes lipid peroxidation and protein carbonylation, causes DNA strand breaks and base alterations, and activates pro-inflammatory signaling pathways that worsen tissue damage [51].
Cells counterbalance these pressures through layered antioxidant networks. Enzymatic defense systems form the main pathway for detoxification [52]. Superoxide dismutases convert superoxide anions into hydrogen peroxide, while catalase and glutathione peroxidases further break down hydrogen peroxide and lipid hydroperoxides into water or their corresponding alcohols [53,54,55]. These reactions rely on catalytic heme groups or glutathione as an electron donor [53,54,55]. Non-enzymatic antioxidants complement these enzymes by buffering electrophiles and free radicals in cellular and extracellular spaces [39]. Key molecules include reduced glutathione (GSH), ascorbate (vitamin C), and α-tocopherol (vitamin E), which participate in tightly coupled recycling cycles that preserve redox homeostasis [39,56].
In the intestine, persistent exposure to luminal microbiota and diet-derived oxidants imposes recurrent redox stress [35]. When ROS generation in inflamed mucosa outpaces local antioxidant capacity, oxidative damage extends beyond the epithelium into deeper layers of the bowel wall [57,58]. The resulting loss of barrier integrity, accumulation of oxidized lipids and proteins, and activation of redox-sensitive inflammatory pathways collectively shift the tissue from controlled signaling to chronic injury, setting the stage for sustained immune activation characteristic of inflammatory bowel disease [27].
3.1. Physiological Roles of Free Radicals and Reactive Species
Although often associated with tissue damage, free radicals are indispensable for maintaining normal physiological functions [51]. When generated at controlled levels, ROS and RNS act as essential mediators in cellular and systemic homeostasis [38,59]. In the immune system, for instance, phagocytes such as neutrophils and macrophages deliberately produce superoxide and other radicals via the NADPH oxidase complex [60]. These reactive species are stored and released during phagocytosis to destroy invading microorganisms [60]. The clinical importance of this mechanism is evident in chronic granulomatous disease, where defective NADPH oxidase activity prevents superoxide formation, resulting in recurrent and persistent infections [61].
Beyond their antimicrobial function, free radicals contribute to intracellular signaling networks. Nonphagocytic isoforms of NADPH oxidase generate low levels of ROS in fibroblasts, endothelial cells, vascular smooth muscle cells, cardiac myocytes, and thyroid tissue, where they serve as second messengers that fine-tune gene expression, proliferation, and adaptive responses [62]. Among these signaling molecules, NO is the most recognized. Produced by nitric oxide synthases, NO regulates vascular tone and platelet aggregation, supports synaptic transmission in the nervous system, and mediates cytotoxic defense against pathogens and tumor cells [63,64]. Furthermore, moderate ROS generation can activate mitogenic pathways, promoting cell growth and tissue repair [65].
Thus, while excessive or chronic radical exposure drives oxidative stress and cellular injury, their physiological presence at low concentrations is vital for immune competence, vascular homeostasis, and signal transduction. The balance between beneficial signaling and pathological oxidation underscores the importance of redox regulation in health and disease.
3.2. Oxidative Stress as a Central Pathogenic Driver in IBD: Molecular Mechanisms and Clinical Relevance
Redox homeostasis is one of the core biological systems disturbed in inflammatory bowel disease [27]. Under physiological conditions, epithelial and immune cells continually balance oxidant production with enzymatic and non-enzymatic antioxidant defenses [66]. In IBD, this equilibrium collapses [27]. The chronically inflamed mucosa is exposed to oxidants originating from diet, microbiota, and infiltrating neutrophils and macrophages, which generate abundant ROS and reactive nitrogen species [27,67,68]. When antioxidant buffering becomes insufficient, the entire redox network tilts toward oxidative and nitrosative stress, creating an environment that favors tissue injury rather than healing [37,69].
A disrupted redox balance undermines the structural and immunological integrity of the intestinal barrier. Excess ROS not only damage lipids, proteins, and DNA but also reshape epithelial signaling programs [27]. Lipid peroxidation products such as malondialdehyde and 4-hydroxynonenal disturb tight junction organization, increase mucosal permeability, and facilitate bacterial translocation [70,71]. As epithelial defenses weaken, luminal antigens gain access to immune compartments, amplifying Toll-like receptor signaling and promoting sustained recruitment of neutrophils and monocytes [27]. This establishes a self-reinforcing inflammatory loop: barrier leakage increases antigen load, immune activation escalates, and ROS generation rises even further.
Protein and DNA oxidation add another layer of dysfunction [72,73]. Oxidative modifications impair protein structure, enzyme activity, and cellular redox sensors, while DNA lesions interfere with cell-cycle regulation and increase mutational burden—mechanisms that partly explain the heightened risk of dysplasia and colorectal cancer in long-standing IBD [27].
At the signaling level, oxidative stress acts as both a trigger and amplifier of inflammation [27]. Nuclear factor κB and MAPKs act as central regulators that translate oxidative signals into gene expression programs, driving the production of cytokines, chemokines, adhesion molecules, and enzymes such as inducible nitric oxide synthase (iNOS) and myeloperoxidase [47,74]. This gene expression pattern sustains recruitment of neutrophils and monocytes and reinforces local production of ROS/RNS, cementing chronic inflammation [74]. At the same time, the cytokine environment shifts toward tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β), and interleukin 8 (IL-8), which further enhance the recruitment of immune cells and intensify epithelial stress [75].
In parallel, the Nrf2 pathway—responsible for upregulating antioxidant and cytoprotective genes—is often insufficiently activated or overwhelmed during active disease, further widening the gap between oxidant load and defensive capacity [76]. The interplay between NF-κB–driven inflammation and inadequate Nrf2-mediated counter-regulation forms a molecular core of redox imbalance in IBD [27].
Evidence from patients supports this mechanism. The activities of superoxide dismutase and glutathione peroxidase are often diminished in both the intestinal mucosa and the blood [77,78,79]. At the same time, levels of non-enzymatic antioxidants such as vitamins A, C, and E, as well as beta-carotene, are reduced, reflecting an overall imbalance in the body’s redox status [66,79]. Oxidative injury markers extend beyond the bowel, with elevated H2O2 detected in circulating monocytes and lymphocytes during active disease [79]. Clinical indicators reflect these biological changes. Composite measures of oxidative stress show a positive correlation with C-reactive protein (CRP) and fibrinogen, while reduced levels of plasma free thiols, which are important substrates for ROS, are associated with more severe inflammation and worse clinical outcomes [78,79]. The cascade of oxidative stress–driven mucosal injury, illustrating the interplay between oxidant overproduction, barrier failure, and immune activation, is summarized in Figure 2.
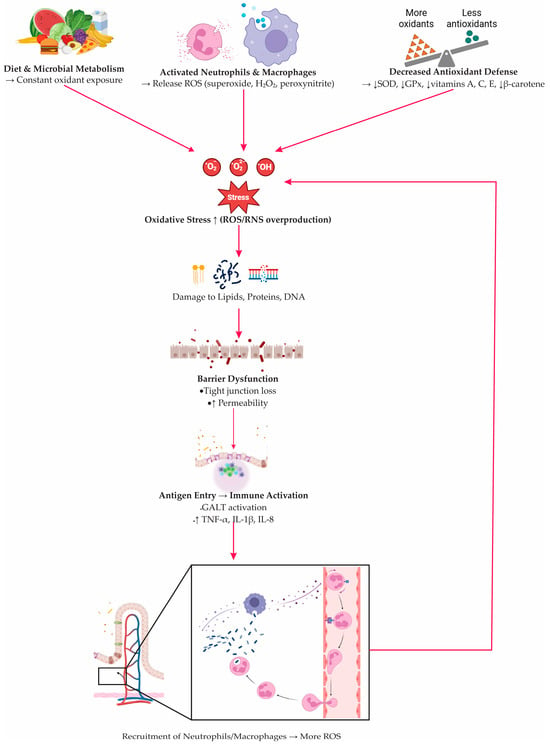
Figure 2.
Oxidative stress–driven epithelial injury and immune activation in inflammatory bowel disease. Dietary and microbial oxidants, together with ROS/RNS released by activated neutrophils and macrophages (ROS: reactive oxygen species; RNS: reactive nitrogen species), overwhelm epithelial redox balance due to reduced antioxidant defenses (SOD: superoxide dismutase; GPx: glutathione peroxidase; vitamins A, C, E, β-carotene). Excess oxidants damage lipids, proteins, and DNA, causing tight-junction loss and increased permeability. Translocated antigens activate GALT (gut-associated lymphoid tissue) and stimulate cytokine production (TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1β; IL-8: interleukin-8), promoting further immune-cell recruitment and reinforcing the oxidative–inflammatory loop.
4. Nrf2–Keap1 Pathway: Central Regulator of Antioxidant and Anti-Inflammatory Defense
The Nuclear factor erythroid 2–related factor 2 (Nrf2 ) is a master transcription factor responsible for coordinating the cellular antioxidant response to oxidative and electrophilic stress [80]. Under normal conditions, Nrf2 is bound in the cytoplasm to Kelch-like ECH-associated protein 1 (Keap1), which targets it for degradation by the proteasome [81]. When cells are exposed to stress signals such as ROS, electrophilic compounds, or metabolites generated during inflammation, structural changes occur in Keap1 that release Nrf2 [82]. The liberated Nrf2 then moves into the nucleus to initiate protective gene expression. There, Nrf2 binds to antioxidant response elements (AREs) in the promoters of target genes, initiating the transcription of cytoprotective molecules [82,83]. The dual role of the Nrf2–Keap1 signaling axis under basal and stress conditions, and its downstream antioxidant and anti-inflammatory targets, is illustrated in Figure 3.
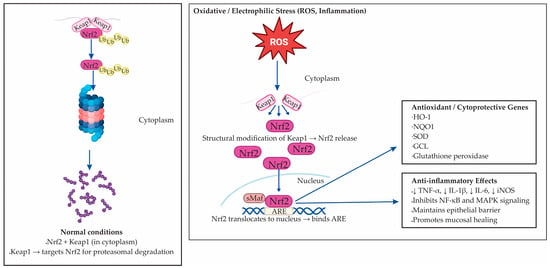
Figure 3.
The Nrf2–Keap1 signaling pathway in oxidative and electrophilic stress. Under normal conditions, nuclear factor erythroid 2–related factor 2 (Nrf2) remains bound to Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm, where Keap1 targets Nrf2 for ubiquitination and proteasomal degradation. Upon exposure to oxidative or electrophilic stress, structural modification of Keap1 cysteine residues causes Nrf2 release and nuclear translocation. In the nucleus, Nrf2 forms heterodimers with small Maf proteins and binds to antioxidant response elements (AREs), initiating transcription of cytoprotective and antioxidant genes, including heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO1), superoxide dismutase (SOD), glutamate–cysteine ligase (GCL), and glutathione peroxidase. Activation of Nrf2 also suppresses pro-inflammatory mediators (TNF-α, IL-1β, IL-6, iNOS), inhibits NF-κB and MAPK signaling, preserves epithelial barrier integrity, and promotes mucosal healing.
Nrf2 activation leads to upregulation of a broad range of enzymes involved in redox balance, detoxification, and cellular repair [82,84]. These include heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO1), superoxide dismutase (SOD), glutamate–cysteine ligase (GCL), and glutathione peroxidase. Together, they re-establish glutathione homeostasis, neutralize ROS, and prevent further oxidative damage [82,84]. In parallel, Nrf2 suppresses the expression of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and iNOS, thereby exerting anti-inflammatory effects [85].
The relevance of Nrf2 extends beyond oxidative detoxification. Experimental evidence shows that activation of Nrf2 can suppress NF-κB signaling, maintain the integrity of the epithelial barrier, and support mucosal regeneration [86,87]. These functions are particularly relevant to the pathogenesis and treatment of IBD [87]. In murine models and human samples, enhanced Nrf2 activity correlates with reduced inflammatory infiltration and improved histological scores [88,89,90]. Conversely, impaired Nrf2 signaling is associated with sustained oxidative damage, increased permeability of the intestinal barrier, and exaggerated immune responses [87,90].
Beyond the gut, the Nrf2 pathway has shown therapeutic promise in other inflammation-driven conditions, including neurodegeneration, aging, and carcinogenesis [91,92,93]. Its dual capacity to neutralize ROS and modulate immune signaling makes Nrf2 a strategic target in diseases characterized by chronic inflammation and redox imbalance [82]. In IBD specifically, pharmacological or dietary activation of Nrf2 may complement conventional immunosuppressive therapies by reinforcing the mucosal antioxidant shield and breaking the vicious cycle of oxidative stress and immune activation [94,95].
4.1. Structural Complexity and Activation Mechanism of Nrf2 in Redox Regulation
Nuclear factor erythroid 2–related factor 2, encoded by the NFE2L2 gene, is a central transcriptional regulator of antioxidant defense and redox balance [96]. Originally cloned from the human leukemia K562 cell line, Nrf2 belongs to the Cap’n’Collar (CNC) subfamily of basic leucine zipper (bZIP) transcription factors, which also includes Nrf1, Nrf3, and the NF-E2 p45 subunit [97,98,99]. The human Nrf2 protein consists of 605 amino acids and contains seven highly conserved functional domains, known as Nrf2 –ECH homology (Neh1–Neh7), each of which plays a specific role in regulating the stability, localization, and transcriptional activity of the protein [100,101]. The modular organization of Nrf2 and its conserved Neh domains, which mediate DNA binding, protein stability, and transcriptional activation, is illustrated in Figure 4.
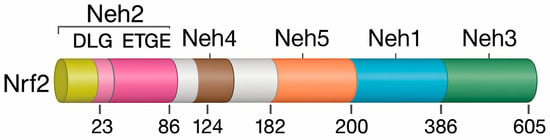
Figure 4.
Structural organization of human Nrf2 and its conserved Neh domains. Nrf2 contains seven Nrf2–ECH homology (Neh) domains with distinct regulatory functions. Neh2 at the N-terminus includes the DLG and ETGE motifs that mediate Keap1 binding. Neh4 and Neh5 function as transactivation modules, while Neh1 contains the bZIP (basic leucine zipper) region required for small Maf heterodimerization and ARE binding. Neh3 at the C-terminus contributes to transcriptional activation. Neh6 regulates Keap1-independent degradation via β-TrCP, and Neh7 represses Nrf2 activity through interaction with RXRα (retinoid X receptor-alpha).
The Neh1 domain contains the bZIP structure required for dimerization with small musculoaponeurotic fibrosarcoma (Maf) proteins and DNA binding [102]. Neh2, located near the N-terminus, is the principal regulatory site for Keap1-dependent ubiquitination via its DLG and ETGE motifs, thereby controlling basal Nrf2 degradation [96,103]. The C-terminal Neh3 domain supports transcriptional activation through interactions with chromatin-remodeling factors such as CHD6, while Neh4 and Neh5 function as transactivation domains that recruit coactivators essential for antioxidant gene induction [96].
Neh6 domain, enriched in serine residues, mediates Keap1-independent proteasomal degradation via β-TrCP [104]. Conversely, Neh7 binds retinoid X receptor-α (RXRα), which suppresses Nrf2 by blocking its transcriptional activity, revealing cross-talk between nuclear receptors and redox pathways [104,105].
Under basal conditions, Nrf2 is sequestered in the cytoplasm by Keap1, which acts as an adaptor for the Cul3–Rbx1 ubiquitin ligase complex that continuously targets Nrf2 for proteasomal degradation [106]. Oxidative or electrophilic stress modifies critical cysteine residues in Keap1, disrupting Keap1–Nrf2 binding and preventing ubiquitination [82,107]. Stabilized Nrf2 accumulates, enters the nucleus, dimerizes with Maf proteins, and binds ARE sequences to initiate transcription [96].
Nrf2 activation induces a broad cytoprotective program, including detoxification enzymes (GST, NQO1), antioxidant systems (SOD, GPx), and HO-1, which collectively enhance resistance to oxidative injury, inflammation, and apoptosis [76,82]. Through this tightly regulated architecture, Nrf2 acts as a central redox-sensing switch that maintains cellular homeostasis—an especially important function in IBD, where Nrf2-mediated defenses may mitigate mucosal damage and chronic inflammation [76].
4.2. Mechanisms of Nrf2 Activation: Keap1-Dependent and Keap1-Independent Pathways
Nrf2–ARE signaling can be activated through both Keap1-dependent and Keap1-independent mechanisms, each converging on the induction of cytoprotective genes [81].Under basal conditions, Keap1 binds Nrf2 via its DLG and ETGE motifs and targets it for Cul3–Rbx1–mediated ubiquitination and proteasomal degradation [108,109]. Oxidative or electrophilic stress modifies key cysteine residues of Keap1—particularly Cys151, Cys273, and Cys288—disrupting its E3 ligase activity and allowing Nrf2 to accumulate, translocate to the nucleus, dimerize with small Maf proteins, and activate ARE-driven transcription [80,82,110,111,112,113].
Nrf2 can also be activated independently of Keap1 through phosphorylation by several kinases [114,115]. Protein kinase C (PKC) promotes Nrf2 release and nuclear import via Ser40 phosphorylation [82,116], while adenosine monophosphate–activated protein kinase (AMPK) enhances Nrf2 nuclear accumulation and antioxidant gene expression through Ser550 modification, contributing to improved stress tolerance [82,116,117,118,119,120,121]. Mitogen-activated protein kinases (MAPKs), including extracellular signal–regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38—further modulate Nrf2 stability and transactivation potential, although their exact phosphorylation sites are still being defined [119,120]. Together, these pathways integrate redox, metabolic, and stress signals to fine-tune Nrf2 activation and antioxidant defense.
Together, the Keap1-dependent sensor system and Keap1-independent kinase pathways allow for flexible, multilayered regulation of Nrf2. This coordination enables cells to quickly respond to various stress signals, from oxidative injury to metabolic disturbances, by activating a comprehensive antioxidant defense program. The central components and activation routes of the Nrf2–Keap1 pathway, along with their molecular effects and therapeutic relevance in inflammatory bowel disease, are summarized in Table 1.

Table 1.
Nrf2–Keap1 pathway: activation mechanisms, molecular targets, and therapeutic significance in inflammatory bowel disease.
5. Physiological Role of Nrf2 in Intestinal Development and Barrier Function
The Nrf2–ARE signaling pathway plays an essential role not only in cellular redox defense but also in intestinal morphogenesis, epithelial homeostasis, and barrier maintenance [121,122]. During embryonic development, Nrf2 expression is spatially and temporally regulated. Elevated levels of Nrf2 mRNA were detected in the hindgut during late gestation, particularly between days 14.5 and 18.5 of mouse embryogenesis, in contrast to declining expression in organs such as the lung and heart [121,123,124]. This suggests a developmentally restricted role of Nrf2 in intestinal formation. Indeed, Nrf2 transcriptional activity is necessary for the establishment of normal epithelial architecture, including villus formation, crypt development, and lineage specification of absorptive and secretory cells [121,125].
At the molecular level, Nrf2 orchestrates intestinal epithelial differentiation by modulating two key developmental pathways: Notch and Wnt [76]. The Notch1 promoter contains a functional ARE, enabling direct transcriptional regulation by Nrf2 [126]. Loss of Nrf2 impairs Notch1 signaling, which disrupts enterocyte maturation [121]. Similarly, the Notch effector Math1 appears to be indirectly repressed by Nrf2, suggesting a balancing role between proliferation and differentiation [76,127]. On the other hand, Wnt signaling interacts bidirectionally with Nrf2, as β-catenin can activate Nrf2, while Nrf2 negatively regulates β-catenin, creating a feedback loop that fine-tunes epithelial cell fate decisions, especially the differentiation into Paneth cells critical for barrier integrity [94,128,129].
In adult tissue, Nrf2 maintains intestinal barrier function by preserving tight junctions (TJs) and limiting epithelial permeability [86,87]. The epithelial layer comprises tight junctions, adherens junctions, and desmosomes. Among TJs, claudins, occludin, and zonula occludens 1 (ZO-1) are critical for sealing the paracellular space [130]. Nrf2 activation enhances the expression of TJ proteins, including Claudin-4 and Occludin, through ARE-mediated transcription [76,131]. For example, activation of the ERK/Nrf2/HO-1 axis leads to increased levels of Occludin and ZO-1, whereas Nrf2-deficient mice exhibit downregulation of Claudin-4 and greater susceptibility to epithelial disruption [76,90].
Beyond transcriptional activation of junctional proteins, Nrf2 contributes to barrier maintenance via indirect mechanisms such as prevention of epithelial apoptosis and promotion of autophagy [76]. Recent studies have demonstrated that Nrf2-induced autophagy facilitates degradation of Claudin-2, a pore-forming TJ protein upregulated in IBD and associated with barrier leakiness [132]. Additionally, Nrf2 protects mitochondrial DNA, suppresses mitochondrial ROS, and reduces epithelial apoptosis in ischemia/reperfusion models, further underscoring its role in preserving epithelial viability and metabolic stability [133].
Nrf2 also regulates intestinal contractility by modulating signaling pathways in smooth muscle cells [134]. It influences acetylcholine receptor signaling, calcium flux, and the activity of second messengers [90,134]. Mice lacking Nrf2 show increased oxidation of tetrahydrobiopterin, reduced nitric oxide bioavailability, and impaired nitrergic relaxation, pointing to a role in neuromuscular coordination and gastric motility. Such changes may predispose to gastroparesis and delayed transit [90,134].
Moreover, recent findings implicate Nrf2 in the regulation of energy metabolism within the gastrointestinal epithelium [135]. In mice lacking Nrf2, the expression of mitochondrial proteins such as cytochrome c oxidase subunit IV (COX IV) and genes responsible for adenosine triphosphate (ATP) production is reduced, impairing the energy-dependent maintenance of tight junctions [136]. This is accompanied by increased DNA oxidative damage, reduced transepithelial electrical resistance (TEER), and enhanced permeability in models of gastroesophageal reflux disease (GERD) [136]. ChIP assays confirmed direct binding of Nrf2 to the Claudin-4 promoter, linking redox control to epithelial bioenergetics and structural integrity [136]. The developmental and physiological roles of Nrf2 in intestinal morphogenesis, barrier maintenance, and metabolic regulation are summarized in Figure 5.
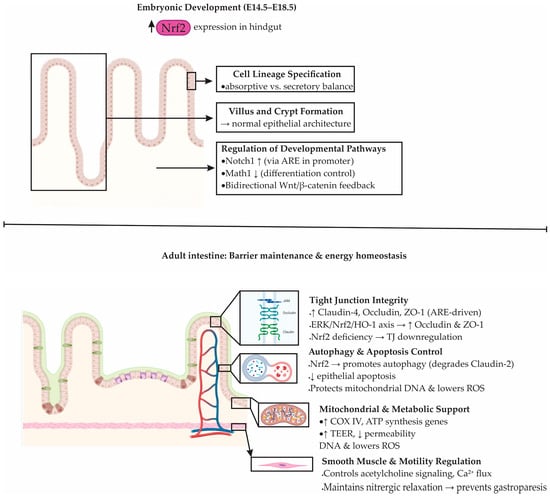
Figure 5.
Physiological role of Nrf2 in intestinal development, epithelial homeostasis, and barrier maintenance. During embryogenesis (E14.5–E18.5), Nrf2 regulates villus–crypt formation, epithelial lineage balance, and developmental pathways including Notch1, Math1, and Wnt/β-catenin. In the adult intestine, Nrf2 maintains homeostasis by supporting tight-junction (TJ) integrity through the ERK/Nrf2/HO-1 pathway (ERK: extracellular signal-regulated kinase; HO-1: heme oxygenase-1), promoting autophagy and limiting apoptosis, protecting mitochondrial DNA, and reducing ROS (reactive oxygen species). Nrf2 also enhances ATP production and transepithelial electrical resistance (TEER) while regulating acetylcholine signaling and Ca2+ flux in smooth muscle to ensure proper motility.
6. Crosstalk Between Nrf2 and Inflammatory Pathways in IBD: Mechanistic Insights and Cellular Context
Chronic intestinal inflammation, as seen in IBDs, is tightly linked to oxidative stress and redox imbalance [27]. The overproduction of ROS, a hallmark of intestinal inflammation, not only contributes to mucosal damage but also reinforces a cycle of persistent inflammation [27]. Reactive oxygen species activate redox-sensitive signaling pathways, including NF-κB, activator protein 1 (AP-1), and kinases such as MAPKs and phosphoinositide 3-kinase (PI3K). This activation stimulates the production of pro-inflammatory cytokines and further intensifies tissue injury [74,137]. This self-perpetuating cycle highlights the need for regulatory systems that can restrain oxidative stress and inflammatory signaling. Among these, the Nrf2-Keap1 pathway has emerged as a pivotal regulator of intestinal redox and immune homeostasis [76].
Upon oxidative insult, Nrf2 dissociates from its inhibitor Keap1 and translocates to the nucleus, where it orchestrates the expression of antioxidant enzymes such as HO-1, NQO1, and glutathione-related enzymes. These enzymes detoxify ROS and re-establish cellular homeostasis [76,138]. Notably, Nrf2 also indirectly suppresses inflammatory cascades by reducing ROS-mediated activation of NF-κB and by modulating upstream regulators such as IKKβ [139]. For example, Keap1 can interact with IKKβ, inhibiting IκBα degradation and preventing NF-κB nuclear translocation, thus attenuating the transcription of pro-inflammatory genes [139,140]. These regulatory loops position Nrf2 as both a sensor and modulator of intestinal inflammation.
In addition to its role in reducing inflammation, Nrf2 also regulates the polarization of immune cells. In macrophages, activation of Nrf2 decreases the pro-inflammatory M1 phenotype by lowering the expression of toll-like receptor 4 (TLR4), forkhead box protein O1 (FOXO1), and cytokines such as interleukin 1 beta (IL-1β) and interferon beta (IFN-β), while promoting the anti-inflammatory M2 phenotype. Through this dual action, Nrf2 helps limit tissue injury and supports the resolution of inflammation [141,142]. In contrast, Nrf2-deficient neutrophils exhibit exaggerated responses to LPS, with elevated levels of monocyte chemoattractant protein 1 (MCP-1), TNF-α, and ROS, further linking Nrf2 to immune regulation at the mucosal interface [142].
In addition to modulating cytokine production, Nrf2 regulates other inflammation-associated molecules, such as iNOS and COX-2, both of which are upregulated during intestinal inflammation. Studies show that Nrf2 activation suppresses the transcription of these enzymes in macrophages and dendritic cells [94,143,144]. Moreover, Nrf2 also affects extracellular matrix remodeling by regulating metalloproteinases (MMPs). Overexpression of NRF2 leads to reduced MMP-13 and MMP-9 levels, which has been linked not only to amelioration of inflammation in arthritis and ocular models but also to decreased cancer cell invasiveness via the Nrf2/HO-1 axis [145].
6.1. Nrf2 as a Key Guardian of the Intestinal Epithelial Barrier
The intestinal mucosal barrier is a complex defense system composed of mucus, epithelial cells, intercellular junctions, and commensal microbiota [130]. This multilayered structure separates luminal microbiota and toxins from host tissues, while still allowing controlled absorption of nutrients and water. Tight junctions, composed of proteins such as occludin, claudins, and members of the zonula occludens family, play a crucial role in controlling epithelial permeability and maintaining barrier integrity [130]. When barrier integrity is compromised, harmful substances and pathogens cross into the lamina propria, triggering immune activation and chronic inflammation. Such events are well documented in UC, where disruption of tight junctions amplifies mucosal injury and drives ulcer formation [146,147].
Mounting evidence shows that Nrf2 is indispensable for maintaining epithelial integrity. In models of DSS-induced colitis, expression of TJ proteins such as ZO-1 and claudin is markedly reduced, leading to increased permeability. Nrf2 activation restores their expression, thereby stabilizing the epithelial barrier [148,149]. Similarly, in LPS-induced epithelial damage, mitochondria-targeted antioxidants like MitoQ protect the barrier by stimulating Nrf2-dependent gene expression and reducing oxidative stress. These protective effects extend beyond the intestine [150]. In reflux esophagitis, Nrf2 binds directly to the claudin-4 promoter, enhancing its expression, whereas Nrf2 deficiency results in mitochondrial dysfunction, reduced claudin-4 levels, and impaired junctional integrity [136].
Nrf2 protects tight junctions not only by transcriptional regulation but also by suppressing epithelial apoptosis and promoting autophagy [94]. Mechanistic studies have shown that activation of the ERK/Nrf2/HO-1 axis triggers mitophagy, which alleviates oxidative stress and reinforces junctional proteins like ZO-1 and occluding [151]. Modulators of autophagy strongly influence this protective response, highlighting the dual role of Nrf2 in redox regulation and cellular quality control [122].
The functional role of Nrf2 in barrier defense has been validated across diverse disease models. In colonic inflammation induced by chronic kidney disease, treatment with the Nrf2 activator dh404 restored levels of occludin, claudin-1, and ZO-1, thereby repairing barrier integrity [152]. Likewise, in experimental colitis and systemic inflammatory models, genetic loss of Nrf2 consistently exacerbates permeability defects, whereas pharmacological or dietary activation of the pathway improves mucosal function [122].
6.2. Nrf2 as a Central Regulator of Intestinal Immune Homeostasis
The intestinal immune system plays a pivotal role in maintaining gut homeostasis, and its dysregulation is a hallmark of IBD [153,154]. Among the key molecular regulators in this context, Nrf2 has emerged as a critical modulator of both redox balance and immune cell function in the gastrointestinal tract [87].
Recent studies indicate that the activity of Nrf2 is closely linked to the differentiation and function of T helper (Th) cells [155,156,157]. When cluster of differentiation 4 positive (CD4+) T cells are activated, they develop into specialized subsets, including T helper 1 (Th1), T helper 2 (Th2), T helper 17 (Th17), and regulatory T cells (Tregs), each playing a distinct role in orchestrating the immune and inflammatory response [158]. Several studies have shown that Nrf2 activation skews this differentiation, favoring Th2 over Th1 responses [155,159,160]. For instance, the synthetic antioxidant tert-butylhydroquinone (tBHQ) suppresses Th1 cytokines like IFN-γ while promoting the expression of Th2-associated cytokines, such as IL-4 and IL-5 [159]. Moreover, Nrf2 activation appears to enhance IL-2 production, which is essential not only for Th2 polarization but also for the survival and stability of Treg cells, which help to maintain mucosal tolerance [160]. This suggests a dual immunoregulatory role for Nrf2, both in limiting excessive inflammation and in reinforcing epithelial tolerance mechanisms. Furthermore, through interaction with the aryl hydrocarbon receptor (AhR) pathway, Nrf2 can modulate IL-22 production in T cells, reinforcing its role in both adaptive immunity and mucosal defense [161].
Dimethyl fumarate (DMF), a known Nrf2 activator, has shown promise in experimental colitis models, where it reduced inflammation and improved histological outcomes [162,163,164]. These effects are believed to stem from simultaneous activation of antioxidant defenses and inhibition of the NF-κB pathway, a major driver of pro-inflammatory gene expression [162,163,164]. In an acute graft-versus-host disease model, DMF promoted the expansion of donor-derived Tregs and mitigated harmful allogeneic responses, further supporting the idea that Nrf2 shapes immune tolerance [165].
The role of Nrf2 is not limited to CD4+ T cells. CD8+ cytotoxic T lymphocytes (CTLs) also depend on Nrf2-mediated redox control for optimal function [166]. Loss of Nrf2 in macrophages impairs the antigen-driven activation of CD8+ T cells by limiting intracellular glutathione and cysteine levels, both essential for T cell metabolism and cytolytic activity [166]. Beyond T cells, Nrf2 also influences B cell function. Evidence indicates that B cells exposed to LPS increase immunoglobulin M (IgM) secretion when Nrf2 is pharmacologically activated [167]. Furthermore, Nrf2 signaling impacts dendritic cells, with its activation associated with suppressed antigen presentation capacity [168]. This dampening of dendritic cell activity indirectly favors T cell tolerance, while Nrf2 deficiency in these cells enhances Th1 polarization and T cell proliferation [168].
The immunoregulatory landscape also includes myeloid-derived suppressor cells (MDSCs), whose expansion and suppressive function are enhanced through Nrf2 activation [169]. In inflamed tissues, this contributes to reduced effector T cell activity and supports mucosal immune tolerance.
However, the immunoregulatory role of Nrf2 is context-dependent. Studies using genetically modified mice with constitutively active Nrf2 expression in epithelial or myeloid cells revealed exacerbated inflammation in acute colitis, but not in chronic models [170]. This paradox underscores the need for tight redox regulation, as both insufficient and excessive antioxidant responses can disturb mucosal immunity.
In clinical settings, findings remain somewhat inconsistent. Early research linked certain Nrf2 gene polymorphisms to UC susceptibility, particularly in females with a chronic disease phenotype [171]. Some studies reported reduced Nrf2 expression in patients with active UC and CD, while others found upregulated Nrf2 protein in inflamed mucosa [87,172]. These discrepancies may reflect differences in disease stage, sampling site, or compensatory responses to chronic oxidative stress. Interestingly, elevated Nrf2 in active disease correlated with increased expression of protective enzymes and inversely with pro-inflammatory IL-17A levels [173].
Current evidence on Nrf2 in CD is still limited. Some studies report that Nrf2 expression is reduced during active disease, while others demonstrate that pharmacological activation of Nrf2, for example with Bryostatin-1, alleviates colitis in interleukin 10 (IL-10)–deficient mice, an experimental model that mimics Crohn’s disease [174,175,176]. These observations suggest that Nrf2 may also hold therapeutic relevance in Crohn’s disease, although further studies are required.
6.3. Nrf2 as a Mediator of Gut Microbiota Balance and Intestinal Homeostasis
The gut microbiota constitutes a complex and dynamic ecosystem that plays a fundamental role in regulating host immunity, metabolism, and the integrity of the intestinal mucosa. In healthy individuals, this microbial community, although diverse in its composition, maintains a functional equilibrium that supports intestinal and systemic homeostasis [177,178]. However, microbial dysbiosis, characterized by disrupted composition and metabolite profiles, has been strongly implicated in the pathogenesis of IBD, colorectal cancer, and other intestinal disorders [179,180,181]. In IBD, dysbiosis is often marked by reduced microbial diversity, decreased abundance of beneficial Firmicutes (e.g., Clostridiales), and increased levels of potentially pathogenic Proteobacteria and Actinobacteria [182,183]. These shifts are associated with disease severity and mucosal barrier dysfunction.
Emerging evidence indicates that Nrf2 signaling exerts a profound influence on the gut microbial ecosystem, serving as a molecular interface between oxidative stress responses and microbial regulation [90,184]. Activation of Nrf2 can both shape microbial composition and mediate host tolerance to microbial metabolites [184]. For instance, oxidative stress in the gut can lead to epithelial barrier compromise, favoring dysbiosis; conversely, Nrf2 activation enhances antioxidant defense, preserving epithelial integrity and microbial balance [67,184,185].
6.4. Probiotics, Nrf2 Activation, and Gut Barrier Preservation
Several probiotic strains have demonstrated the ability to modulate the Nrf2 pathway, thereby exerting protective effects on the gut epithelium. For example, Lactobacillus fermentum Lf1 increased the expression of Nrf2 and downstream antioxidant enzymes such as SOD2 and thioredoxin reductase 1 (TrxR-1), while reducing lipid peroxidation in a DSS-induced colitis model [186]. Similarly, Lactobacillus casei Shirota protected epithelial cells against oxidative stress by activating both Nrf2 and NF-κB signaling pathways [187].
Other strains, such as L. reuteri, have been shown to enhance tight junction protein expression (e.g., occludin, claudin-1) and maintain mucosal barrier integrity via Nrf2/HO-1 axis activation [188]. In parallel, Saccharomyces boulardii alleviated colonic oxidative damage in murine colitis models through upregulation of SOD and catalase via Nrf2-mediated mechanisms [189]. These findings emphasize the dual role of probiotics in maintaining intestinal health. They not only modulate the redox balance through activation of antioxidant pathways but also reshape microbial communities, thereby helping to prevent or reduce colonic inflammation.
6.5. Dietary Compounds and Microbial Intermediates Acting Through Nrf2
Beyond live bacteria, bacterial metabolites and diet-derived phytochemicals also interface with the Nrf2 pathway [144]. For instance, Urolithin A (UroA), a microbial metabolite from ellagitannins in pomegranate and berries, activates the AhR-Nrf2 axis, leading to increased expression of antioxidant enzymes such as HO-1 and NQO1, and improved gut barrier function [184]. Similarly, Indole-3-lactic acid (ILA), produced by Bifidobacterium longum subsp. infantis, has been shown to reduce inflammation in epithelial cells through co-activation of AhR and Nrf2, enhancing the expression of target genes like GPX2, SOD2, and NQO1 [190].
Selenium nanoparticles (SeNPs) biosynthesized by Lactobacillus casei ATCC 393 further exemplify the therapeutic potential of microbiota–Nrf2 crosstalk [191]. These SeNPs improved epithelial antioxidant responses and tight junction integrity both in vitro and in DSS-induced injury models, underscoring a promising bioengineered approach to intestinal redox regulation [191].
Furthermore, Lactobacillus helveticus, L. mali, and other Lactobacillus strains have shown systemic effects beyond the gut, modulating hepatic oxidative stress and influencing the Firmicutes/Bacteroidetes ratio in high-fat diet models through Nrf2-related signaling [192,193]. The synergistic action of rice protein peptides and phytochemicals such as those from Bruguiera gymnorrhiza and Tetrastigma hemsleyanum have also been shown to activate the Keap1-Nrf2 pathway, enrich beneficial microbial taxa (e.g., Akkermansia, Bifidobacterium, Anaerotruncus), and enhance mucosal tight junction protein expression, suggesting diet–microbiota–host triad interactions mediated by redox-sensitive transcription factors [194].
To clarify these diverse functions, Table 2 summarizes the major Nrf2-dependent pathways implicated in IBD pathogenesis, emphasizing their cellular context and therapeutic relevance.
Table 2.
Key Nrf2-Dependent Pathways in Intestinal Inflammation, Barrier Regulation, and Microbial Homeostasis.
7. Nrf2 in IBD-Associated Tumorigenesis: A Double-Edged Sword
Nrf2 is a central transcription factor controlling the expression of genes involved in antioxidant defense, detoxification, xenobiotic metabolism, and cellular stress responses [82]. Through these functions, Nrf2 preserves epithelial integrity and modulates inflammation, but its role in cancer biology is complex [195,196]. In the intestine, the nature and timing of Nrf2 activation largely determine whether it acts as a tumor suppressor or a tumor promoter [197]. Basal Nrf2 activity protects tissues from genotoxic insults, while sustained or dysregulated activation can favor tumor progression and chemoresistance [94,197,198].
Colorectal cancer (CRC) is one of the most common malignancies worldwide, and its incidence is significantly increased in patients with long-standing ulcerative colitis [199]. The chronic inflammatory microenvironment characteristic of IBD, rich in ROS, cytokines, and infiltrating immune cells, fosters mutagenesis and epigenetic changes that drive malignant transformation [200,201]. Nrf2 regulates many cytoprotective genes that mitigate oxidative and proteotoxic stress, and in early stages this activity may counteract DNA damage and reduce tumor initiation [197,202]. However, persistent Nrf2 activation may inadvertently support the survival of dysplastic or transformed cells by suppressing apoptosis, maintaining proteasome activity, and enhancing DNA repair [203,204].
Experimental models illustrate this duality. In DSS-induced colitis-associated cancer, Nrf2 deficiency produced opposing outcomes depending on disease stage. Some studies showed that Nrf2 knockout mice developed more numerous and larger tumors with increased inflammatory mediators such as COX-2, lipoxygenases, and prostaglandins, highlighting its protective function during early carcinogenesis [89,205]. In contrast, other models demonstrated that ablation of Nrf2 reduced aggressive tumor formation and upregulated tumor suppressors such as 15-hydroxyprostaglandin dehydrogenase (15-PGDH), suggesting that in later stages Nrf2 may facilitate tumor growth [89,205]. These conflicting results underscore that the impact of Nrf2 on tumorigenesis is context-dependent.
Clinical and mechanistic data further support this dual role. Polymorphisms in the Nrf2 promoter region have been linked to higher CRC risk, while high nuclear expression of Nrf2 correlates with increased antioxidant enzyme levels and lower inflammatory markers in colonic mucosa [206,207]. Conversely, cytoplasmic retention of Nrf2 (cNrf2) in tumor cells is associated with poor prognosis, increased metastasis, and resistance to chemotherapy [208]. Mechanistically, cNrf2 interacts with PSMD4 to promote nuclear export and activate downstream pathways such as NF-κB, AKT, and β-catenin, conferring resistance to 5-fluorouracil (5-FU) and oxaliplatin [208]. Epigenetic changes, such as DNA demethylation, may also upregulate Nrf2 and HO-1 expression in drug-resistant CRC cells, further contributing to chemoresistance [209,210].
Nrf2 also influences key processes in metastasis. Its overexpression in highly invasive CRC correlates with advanced Duke’s stage and increased vascular endothelial growth factor (VEGF) signaling [211]. Nrf2 enhances angiogenesis under hypoxic conditions by stabilizing hypoxia-inducible factor 1 alpha (HIF-1α) and inducing VEGF expression, whereas inhibition of Nrf2 reduces tumor vascularization and growth in xenograft models [212]. These findings suggest that while Nrf2 activation may be beneficial in preventing oxidative damage in premalignant tissues, its persistent activation in established tumors can facilitate angiogenesis, invasion, and therapy resistance.
8. Nrf2-Linked Regulation of Intestinal Fibrosis
Fibrosis represents one of the most challenging complications in inflammatory bowel diseases, particularly CD, where it leads to stricture formation and bowel obstruction [213]. Unlike ulcerative colitis, where fibrosis is mostly confined to the mucosa and submucosa, in CD the remodeling process involves the entire bowel wall [213]. This remodeling is driven by chronic inflammation, persistent epithelial injury, and an imbalance between extracellular matrix (ECM) deposition and degradation [214]. Excessive accumulation of ECM proteins such as collagen thickens and stiffens the intestinal wall, contributing to stricture development and functional impairment [215].
At the molecular level, TGF-β is recognized as a central mediator of fibrogenesis. It stimulates fibroblast-to-myofibroblast transition, activates collagen-producing cells, and orchestrates ECM accumulation [215,216]. TGF-β signaling operates mainly through the Smad pathway but also intersects with MAPK cascades [217]. Importantly, the activity of TGF-β is closely tied to redox homeostasis. Elevated levels of ROS can amplify TGF-β/Smad signaling, creating a feed-forward loop that enhances fibrotic responses [218,219].
Nrf2 plays a critical role in counteracting these processes. By inducing antioxidant enzymes such as HO-1, NQO1, and SOD, Nrf2 limits oxidative stress and suppresses pro-fibrotic signaling [76]. Experimental studies in TNBS-induced colitis have shown that pharmacological activation of Nrf2 with tBHQ reduces fibrosis by inhibiting the TGF-β/Smad pathway [220,221]. In contrast, Nrf2 deficiency enhances fibroblast sensitivity to TGF-β, promoting their differentiation into myofibroblasts and driving ECM overproduction [220]. This demonstrates a protective, anti-fibrotic role of Nrf2 through regulation of the ROS–TGF-β–Smad axis.
In addition to TGF-β signaling, the balance between MMPs and their inhibitors (TIMPs) is crucial for ECM turnover. Dysregulation of this system accelerates fibrosis [215]. Among MMPs, MMP-7 and MMP-3 appear particularly relevant in IBD [222]. The Nrf2/HO-1 pathway has been shown to suppress MMP-7 activity in intestinal epithelial cells, suggesting a direct mechanism by which Nrf2 curtails fibrotic remodeling [87,223]. Elevated MMP-3 levels have been detected in both CD and UC patients, with concentrations correlating with disease severity and response to anti-TNF therapy [224,225]. Notably, Nrf2-deficient mice exhibit exaggerated MMP-3 expression, linking oxidative stress with impaired ECM regulation [226].
Although fibrosis is less common in UC, evidence suggests that submucosal thickening and muscularis mucosae remodeling can occur, particularly in patients with long-standing disease. These findings underscore the need to better understand the drivers of fibrosis in both forms of IBD and highlight the potential of targeting redox-sensitive pathways. Nrf2, by integrating ROS detoxification with regulation of fibrotic signaling, emerges as a promising therapeutic node. Agents capable of fine-tuning Nrf2 activation may help restore ECM balance, limit fibrotic complications, and ultimately improve long-term outcomes in IBD. Table 3 summarizes the principal Nrf2-dependent pathways implicated in IBD-associated tumorigenesis and intestinal fibrosis, highlighting their molecular targets and clinical relevance
Table 3.
Key Nrf2-Dependent Mechanisms in IBD-Associated Tumorigenesis and Fibrosis.
9. Pharmacological and Nutritional Modulation of the Nrf2/Keap1 Pathway in the Treatment of Inflammatory Bowel Disease
Over recent years, the Nrf2/Keap1 signaling axis has emerged as a promising therapeutic target in IBD, including both Crohn’s disease and ulcerative colitis [94,227]. This transcriptional system regulates cellular responses to oxidative stress by inducing a battery of cytoprotective genes involved in antioxidant defense, inflammation control, and epithelial integrity maintenance [142,198]. Regulation of this pathway, whether through pharmacological agents or naturally occurring bioactive compounds, has demonstrated beneficial effects in experimental models of colitis [227,228].
Among clinically approved drugs, 5-aminosalicylic acid (5-ASA) remains a cornerstone in IBD therapy [229]. Beyond its anti-inflammatory activity, 5-ASA enhances Nrf2 nuclear translocation in inflamed colonic mucosa, leading to increased expression of HO-1 and suppression of TNF-α [230,231]. This redox-sensitive activation appears to occur selectively under inflammatory conditions, suggesting that 5-ASA may function as a prodrug whose efficacy depends on local oxidative stress. Dimethyl fumarate (DMF), currently used for multiple sclerosis, has similarly demonstrated the ability to activate Nrf2 and suppress colonic inflammation in animal models [162].
Other repurposed agents such as telmisartan, olmesartan, and dapagliflozin also exert protective effects in experimental colitis via Nrf2 activation [94]. These drugs reduce oxidative injury and inflammatory cytokine production while promoting the expression of detoxifying enzymes [94]. Notably, metformin, traditionally used in type 2 diabetes, enhances Nrf2 activity through AMPK-dependent mechanisms, reinforcing the interconnectedness between metabolic and redox pathways in IBD pathology [232]. Novel compounds like CPUY192018 directly target Keap1 to stabilize Nrf2 and are currently under investigation as potential disease-modifying agents [233].
In parallel, numerous naturally derived compounds have gained attention as Nrf2 inducers with therapeutic potential in IBD. Flavonoids such as luteolin, galangin, genistein, and puerarin have shown to upregulate HO-1 and NQO1 in DSS-induced colitis, resulting in reduced oxidative stress and improved mucosal architecture [144]. Terpenoids, particularly ginsenosides and compounds such as dehydrocostus lactone, modulate the Nrf2/HO-1 axis while suppressing NF-κB and apoptotic mediators, thereby preserving epithelial barrier function [234]. Alkaloids like berberine and rutaecarpine demonstrate similar benefits, acting through Nrf2-mediated regulation of P-glycoprotein and antioxidant enzymes [235,236].
Polysaccharides from herbal sources such as Astragalus membranaceus and Aloe vera have also shown strong efficacy in restoring epithelial integrity and lowering oxidative markers in preclinical models [94]. These complex carbohydrates activate Nrf2 signaling and suppress ferroptosis and pyroptosis, two forms of regulated cell death that are increasingly recognized as key contributors to the development of inflammatory bowel disease IBD [94].
Several medicinal plant extracts, including Perilla frutescens, Forsythia suspensa, Artemisia argyi, and Vaccinium species, have demonstrated the capacity to activate Nrf2, often in conjunction with suppression of NF-κB and MAPK pathways [144]. These extracts not only modulate inflammation but also improve tight junction integrity and microbiota composition.
Traditional Chinese medicine (TCM) formulations, such as Huang-Lian-Jie-Du decoction and Banxia Xiexin decoction, offer multi-target therapeutic strategies [149,237]. Their synergistic components activate Nrf2 while simultaneously suppressing pro-inflammatory transcription factors, offering an integrative approach to IBD therapy.
10. Comparative Perspective: Nrf2 Versus Galectin-3 as Emerging Biomarkers in IBD
Nrf2 and galectin-3 (Gal-3) represent two mechanistically distinct biomarker families that capture different pathogenic dimensions of inflammatory bowel disease [87,238]. Nrf2 functions as a transcriptional regulator of antioxidant and cytoprotective genes, offering an upstream readout of redox imbalance and epithelial stress [87]. By contrast, Gal-3 is a secreted lectin measurable in serum and tissue that reflects innate immune activation, macrophage activity, and early fibrotic remodeling [238].
Clinically, Nrf2-related signatures correlate with oxidative stress, impaired barrier function, and shifts in mucosal antioxidant capacity—parameters not captured by routine inflammatory markers such as CRP or fecal calprotectin [87]. Gal-3, on the other hand, rises with endoscopic severity, predicts fibrotic complications, and associates with response to anti-TNF therapy, highlighting its translational usefulness as a soluble biomarker [238].
Mechanistically, Nrf2 suppresses ROS-driven NF-κB activation, stabilizes tight junctions, and modulates immune polarization, positioning it as a regulator of upstream oxidative and inflammatory loops [87]. Gal-3 primarily amplifies leukocyte recruitment, macrophage activation, and fibroblast proliferation, linking it more directly to chronic inflammation and tissue remodeling [238].
Therapeutically, Nrf2 is already a validated drug target, with activators such as dimethyl fumarate showing efficacy in experimental colitis. [87] Gal-3 inhibitors are emerging but remain earlier in development and less explored in IBD [238].
Together, Nrf2 and Gal-3 highlight complementary aspects of IBD pathogenesis—redox imbalance versus lectin-mediated inflammation—and combined assessment may ultimately offer improved disease stratification and prediction of complications.
11. Future Directions in Nrf2 Modulation and Its Therapeutic Potential in IBD
Despite the strong preclinical evidence supporting Nrf2 as a therapeutic target in IBD, multiple challenges limit the clinical translation of Nrf2-modulating strategies. One of the key barriers is the context-dependent nature of Nrf2 activation, which can be protective in early inflammation but potentially detrimental in chronic disease stages or in the presence of dysplastic lesions. Sustained or excessive Nrf2 activation may promote epithelial survival, impair apoptosis of damaged cells, and contribute to chemoresistance or tumor progression. This duality underscores the need to define disease stage–specific therapeutic windows and to develop pharmacologic agents capable of precise, titratable Nrf2 activation.
Another major challenge is the lack of reliable biomarkers for monitoring Nrf2 pathway activity in vivo. Current studies rely on transcript levels of downstream enzymes such as HO-1 or NQO1, but these readouts lack temporal sensitivity and may be influenced by parallel inflammatory pathways. To implement Nrf2-directed therapies safely, future studies should identify non-invasive biomarkers (e.g., metabolomic signatures, redox-derived lipids, circulating antioxidant capacity markers) that reflect real-time pathway activation and help stratify patients who may benefit from targeted interventions.
A further obstacle concerns the heterogeneity of IBD phenotypes, which may result in differential Nrf2 responses across patients. Variants in the NFE2L2 promoter, differences in microbiota composition, and variability in epithelial redox status may all influence responsiveness to Nrf2-activating drugs. Precision-medicine approaches integrating genetic stratification, microbiome profiling, and redox phenotyping will be essential to determine which patient subgroups derive the greatest benefit.
Additionally, while numerous phytochemicals and probiotics show Nrf2-activating properties, their bioavailability, metabolic stability, and tissue-specific targeting remain inconsistently characterized. The development of optimized formulations—such as nanoencapsulation, targeted delivery systems, or engineered probiotic strains—could enhance mucosal uptake and reduce systemic off-target effects. Similarly, selective Keap1 inhibitors and next-generation small molecules should be evaluated in long-term models to assess potential safety concerns, particularly regarding tumorigenesis.
Finally, further work is needed to clarify how Nrf2 interacts with other redox-sensitive pathways, including NF-κB, STAT3, ferroptosis regulators, and mitochondrial quality-control mechanisms. Understanding these interactions may allow design of rational combination therapies, where Nrf2 activation is paired with immunomodulators, microbiota-directed therapies, or anti-fibrotic agents. Longitudinal human studies integrating multi-omics, functional imaging, and organoid systems will be instrumental in defining how the Nrf2 network evolves across disease stages and treatment responses.
Together, these lines of inquiry point to an urgent need for biomarker-guided, context-specific, and precision-engineered approaches to safely leverage Nrf2 signaling as a therapeutic axis in IBD.
12. Conclusions
The Nrf2/Keap1 signaling pathway plays a multifaceted and dynamic role in the pathophysiology of inflammatory bowel disease. As a master regulator of antioxidant defense, Nrf2 not only neutralizes reactive oxygen species and limits oxidative injury but also orchestrates immune responses, reinforces the epithelial barrier, and shapes the intestinal microbiome. Its protective functions are context-dependent—conferring cytoprotection in early disease stages, yet potentially promoting tumorigenesis and fibrosis under conditions of sustained or dysregulated activation. Preclinical and clinical data underscore the translational potential of targeted Nrf2 modulation, whether through pharmacological activators, repurposed drugs, or dietary and probiotic interventions. However, fine-tuning the intensity and timing of Nrf2 activation remains essential to avoid adverse effects and achieve therapeutic precision. Future research should focus on biomarker-guided patient stratification and the development of Nrf2-based combinatorial therapies that complement existing immunomodulatory approaches in IBD.
Author Contributions
Conceptualization, B.S., I.M.B., M.D.S. and B.S.S.; methodology, M.D.S., I.R. and M.J.; investigation, B.S., I.M.B., M.D.S. and S.L. (Snezana Lazarevic); validation, B.S.S., B.M. and D.T.-U.; resources, M.D.S., N.M., B.S.S. and D.P.; writing—original draft preparation, B.S., M.M., I.C., F.M., M.D.S., S.L. (Snezna Lukic) and B.S.S.; writing—review and editing, S.L. (Snezana Lazarevic), M.M., M.J., Đ.T. and S.L. (Snezna Lukic); visualization, B.S. and I.M.B.; supervision, I.R., M.J. and S.L. (Snezna Lukic). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors sincerely thank the Faculty of Medical Sciences, University of Kragujevac, Serbia, for their continuous support.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 5-ASA | 5-aminosalicylic acid |
| 5-FU | fluorouracil |
| α-SMA | alpha-smooth muscle actin |
| AhR | aryl hydrocarbon receptor |
| AMPK | adenosine monophosphate–activated protein kinase |
| AP-1 | activator protein 1 |
| AREs | antioxidant response elements |
| ATP | adenosine triphosphate |
| bZIP | of basic leucine zipper |
| CBP | CREB-binding protein |
| CD | Crohn’s disease |
| CD4+ | cluster of differentiation 4 positive |
| CNC | the Cap’n’Collar |
| COX IV | cytochrome c oxidase subunit IV |
| COX-2 | cyclooxygenase-2 |
| CRC | Colorectal cancer |
| CTLs | cytotoxic T lymphocytes |
| cNrf2 | cytoplasmic retention of Nrf2 |
| Cul3 | cullin-3 |
| DMF | Dimethyl fumarate |
| ECM | extracellular matrix |
| ERK | extracellular signal–regulated kinase |
| FOXO1 | forkhead box protein O1 |
| GCL | glutamate–cysteine ligase |
| GSH | glutathione |
| GST | glutathione S-transferase |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HO-1 | heme oxygenase-1 |
| HOCl | hypochlorous acid |
| IBD | Inflammatory bowel disease |
| IFN-β | interferon beta |
| IgM | immunoglobulin M |
| IKKβ | I kappa B kinase beta |
| ILA | Indole-3-lactic acid |
| IL-1β | interleukin 1 beta |
| IL-8 | interleukin 8 |
| iNOS | inducible nitric oxide synthase |
| JNK | c-Jun N-terminal kinase |
| Keap1 | Kelch-like ECH-associated protein 1 |
| Maf | musculoaponeurotic fibrosarcoma |
| MAPK | mitogen-activated protein kinase |
| MAPKs | mitogen-activated protein kinases |
| Mkp-1 | mitogen-activated protein kinase phosphatase 1 |
| MMPs | metalloproteinases |
| NF-κB | nuclear factor kappa B |
| NLRP3 | NLR family pyrin domain containing 3 |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| O3 | ozone |
| PI3K | phosphoinositide 3-kinase |
| Pink1 | phosphatase and tensin homolog–induced putative kinase 1 |
| PKC | protein kinase C |
| Rbx1 | RING-box protein 1 |
| ROS | reactive oxygen species |
| SeNPs | Selenium nanoparticles |
| SOD | superoxide dismutase |
| STAT3 | signal transducer and activator of transcription 3 |
| TGF-β1 | transforming growth factor beta 1 |
| Th | T helper |
| TIMP-1 | tissue inhibitor of metalloproteinases 1 |
| TJs | tight junctions |
| TLR4 | toll-like receptor 4 |
| TNBS | trinitrobenzene sulfonic acid |
| TNF-α | tumor necrosis factor alpha |
| TrxR-1 | thioredoxin reductase 1 |
| Tregs | regulatory T cells |
| tBHQ | tert-butylhydroquinone |
| UC | ulcerative colitis |
| UGT1A1 | uridine diphosphate glucuronosyltransferase 1A1 |
| UroA | Urolithin A |
| VEGF | vascular endothelial growth factor |
| ZnO | Zinc oxide |
| ZO-1 | zonula occludens 1 |
References
- Silaghi, A.; Constantin, V.D.; Socea, B.; Banu, P.; Sandu, V.; Andronache, L.F.; Dumitriu, A.S.; Paunica, S. Inflammatory Bowel Disease: Pathogenesis, Diagnosis and Current Therapeutic Approach. J. Mind Med. Sci. 2022, 9, 56–77. [Google Scholar] [CrossRef]
- Mak, W.Y.; Hart, A.L.; Ng, S.C. Crohn’s disease. Medicine 2019, 47, 377–387. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef] [PubMed]
- Cicerone, C.; D’Amico, F.; Allocca, M.; Zilli, A.; Parigi, T.L.; Danese, S.; Furfaro, F. A Comprehensive Multidisciplinary Approach to Diagnosing Chronic Inflammatory Bowel Diseases: Integration of Clinical, Endoscopic, and Imaging Modalities. Diagnostics 2024, 14, 1530. [Google Scholar] [CrossRef] [PubMed]
- Solitano, V.; Vuyyuru, S.K.; Aruljothy, A.; Alkhattabi, M.; Zou, J.; Beaton, M.; Gregor, J.; Kassam, Z.; Sedano, R.; Marshall, H.; et al. Endoscopic Skipping, Stricturing, and Penetrating Complications in Crohn’s Disease on Tandem Ileo-colonoscopy and Cross-sectional Imaging: A Retrospective Cohort Study. Inflamm. Bowel Dis. 2025, 31, 1529–1536. [Google Scholar] [CrossRef]
- Daperno, M. Endoscopy in IBD: When and How? Diagnostics 2023, 13, 3423. [Google Scholar] [CrossRef]
- Rodríguez-Lago, I.; Aduna, M.; Ramírez de la Piscina, P.; Merino, O.; Carrascosa, J.; Higuera, R.; Maíz, A.; Zapata, E.; Cabriada, J.L.; Barreiro-de Acosta, M. Transmural cross-sectional findings and bowel damage assessment in preclinical Crohn’s disease: A case-control study. Int. J. Color. Dis. 2024, 39, 92. [Google Scholar] [CrossRef]
- Faggiani, I.; Fanizza, J.; D’Amico, F.; Allocca, M.; Zilli, A.; Parigi, T.L.; Barchi, A.; Danese, S.; Furfaro, F. Extraintestinal Manifestations in Inflammatory Bowel Disease: From Pathophysiology to Treatment. Biomedicines 2024, 12, 1839. [Google Scholar] [CrossRef]
- Kellermann, L.; Riis, L.B. A close view on histopathological changes in inflammatory bowel disease, a narrative review. Dig. Med. Res. 2021, 4, 3. [Google Scholar] [CrossRef]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory Bowel Disease in Children and Adolescents. JAMA Pediatr. 2015, 169, 1053–1060. [Google Scholar] [CrossRef]
- Cross, E.; Saunders, B.; Farmer, A.D.; Prior, J.A. Diagnostic delay in adult inflammatory bowel disease: A systematic review. Indian J. Gastroenterol. 2023, 42, 40–52. [Google Scholar] [CrossRef]
- Altaş, U.; Ertem, D. Evaluation of Growth in Children with Inflammatory Bowel Disease. Children 2024, 11, 1038. [Google Scholar] [CrossRef]
- Hracs, L.; Windsor, J.W.; Gorospe, J.; Cummings, M.; Coward, S.; Buie, M.J.; Quan, J.; Goddard, Q.; Caplan, L.; Markovinović, A.; et al. Global evolution of inflammatory bowel disease across epidemiologic stages. Nature 2025, 642, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Bhayani, P.; Natarajan, K.; Coelho-Prabhu, N. Rising Incidence of Inflammatory Bowel Disease in the Asian Subcontinent—An Exploration of Causative Factors. Gastrointest. Disord. 2024, 6, 549–556. [Google Scholar] [CrossRef]
- Ren, X.; Xu, J.; Zhao, X. Global Trends and Future Projections in the Burden of Inflammatory Bowel Disease Among Adolescents and Young Adults (15–49 Years) from 1990 to 2021. JGH Open 2025, 9, e70282. [Google Scholar] [CrossRef] [PubMed]
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Diez-Martin, E.; Hernandez-Suarez, L.; Muñoz-Villafranca, C.; Martin-Souto, L.; Astigarraga, E.; Ramirez-Garcia, A.; Barreda-Gómez, G. Inflammatory Bowel Disease: A Comprehensive Analysis of Molecular Bases, Predictive Biomarkers, Diagnostic Methods, and Therapeutic Options. Int. J. Mol. Sci. 2024, 25, 7062. [Google Scholar] [CrossRef]
- Chiarello, M.M.; Cariati, M.; Brisinda, G. Colonic Crohn’s disease—Decision is more important than incision: A surgical dilemma. World J. Gastrointest. Surg. 2021, 13, 1–6. [Google Scholar] [CrossRef]
- Follin-Arbelet, B.; Cvancarova Småstuen, M.; Hovde, Ø.; Jelsness-Jørgensen, L.-P.; Moum, B. Mortality in Patients with Inflammatory Bowel Disease: Results from 30 Years of Follow-up in a Norwegian Inception Cohort (the IBSEN study). J. Crohn’s Colitis 2023, 17, 497–503. [Google Scholar] [CrossRef]
- Liu, S.; Eisenstein, S. State-of-the-art surgery for ulcerative colitis. Langenbecks Arch. Surg. 2021, 406, 1751–1761. [Google Scholar] [CrossRef]
- Laredo, V.; García-Mateo, S.; Martínez-Domínguez, S.J.; López de la Cruz, J.; Gargallo-Puyuelo, C.J.; Gomollón, F. Risk of Cancer in Patients with Inflammatory Bowel Diseases and Keys for Patient Management. Cancers 2023, 15, 871. [Google Scholar] [CrossRef]
- Schiavone, S.C.; Biancone, L.; Fiorillo, M.; Divizia, A.; Mancone, R.; Neri, B. Colitis-Associated Dysplasia in Inflammatory Bowel Disease: Features and Endoscopic Management. Cancers 2025, 17, 784. [Google Scholar] [CrossRef]
- Hong, S.M.; Baek, D.H. Diagnostic Procedures for Inflammatory Bowel Disease: Laboratory, Endoscopy, Pathology, Imaging, and Beyond. Diagnostics 2024, 14, 1384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shi, Y.; Lin, C.; He, C.; Wang, S.; Li, Q.; Sun, Y.; Li, M. Overcoming cancer risk in inflammatory bowel disease: New insights into preventive strategies and pathogenesis mechanisms including interactions of immune cells, cancer signaling pathways, and gut microbiota. Front. Immunol. 2023, 14, 1338918. [Google Scholar] [CrossRef] [PubMed]
- Calvez, V.; Puca, P.; Di Vincenzo, F.; Del Gaudio, A.; Bartocci, B.; Murgiano, M.; Iaccarino, J.; Parand, E.; Napolitano, D.; Pugliese, D.; et al. Novel Insights into the Pathogenesis of Inflammatory Bowel Diseases. Biomedicines 2025, 13, 305. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Zhang, S.; Qi, C.; Zhang, Z.; Ma, R.; Xiang, L.; Chen, L.; Zhu, Y.; Tang, C.; et al. Oxidative stress gene expression, DNA methylation, and gut microbiota interaction trigger Crohn’s disease: A multi-omics Mendelian randomization study. BMC Med. 2023, 21, 179. [Google Scholar] [CrossRef]
- Muro, P.; Zhang, L.; Li, S.; Zhao, Z.; Jin, T.; Mao, F.; Mao, Z. The emerging role of oxidative stress in inflammatory bowel disease. Front. Endocrinol. 2024, 15, 1390351. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Kloska, D.; Grochot-Przęczek, A.; Feelisch, M.; Cuadrado, A.; van Goor, H. Personalized redox medicine in inflammatory bowel diseases: An emerging role for HIF-1α and NRF2 as therapeutic targets. Redox Biol. 2023, 60, 102603. [Google Scholar] [CrossRef]
- Tie, Y.; Huang, Y.; Chen, R.; Li, L.; Chen, M.; Zhang, S. Current insights on the roles of gut microbiota in inflammatory bowel disease-associated extra-intestinal manifestations: Pathophysiology and therapeutic targets. Gut Microbes 2023, 15, 2265028. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, F.; D’Amico, F.; Bencardino, S.; Faggiani, I.; Fanizza, J.; Zilli, A.; Parigi, T.L.; Allocca, M.; Danese, S.; Furfaro, F. Gut Microbiota Metabolites: Unveiling Their Role in Inflammatory Bowel Diseases and Fibrosis. Pharmaceuticals 2024, 17, 347. [Google Scholar] [CrossRef]
- Mak, J.W.Y.; Lo, A.T.W.; Ng, S.C. Early life factors, diet and microbiome, and risk of inflammatory bowel disease. J. Can. Assoc. Gastroenterol. 2025, 8, S44–S50. [Google Scholar] [CrossRef]
- Yan, J.; Wang, L.; Gu, Y.; Hou, H.; Liu, T.; Ding, Y.; Cao, H. Dietary Patterns and Gut Microbiota Changes in Inflammatory Bowel Disease: Current Insights and Future Challenges. Nutrients 2022, 14, 4003. [Google Scholar] [CrossRef] [PubMed]
- Törüner, M.; Ünal, N.G. Epigenetics of Inflammatory Bowel Diseases. Turk. J. Gastroenterol. 2023, 34, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Song, A.; Son, A.; Shin, Y. Gut microbiota and epigenetic choreography: Implications for human health: A review. Medicine 2024, 103, e39051. [Google Scholar] [CrossRef]
- Shoham, S.; Pintel, N.; Avni, D. Oxidative Stress, Gut Bacteria, and Microalgae: A Holistic Approach to Manage Inflammatory Bowel Diseases. Antioxidants 2025, 14, 697. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies. Oxidative Med. Cell. Longev. 2017, 2017, 4535194. [Google Scholar] [CrossRef]
- Manful, C.F.; Fordjour, E.; Subramaniam, D.; Sey, A.A.; Abbey, L.; Thomas, R. Antioxidants and Reactive Oxygen Species: Shaping Human Health and Disease Outcomes. Int. J. Mol. Sci. 2025, 26, 7520. [Google Scholar] [CrossRef]
- Gulcin, İ. Antioxidants: A comprehensive review. Arch. Toxicol. 2025, 99, 1893–1997. [Google Scholar] [CrossRef]
- Noviello, D.; Amoroso, C.; Vecchi, M.; Facciotti, F.; Caprioli, F. Curing inflammatory bowel diseases: Breaking the barriers of current therapies- emerging strategies for a definitive treatment. Curr. Opin. Immunol. 2025, 95, 102593. [Google Scholar] [CrossRef]
- Appiah, J.K.; Hayat, U.; Garg, N.; Asante, R.; Donneyong, E.; Haider, M.U.; Patel, P.; Khan, Z.; Siddiqui, A.A. Emerging Therapies in Inflammatory Bowel Disease: A Comprehensive Review. J. Clin. Med. 2025, 14, 6119. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Jiang, M.; Li, X.; Yuan, J.; Zhang, H. Precision medicine in inflammatory bowel disease. Precis. Clin. Med. 2023, 6, pbad033. [Google Scholar] [CrossRef]
- Stojanovic, B.; Jovanovic, I.; Dimitrijevic Stojanovic, M.; Stojanovic, B.S.; Kovacevic, V.; Radosavljevic, I.; Jovanovic, D.; Miletic Kovacevic, M.; Zornic, N.; Arsic, A.A.; et al. Oxidative Stress-Driven Cellular Senescence: Mechanistic Crosstalk and Therapeutic Horizons. Antioxidants 2025, 14, 987. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, B.; Milivojcevic Bevc, I.; Dimitrijevic Stojanovic, M.; Stojanovic, B.S.; Lazarevic, T.; Spasic, M.; Petrovic, M.; Stefanovic, I.; Markovic, M.; Nesic, J.; et al. Oxidative Stress, Inflammation, and Cellular Senescence in Neuropathic Pain: Mechanistic Crosstalk. Antioxidants 2025, 14, 1166. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef] [PubMed]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Morris, G.; Gevezova, M.; Sarafian, V.; Maes, M. Redox regulation of the immune response. Cell. Mol. Immunol. 2022, 19, 1079–1101. [Google Scholar] [CrossRef]
- Ukaegbu, K.; Allen, E.; Svoboda, K.K.H. Reactive Oxygen Species and Antioxidants in Wound Healing: Mechanisms and Therapeutic Potential. Int. Wound J. 2025, 22, e70330. [Google Scholar] [CrossRef]
- Wang, G.; Yang, F.; Zhou, W.; Xiao, N.; Luo, M.; Tang, Z. The initiation of oxidative stress and therapeutic strategies in wound healing. Biomed. Pharmacother. 2023, 157, 114004. [Google Scholar] [CrossRef]
- Jena, A.B.; Samal, R.R.; Bhol, N.K.; Duttaroy, A.K. Cellular Red-Ox system in health and disease: The latest update. Biomed. Pharmacother. 2023, 162, 114606. [Google Scholar] [CrossRef]
- Chandimali, N.; Bak, S.G.; Park, E.H.; Lim, H.-J.; Won, Y.-S.; Kim, E.-K.; Park, S.-I.; Lee, S.J. Free radicals and their impact on health and antioxidant defenses: A review. Cell Death Discov. 2025, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Anwar, S.; Sarwar, T.; Khan, A.A.; Rahmani, A.H. Therapeutic Applications and Mechanisms of Superoxide Dismutase (SOD) in Different Pathogenesis. Biomolecules 2025, 15, 1130. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several lines of antioxidant defense against oxidative stress: Antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch. Toxicol. 2024, 98, 1323–1367. [Google Scholar] [CrossRef]
- Kozlov, A.V.; Javadov, S.; Sommer, N. Cellular ROS and Antioxidants: Physiological and Pathological Role. Antioxidants 2024, 13, 602. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Kuiper, I.; Lamers, C.B.; Verspaget, H.W. Intestinal oxidative damage in inflammatory bowel disease: Semi-quantification, localization, and association with mucosal antioxidants. J. Pathol. 2003, 201, 28–36. [Google Scholar] [CrossRef]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The Impact of Oxidative Stress in Human Pathology: Focus on Gastrointestinal Disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef]
- de Almeida, A.; de Oliveira, J.; da Silva Pontes, L.V.; de Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, I.A. ROS: Basic Concepts, Sources, Cellular Signaling, and its Implications in Aging Pathways. Oxidative Med. Cell. Longev. 2022, 2022, 1225578. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Deffert, C.; Cachat, J.; Krause, K.-H. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell. Microbiol. 2014, 16, 1168–1178. [Google Scholar] [CrossRef]
- Kračun, D.; Lopes, L.R.; Cifuentes-Pagano, E.; Pagano, P.J. NADPH oxidases: Redox regulation of cell homeostasis and disease. Physiol. Rev. 2025, 105, 1291–1428. [Google Scholar] [CrossRef]
- Kim, M.E.; Lee, J.S. Advances in the Regulation of Inflammatory Mediators in Nitric Oxide Synthase: Implications for Disease Modulation and Therapeutic Approaches. Int. J. Mol. Sci. 2025, 26, 1204. [Google Scholar] [CrossRef] [PubMed]
- Mazuryk, O.; Gurgul, I.; Oszajca, M.; Polaczek, J.; Kieca, K.; Bieszczad-Żak, E.; Martyka, T.; Stochel, G. Nitric Oxide Signaling and Sensing in Age-Related Diseases. Antioxidants 2024, 13, 1213. [Google Scholar] [CrossRef]
- Brandl, N.; Seitz, R.; Sendtner, N.; Müller, M.; Gülow, K. Living on the Edge: ROS Homeostasis in Cancer Cells and Its Potential as a Therapeutic Target. Antioxidants 2025, 14, 1002. [Google Scholar] [CrossRef] [PubMed]
- Esworthy, R.S. Enzymatic Oxidants, Antioxidants, and Inflammatory Bowel Disease. Appl. Biosci. 2025, 4, 19. [Google Scholar] [CrossRef]
- Li, L.; Peng, P.; Ding, N.; Jia, W.; Huang, C.; Tang, Y. Oxidative Stress, Inflammation, Gut Dysbiosis: What Can Polyphenols Do in Inflammatory Bowel Disease? Antioxidants 2023, 12, 967. [Google Scholar] [CrossRef]
- Ortega-Zapero, M.; Gomez-Bris, R.; Pascual-Laguna, I.; Saez, A.; Gonzalez-Granado, J.M. Neutrophils and NETs in Pathophysiology and Treatment of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2025, 26, 7098. [Google Scholar] [CrossRef]
- Remigante, A.; Morabito, R. Cellular and Molecular Mechanisms in Oxidative Stress-Related Diseases 2.0/3.0. Int. J. Mol. Sci. 2023, 24, 16018. [Google Scholar] [CrossRef]
- Luceri, C.; Bigagli, E.; Agostiniani, S.; Giudici, F.; Zambonin, D.; Scaringi, S.; Ficari, F.; Lodovici, M.; Malentacchi, C. Analysis of Oxidative Stress-Related Markers in Crohn’s Disease Patients at Surgery and Correlations with Clinical Findings. Antioxidants 2019, 8, 378. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Wang, G.; Yuan, J.; Luo, J.; Ocansey, D.K.W.; Zhang, X.; Qian, H.; Xu, W.; Mao, F. Emerging role of protein modification in inflammatory bowel disease. J. Zhejiang Univ. Sci. B 2022, 23, 173–188. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Coda, A.R.D.; Trecca, M.I.; Lo Buglio, A.; Serviddio, G.; Vendemiale, G. Redox Imbalance in Inflammation: The Interplay of Oxidative and Reductive Stress. Antioxidants 2025, 14, 656. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Junior, J.I.; Vasconcelos, J.K.; Xavier, L.E.; Gomes, A.D.; Santos, J.C.; Campos, S.B.; Martins, A.S.; Goulart, M.O.; Moura, F.A. Antioxidant Therapy in Inflammatory Bowel Disease: A Systematic Review and a Meta-Analysis of Randomized Clinical Trials. Pharmaceuticals 2023, 16, 1374. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Wang, Y.; Li, N.; Yang, X.; Sun, X.; Tian, H.e.; Zhang, Y. Mechanism of Action and Therapeutic Implications of Nrf2/HO-1 in Inflammatory Bowel Disease. Antioxidants 2024, 13, 1012. [Google Scholar] [CrossRef] [PubMed]
- Tratenšek, A.; Locatelli, I.; Grabnar, I.; Drobne, D.; Vovk, T. Oxidative stress-related biomarkers as promising indicators of inflammatory bowel disease activity: A systematic review and meta-analysis. Redox Biol. 2024, 77, 103380. [Google Scholar] [CrossRef]
- Tratenšek, A.; Grabnar, I.; Drobne, D.; Vovk, T. Oxidative Stress-Related Biomarkers in Inflammatory Bowel Disease: Dual Tools for Remission Assessment and Prediction of Treatment Outcome. Antioxidants 2025, 14, 1183. [Google Scholar] [CrossRef]
- Alemany-Cosme, E.; Sáez-González, E.; Moret, I.; Mateos, B.; Iborra, M.; Nos, P.; Sandoval, J.; Beltrán, B. Oxidative Stress in the Pathogenesis of Crohn’s Disease and the Interconnection with Immunological Response, Microbiota, External Environmental Factors, and Epigenetics. Antioxidants 2021, 10, 64. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Bonay, M. Molecular Targets of Oxidative Stress: Focus on the Nrf2 Signaling Pathway in Health and Disease. Antioxidants 2024, 13, 262. [Google Scholar] [CrossRef]
- Hammad, M.; Raftari, M.; Cesário, R.; Salma, R.; Godoy, P.; Emami, S.N.; Haghdoost, S. Roles of Oxidative Stress and Nrf2 Signaling in Pathogenic and Non-Pathogenic Cells: A Possible General Mechanism of Resistance to Therapy. Antioxidants 2023, 12, 1371. [Google Scholar] [CrossRef] [PubMed]
- Tonolo, F.; Folda, A.; Scalcon, V.; Marin, O.; Bindoli, A.; Rigobello, M.P. Nrf2-Activating Bioactive Peptides Exert Anti-Inflammatory Activity through Inhibition of the NF-κB Pathway. Int. J. Mol. Sci. 2022, 23, 4382. [Google Scholar] [CrossRef]
- Liu, X.; Liang, F.; Song, W.; Diao, X.; Zhu, W.; Yang, J. Effect of Nrf2 signaling pathway on the improvement of intestinal epithelial barrier dysfunction by hyperbaric oxygen treatment after spinal cord injury. Cell Stress Chaperones 2021, 26, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Shen, L.; Yu, X.; Zhang, L.; Xu, K.; Xia, Y.; Zha, L.; Wu, J.; Luo, H. The role of Nrf2 in the pathogenesis and treatment of ulcerative colitis. Front. Immunol. 2023, 14, 1200111. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Huang, M.T.; Prawan, A.; Liu, Y.; Hao, X.; Yu, S.; Cheung, W.K.; Chan, J.Y.; Reddy, B.S.; Yang, C.S.; et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. 2008, 1, 187–191. [Google Scholar] [CrossRef]
- Piotrowska, M.; Swierczynski, M.; Fichna, J.; Piechota-Polanczyk, A. The Nrf2 in the pathophysiology of the intestine: Molecular mechanisms and therapeutic implications for inflammatory bowel diseases. Pharmacol. Res. 2021, 163, 105243. [Google Scholar] [CrossRef]
- Rybarczyk, A.; Majchrzak-Celińska, A.; Krajka-Kuźniak, V. Targeting Nrf2 Signaling Pathway in Cancer Prevention and Treatment: The Role of Cannabis Compounds. Antioxidants 2023, 12, 2052. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Riera-Ponsati, L.; Kauppinen, S.; Klitgaard, H.; Erler, J.T.; Hansen, S.N. Targeting the NRF2 pathway for disease modification in neurodegenerative diseases: Mechanisms and therapeutic implications. Front. Pharmacol. 2024, 15, 1437939. [Google Scholar] [CrossRef] [PubMed]
- Matsumaru, D.; Motohashi, H. The KEAP1-NRF2 System in Healthy Aging and Longevity. Antioxidants 2021, 10, 1929. [Google Scholar] [CrossRef]
- Li, B.; Wang, Y.; Jiang, X.; Du, H.; Shi, Y.; Xiu, M.; Liu, Y.; He, J. Natural products targeting Nrf2/ARE signaling pathway in the treatment of inflammatory bowel disease. Biomed. Pharmacother. 2023, 164, 114950. [Google Scholar] [CrossRef]
- Yu, X.; Li, X.; Xu, Y.; Li, Y.; Zhou, Y.; Zhang, J.; Guo, L. Resveratrol ameliorates ulcerative colitis by upregulating Nrf2/HO--1 pathway activity: Integrating animal experiments and network pharmacology. Mol. Med. Rep. 2024, 29, 77. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci. 2022, 291, 120111. [Google Scholar] [CrossRef]
- Zhang, D.D.; Chapman, E. The role of natural products in revealing NRF2 function. Nat. Prod. Rep. 2020, 37, 797–826. [Google Scholar] [CrossRef]
- Medoro, A.; Saso, L.; Scapagnini, G.; Davinelli, S. NRF2 signaling pathway and telomere length in aging and age-related diseases. Mol. Cell. Biochem. 2024, 479, 2597–2613. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef]
- Song, M.-Y.; Lee, D.-Y.; Chun, K.-S.; Kim, E.-H. The Role of NRF2/KEAP1 Signaling Pathway in Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [Google Scholar] [CrossRef]
- Crisman, E.; Duarte, P.; Dauden, E.; Cuadrado, A.; Rodríguez-Franco, M.I.; López, M.G.; León, R. KEAP1-NRF2 protein-protein interaction inhibitors: Design, pharmacological properties and therapeutic potential. Med. Res. Rev. 2023, 43, 237–287. [Google Scholar] [CrossRef]
- McCord, J.M.; Gao, B.; Hybertson, B.M. The Complex Genetic and Epigenetic Regulation of the Nrf2 Pathways: A Review. Antioxidants 2023, 12, 366. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Xiao, J.H. The Keap1-Nrf2 System: A Mediator between Oxidative Stress and Aging. Oxidative Med. Cell. Longev. 2021, 2021, 6635460. [Google Scholar] [CrossRef] [PubMed]
- Oskomić, M.; Tomić, A.; Barbarić, L.; Matić, A.; Kindl, D.C.; Matovina, M. KEAP1-NRF2 Interaction in Cancer: Competitive Interactors and Their Role in Carcinogenesis. Cancers 2025, 17, 447. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, H.; Li, S.; Liu, Z.; Wang, C. New insights into the mechanism of Keap1-Nrf2 interaction based on cancer-associated mutations. Life Sci. 2021, 282, 119791. [Google Scholar] [CrossRef]
- Sato, M.; Yaguchi, N.; Iijima, T.; Muramatsu, A.; Baird, L.; Suzuki, T.; Yamamoto, M. Sensor systems of KEAP1 uniquely detecting oxidative and electrophilic stresses separately In vivo. Redox Biol. 2024, 77, 103355. [Google Scholar] [CrossRef]
- Song, Y.; Qu, Y.; Mao, C.; Zhang, R.; Jiang, D.; Sun, X. Post-translational modifications of Keap1: The state of the art. Front. Cell Dev. Biol. 2023, 11, 1332049. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef]
- Li, L.; Liu, X.; Si, Z.; Wang, X. Epigenetic Mechanisms Governing Nrf2 Expression and Its Role in Ferroptosis. Biomedicines 2025, 13, 1913. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Warabi, E.; Mann, G.E. Stress Activated MAP Kinases and Cyclin-Dependent Kinase 5 Mediate Nuclear Translocation of Nrf2 via Hsp90α-Pin1-Dynein Motor Transport Machinery. Antioxidants 2023, 12, 274. [Google Scholar] [CrossRef] [PubMed]
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Johnston, L.J.; Wang, F.; Ma, X. Triggers for the Nrf2/ARE Signaling Pathway and Its Nutritional Regulation: Potential Therapeutic Applications of Ulcerative Colitis. Int. J. Mol. Sci. 2021, 22, 11411. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef]
- Liu, N.; Liang, Y.; Wei, T.; Huang, X.; Zhang, T.; Tang, M. ROS-mediated NRF2/p-ERK1/2 signaling-involved mitophagy contributes to macrophages activation induced by CdTe quantum dots. Toxicology 2024, 505, 153825. [Google Scholar] [CrossRef]
- Jasek-Gajda, E.; Jurkowska, H.; Jasińska, M.; Lis, G.J. Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells. Antioxidants 2020, 9, 633. [Google Scholar] [CrossRef]
- Kopacz, A.; Kloska, D.; Klimczyk, D.; Kopec, M.; Jozkowicz, A.; Piechota-Polanczyk, A. Nrf2 Transcriptional Activity Governs Intestine Development. Int. J. Mol. Sci. 2022, 23, 6175. [Google Scholar] [CrossRef]
- Wen, Z.; Liu, W.; Li, X.; Chen, W.; Liu, Z.; Wen, J.; Liu, Z. A Protective Role of the NRF2-Keap1 Pathway in Maintaining Intestinal Barrier Function. Oxidative Med. Cell. Longev. 2019, 2019, 1759149. [Google Scholar] [CrossRef]
- He, M.Y.; Zhang, P.; Shi, N.; Li, T.; Wang, J.; He, L.; Liang, Z.; Song, L.; Wang, G.; Yang, X. Nrf2 Is Involved in Hyperglycemia-induced Abnormal Lung Development through both Antioxidation-Dependent and Antioxidation-Independent Activation. Am. J. Respir. Cell Mol. Biol. 2023, 69, 197–209. [Google Scholar] [CrossRef]
- Fasipe, B.; Li, S.; Laher, I. Harnessing the cardiovascular benefits of exercise: Are Nrf2 activators useful? Sports Med. Health Sci. 2021, 3, 70–79. [Google Scholar] [CrossRef]
- Piechota-Polanczyk, A.; Mariwani, Z.; Fichna, J.; Polanczyk, A.; Jozkowicz, A. Chemical Inhibition of NRF2 Transcriptional Activity Influences Colon Function and Oestrogen Receptor Expression in Mice at Different Ages. Int. J. Mol. Sci. 2024, 25, 13647. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Shin, S.; Slocum, S.L.; Agoston, E.S.; Wakabayashi, J.; Kwak, M.K.; Misra, V.; Biswal, S.; Yamamoto, M.; Kensler, T.W. Regulation of notch1 signaling by nrf2: Implications for tissue regeneration. Sci. Signal 2010, 3, ra52. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; McCallum, M.L.; Kensler, T.W.; Wakabayashi, N. Constitutive Activation of Nrf2 in Mice Expands Enterogenesis in Small Intestine Through Negative Regulation of Math1. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 503–524. [Google Scholar] [CrossRef] [PubMed]
- Gasnier, M.; Lim, H.Y.G.; Barker, N. Chapter Ten—Role of Wnt signaling in the maintenance and regeneration of the intestinal epithelium. In Current Topics in Developmental Biology; Yamaguchi, T.P., Willert, K., Eds.; Academic Press: Cambridge, MA, USA, 2023; Volume 153, pp. 281–326. [Google Scholar]
- Gregorieff, A.; Clevers, H. Wnt signaling in the intestinal epithelium: From endoderm to cancer. Genes Dev. 2005, 19, 877–890. [Google Scholar] [CrossRef]
- Moonwiriyakit, A.; Pathomthongtaweechai, N.; Steinhagen, P.R.; Chantawichitwong, P.; Satianrapapong, W.; Pongkorpsakol, P. Tight junctions: From molecules to gastrointestinal diseases. Tissue Barriers 2023, 11, 2077620. [Google Scholar] [CrossRef]
- Xu, B.; Zhuang, Y.; Zhang, Y.; Liu, S.; Fan, R.; Jiang, W. Apigenin Alleviates Intestinal Ischemia/Reperfusion Injury via Upregulating Nrf2-Mediated Tight Junction Integrity. Mol. Nutr. Food Res. 2025, 69, e70043. [Google Scholar] [CrossRef]
- Ganapathy, A.S.; Saha, K.; Wang, A.; Arumugam, P.; Dharmaprakash, V.; Yochum, G.; Koltun, W.; Nighot, M.; Perdew, G.; Thompson, T.A.; et al. Alpha-tocopherylquinone differentially modulates claudins to enhance intestinal epithelial tight junction barrier via AhR and Nrf2 pathways. Cell Rep. 2023, 42, 112705. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Li, G.; Wu, J.; Wu, X.; Wang, G.; Gu, G.; Ren, H.; Hong, Z.; Li, J. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018, 9, 403. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Sekhar, K.R.; Hale, A.B.; Channon, K.M.; Farrugia, G.; Freeman, M.L.; Gangula, P.R. Loss of NRF2 impairs gastric nitrergic stimulation and function. Free Radic. Biol. Med. 2011, 51, 619–625. [Google Scholar] [CrossRef]
- Luchkova, A.; Mata, A.; Cadenas, S. Nrf2 as a regulator of energy metabolism and mitochondrial function. FEBS Lett. 2024, 598, 2092–2105. [Google Scholar] [CrossRef]
- Chen, H.; Hu, Y.; Fang, Y.; Djukic, Z.; Yamamoto, M.; Shaheen, N.J.; Orlando, R.C.; Chen, X. Nrf2 deficiency impairs the barrier function of mouse oesophageal epithelium. Gut 2014, 63, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Averill-Bates, D. Reactive oxygen species and cell signaling. Review. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2024, 1871, 119573. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wu, Q.; Lu, F.; Lei, J.; Zhou, Y.; Liu, Y.; Zhu, N.; Yu, Y.; Ning, Z.; She, T.; et al. Nrf2 signaling pathway: Current status and potential therapeutic targetable role in human cancers. Front. Oncol. 2023, 13, 1184079. [Google Scholar] [CrossRef]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.K.; Yan, M. Dissecting the Crosstalk Between Nrf2 and NF-κB Response Pathways in Drug-Induced Toxicity. Front. Cell Dev. Biol. 2021, 9, 809952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, T.; Liu, J.; Sun, J.; Niu, J.; Ren, D.; Ma, Y.; He, Y.; Liu, S.; Wang, Q. The Regulation of the NF-κB p65 and Nrf2/HO-1 Signaling Pathways by Fucoxanthin in Human THP-1 Monocyte Macrophages Under a Lipopolysaccharide-Induced Inflammation Model. Foods 2025, 14, 1746. [Google Scholar] [CrossRef]
- Han, H.; Gao, Y.; Chen, B.; Xu, H.; Shi, C.; Wang, X.; Liang, Y.; Wu, Z.; Wang, Z.; Bai, Y.; et al. Nrf2 inhibits M1 macrophage polarization to ameliorate renal ischemia-reperfusion injury through antagonizing NF-κB signaling. Int. Immunopharmacol. 2024, 143, 113310. [Google Scholar] [CrossRef]
- Kim, M.-J.; Jeon, J.-H. Recent Advances in Understanding Nrf2 Agonism and Its Potential Clinical Application to Metabolic and Inflammatory Diseases. Int. J. Mol. Sci. 2022, 23, 2846. [Google Scholar] [CrossRef]
- Rahman, M.S.; Alam, M.B.; Kim, Y.K.; Madina, M.H.; Fliss, I.; Lee, S.H.; Yoo, J.C. Activation of Nrf2/HO-1 by Peptide YD1 Attenuates Inflammatory Symptoms through Suppression of TLR4/MYyD88/NF-κB Signaling Cascade. Int. J. Mol. Sci. 2021, 22, 5161. [Google Scholar] [CrossRef]
- Laurindo, L.F.; de Maio, M.C.; Minniti, G.; de Góes Corrêa, N.; Barbalho, S.M.; Quesada, K.; Guiguer, E.L.; Sloan, K.P.; Detregiachi, C.R.P.; Araújo, A.C.; et al. Effects of Medicinal Plants and Phytochemicals in Nrf2 Pathways during Inflammatory Bowel Diseases and Related Colorectal Cancer: A Comprehensive Review. Metabolites 2023, 13, 243. [Google Scholar] [CrossRef]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Dunleavy, K.A.; Raffals, L.E.; Camilleri, M. Intestinal Barrier Dysfunction in Inflammatory Bowel Disease: Underpinning Pathogenesis and Therapeutics. Dig. Dis. Sci. 2023, 68, 4306–4320. [Google Scholar] [CrossRef] [PubMed]
- Lechuga, S.; Braga-Neto, M.B.; Naydenov, N.G.; Rieder, F.; Ivanov, A.I. Understanding disruption of the gut barrier during inflammation: Should we abandon traditional epithelial cell lines and switch to intestinal organoids? Front. Immunol. 2023, 14, 1108289. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J. Honokiol Ameliorates DSS-Induced Mouse Colitis by Inhibiting Inflammation and Oxidative Stress and Improving the Intestinal Barrier. Oxidative Med. Cell. Longev. 2022, 2022, 1755608. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Yang, L.; Zhang, X.; Ji, P.; Hua, Y.; Wei, Y. Huang-Lian-Jie-Du Decoction Ameliorates Acute Ulcerative Colitis in Mice via Regulating NF-κB and Nrf2 Signaling Pathways and Enhancing Intestinal Barrier Function. Front. Pharmacol. 2019, 10, 1354. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Q.; Li, Y.; Zhang, Y.; Wu, Y. MitoQ Modulates Lipopolysaccharide-Induced Intestinal Barrier Dysfunction via Regulating Nrf2 Signaling. Mediat. Inflamm. 2020, 2020, 3276148. [Google Scholar] [CrossRef]
- Liu, Y.; Bao, Z.; Xu, X.; Chao, H.; Lin, C.; Li, Z.; Liu, Y.; Wang, X.; You, Y.; Liu, N.; et al. Extracellular Signal-Regulated Kinase/Nuclear Factor-Erythroid2-like2/Heme Oxygenase-1 Pathway-Mediated Mitophagy Alleviates Traumatic Brain Injury-Induced Intestinal Mucosa Damage and Epithelial Barrier Dysfunction. J. Neurotrauma 2017, 34, 2119–2131. [Google Scholar] [CrossRef]
- Lau, W.L.; Liu, S.M.; Pahlevan, S.; Yuan, J.; Khazaeli, M.; Ni, Z.; Chan, J.Y.; Vaziri, N.D. Role of Nrf2 dysfunction in uremia-associated intestinal inflammation and epithelial barrier disruption. Dig. Dis. Sci. 2015, 60, 1215–1222. [Google Scholar] [CrossRef]
- Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 7618. [Google Scholar] [CrossRef]
- Petit, C.; Rozières, A.; Boschetti, G.; Viret, C.; Faure, M.; Nancey, S.; Duclaux-Loras, R. Advances in Understanding Intestinal Homeostasis: Lessons from Inflammatory Bowel Disease and Monogenic Intestinal Disorder Pathogenesis. Int. J. Mol. Sci. 2025, 26, 6133. [Google Scholar] [CrossRef]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J. Immunol. 2012, 188, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Pant, A.; Dasgupta, D.; Tripathi, A.; Pyaram, K. Beyond Antioxidation: Keap1–Nrf2 in the Development and Effector Functions of Adaptive Immune Cells. Immunohorizons 2023, 7, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Dasgupta, D.; Dahabieh, M.S.; Griffard-Smith, R.; Pant, A.; Bugbee, A.; Yellapu, N.K.; Burt, E.; Choi, B.H.Y.; Ellis, A.E.; et al. Nrf2 drives activation-driven expansion of CD4+T cells by modulating glucose and glutamine metabolism. Cell Rep. 2025, 44, 116177. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int. J. Mol. Sci. 2020, 21, 8011. [Google Scholar] [CrossRef] [PubMed]
- Turley, A.E.; Zagorski, J.W.; Rockwell, C.E. The Nrf2 activator tBHQ inhibits T cell activation of primary human CD4 T cells. Cytokine 2015, 71, 289–295. [Google Scholar] [CrossRef]
- Zagorski, J.W.; Turley, A.E.; Freeborn, R.A.; VanDenBerg, K.R.; Dover, H.E.; Kardell, B.R.; Liby, K.T.; Rockwell, C.E. Differential effects of the Nrf2 activators tBHQ and CDDO-Im on the early events of T cell activation. Biochem. Pharmacol. 2018, 147, 67–76. [Google Scholar] [CrossRef]
- Lin, X.; Tawch, S.; Wong, H.T.; Roy, S.; Gaudino, S.; Castillo, P.; Elsegeiny, W.; Wakabayashi, N.; Oury, T.D.; Pociask, D.; et al. Nrf2 through Aryl Hydrocarbon Receptor Regulates IL-22 Response in CD4(+) T Cells. J. Immunol. 2021, 206, 1540–1548. [Google Scholar] [CrossRef]
- Casili, G.; Cordaro, M.; Impellizzeri, D.; Bruschetta, G.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Dimethyl Fumarate Reduces Inflammatory Responses in Experimental Colitis. J. Crohns Colitis 2016, 10, 472–483. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, W.; Zhang, X.; Lu, P.; Du, Q.; Tao, L.; Ding, Y.; Wang, Y.; Hu, R. Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem. Pharmacol. 2016, 112, 37–49. [Google Scholar] [CrossRef]
- Li, S.; Takasu, C.; Lau, H.; Robles, L.; Vo, K.; Farzaneh, T.; Vaziri, N.D.; Stamos, M.J.; Ichii, H. Dimethyl Fumarate Alleviates Dextran Sulfate Sodium-Induced Colitis, through the Activation of Nrf2-Mediated Antioxidant and Anti-inflammatory Pathways. Antioxidants 2020, 9, 354. [Google Scholar] [CrossRef]
- Han, J.; Ma, S.; Gong, H.; Liu, S.; Lei, L.; Hu, B.; Xu, Y.; Liu, H.; Wu, D. Inhibition of Acute Graft-versus-Host Disease with Retention of Graft-versus-Tumor Effects by Dimethyl Fumarate. Front. Immunol. 2017, 8, 1605. [Google Scholar] [CrossRef]
- Sha, L.K.; Sha, W.; Kuchler, L.; Daiber, A.; Giegerich, A.K.; Weigert, A.; Knape, T.; Snodgrass, R.; Schröder, K.; Brandes, R.P.; et al. Loss of Nrf2 in bone marrow-derived macrophages impairs antigen-driven CD8(+) T cell function by limiting GSH and Cys availability. Free Radic. Biol. Med. 2015, 83, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Bursley, J.K.; Rockwell, C.E. Nrf2-dependent and -independent effects of tBHQ in activated murine B cells. Food Chem. Toxicol. 2020, 145, 111595. [Google Scholar] [CrossRef]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef]
- Beury, D.W.; Carter, K.A.; Nelson, C.; Sinha, P.; Hanson, E.; Nyandjo, M.; Fitzgerald, P.J.; Majeed, A.; Wali, N.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol. 2016, 196, 3470–3478. [Google Scholar] [CrossRef]
- Gerstgrasser, A.; Melhem, H.; Leonardi, I.; Atrott, K.; Schäfer, M.; Werner, S.; Rogler, G.; Frey-Wagner, I. Cell-specific Activation of the Nrf2 Antioxidant Pathway Increases Mucosal Inflammation in Acute but Not in Chronic Colitis. J. Crohns Colitis 2017, 11, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Arisawa, T.; Tahara, T.; Shibata, T.; Nagasaka, M.; Nakamura, M.; Kamiya, Y.; Fujita, H.; Yoshioka, D.; Okubo, M.; Sakata, M.; et al. Nrf2 gene promoter polymorphism is associated with ulcerative colitis in a Japanese population. Hepatogastroenterology 2008, 55, 394–397. [Google Scholar] [PubMed]
- Tang, C.-T.; Liu, Z.-d.; Wang, P.; Zeng, C.-Y.; Chen, Y.-X. Lipopolysaccharide-regulated RNF31/NRF2 axis in colonic epithelial cells mediates homeostasis of the intestinal barrier in ulcerative colitis. Cell. Signal. 2024, 124, 111480. [Google Scholar] [CrossRef] [PubMed]
- Sabzevary-Ghahfarokhi, M.; Shohan, M.; Shirzad, H.; Rahimian, G.; Soltani, A.; Ghatreh-Samani, M.; Deris, F.; Bagheri, N.; Shafigh, M.; Tahmasbi, K. The regulatory role of Nrf2 in antioxidants phase2 enzymes and IL-17A expression in patients with ulcerative colitis. Pathol. Res. Pract. 2018, 214, 1149–1155. [Google Scholar] [CrossRef]
- Kruse, M.-L.; Friedrich, M.; Arlt, A.; Röcken, C.; Egberts, J.-H.; Sebens, S.; Schäfer, H. Colonic Lamina Propria Inflammatory Cells from Patients with IBD Induce the Nuclear Factor-E2 Related Factor-2 Thereby Leading to Greater Proteasome Activity and Apoptosis Protection in Human Colonocytes. Inflamm. Bowel Dis. 2016, 22, 2593–2606. [Google Scholar] [CrossRef] [PubMed]
- Stachel, I.; Geismann, C.; Aden, K.; Deisinger, F.; Rosenstiel, P.; Schreiber, S.; Sebens, S.; Arlt, A.; Schäfer, H. Modulation of nuclear factor E2-related factor-2 (Nrf2) activation by the stress response gene immediate early response-3 (IER3) in colonic epithelial cells: A novel mechanism of cellular adaption to inflammatory stress. J. Biol. Chem. 2014, 289, 1917–1929. [Google Scholar] [CrossRef]
- Zuo, L.; Li, J.; Ge, S.; Ge, Y.; Shen, M.; Wang, Y.; Zhou, C.; Wu, R.; Hu, J. Bryostatin-1 ameliorated experimental colitis in Il-10(-/-) Mice by protecting the intestinal barrier and limiting immune dysfunction. J. Cell Mol. Med. 2019, 23, 5588–5599. [Google Scholar] [CrossRef]
- Wang, J.; He, M.; Yang, M.; Ai, X. Gut microbiota as a key regulator of intestinal mucosal immunity. Life Sci. 2024, 345, 122612. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; Kandalgaonkar, M.R.; Golonka, R.M.; Yeoh, B.S.; Vijay-Kumar, M.; Saha, P. Crosstalk between Gut Microbiota and Host Immunity: Impact on Inflammation and Immunotherapy. Biomedicines 2023, 11, 294. [Google Scholar] [CrossRef]
- Ning, L.; Zhou, Y.-L.; Sun, H.; Zhang, Y.; Shen, C.; Wang, Z.; Xuan, B.; Zhao, Y.; Ma, Y.; Yan, Y.; et al. Microbiome and metabolome features in inflammatory bowel disease via multi-omics integration analyses across cohorts. Nat. Commun. 2023, 14, 7135. [Google Scholar] [CrossRef]
- Garvey, M. Intestinal Dysbiosis: Microbial Imbalance Impacts on Colorectal Cancer Initiation, Progression and Disease Mitigation. Biomedicines 2024, 12, 740. [Google Scholar] [CrossRef]
- Giambra, V.; Pagliari, D.; Rio, P.; Totti, B.; Di Nunzio, C.; Bosi, A.; Giaroni, C.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Gut Microbiota, Inflammatory Bowel Disease, and Cancer: The Role of Guardians of Innate Immunity. Cells 2023, 12, 2654. [Google Scholar] [CrossRef]
- Santana, P.T.; Rosas, S.L.B.; Ribeiro, B.E.; Marinho, Y.; de Souza, H.S.P. Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 3464. [Google Scholar] [CrossRef]
- Quaglio, A.E.V.; Grillo, T.G.; De Oliveira, E.C.S.; Di Stasi, L.C.; Sassaki, L.Y. Gut microbiota, inflammatory bowel disease and colorectal cancer. World J. Gastroenterol. 2022, 28, 4053–4060. [Google Scholar] [CrossRef]
- Singh, R.; Chandrashekharappa, S.; Bodduluri, S.R.; Baby, B.V.; Hegde, B.; Kotla, N.G.; Hiwale, A.A.; Saiyed, T.; Patel, P.; Vijay-Kumar, M.; et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat. Commun. 2019, 10, 89. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Li, L.; Zhong, C.; Zhang, Y.; Yang, X.; Li, M.; Yang, C. The role of gut microbiota in intestinal disease: From an oxidative stress perspective. Front. Microbiol. 2024, 15, 1328324. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.; Sudhakaran Vasanthakumari, A.; Panwar, H.; Mallapa, R.H.; Duary, R.K.; Batish, V.K.; Grover, S. Amelioration of Colitis in Mouse Model by Exploring Antioxidative Potentials of an Indigenous Probiotic Strain of Lactobacillus fermentum Lf1. BioMed Res. Int. 2014, 2014, 206732. [Google Scholar] [CrossRef] [PubMed]
- Finamore, A.; Ambra, R.; Nobili, F.; Garaguso, I.; Raguzzini, A.; Serafini, M. Redox Role of Lactobacillus casei Shirota Against the Cellular Damage Induced by 2,2′-Azobis (2-Amidinopropane) Dihydrochloride-Induced Oxidative and Inflammatory Stress in Enterocytes-Like Epithelial Cells. Front. Immunol. 2018, 9, 1131. [Google Scholar] [CrossRef]
- Zhou, Q.; Wu, F.; Chen, S.; Cen, P.; Yang, Q.; Guan, J.; Cen, L.; Zhang, T.; Zhu, H.; Chen, Z. Lactobacillus reuteri improves function of the intestinal barrier in rats with acute liver failure through Nrf-2/HO-1 pathway. Nutrition 2022, 99–100, 111673. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Li, Y.; Sun, J.; Xu, H.; Wang, M.; Zuo, X.; Fu, Q.; Guo, Y.; Chen, Z.; Zhang, P.; et al. Saccharomyces boulardii Ameliorates Dextran Sulfate Sodium-Induced Ulcerative Colitis in Mice by Regulating NF-κB and Nrf2 Signaling Pathways. Oxidative Med. Cell. Longev. 2021, 2021, 1622375. [Google Scholar] [CrossRef]
- Ehrlich, A.M.; Pacheco, A.R.; Henrick, B.M.; Taft, D.; Xu, G.; Huda, M.N.; Mishchuk, D.; Goodson, M.L.; Slupsky, C.; Barile, D.; et al. Indole-3-lactic acid associated with Bifidobacterium-dominated microbiota significantly decreases inflammation in intestinal epithelial cells. BMC Microbiol. 2020, 20, 357. [Google Scholar] [CrossRef]
- Xu, C.; Qiao, L.; Ma, L.; Guo, Y.; Dou, X.; Yan, S.; Zhang, B.; Roman, A. Biogenic selenium nanoparticles synthesized by lactobacillus casei ATCC 393 alleviate intestinal epithelial barrier dysfunction caused by oxidative stress via nrf2 signaling-mediated mitochondrial pathway. Int. J. Nanomed. 2019, 14, 4491–4502. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Liu, K.H.; Owens, J.A.; Hunter-Chang, S.; Camacho, M.C.; Eboka, R.U.; Chandrasekharan, B.; Baker, N.F.; Darby, T.M.; Robinson, B.S.; et al. Gut-Resident Lactobacilli Activate Hepatic Nrf2 and Protect Against Oxidative Liver Injury. Cell Metab. 2020, 31, 956–968. [Google Scholar] [CrossRef]
- Chen, Y.T.; Lin, Y.C.; Lin, J.S.; Yang, N.S.; Chen, M.J. Sugary Kefir Strain Lactobacillus mali APS1 Ameliorated Hepatic Steatosis by Regulation of SIRT-1/Nrf-2 and Gut Microbiota in Rats. Mol. Nutr. Food Res. 2018, 62, e1700903. [Google Scholar] [CrossRef]
- Martin, D.A.; Bolling, B.W. A review of the efficacy of dietary polyphenols in experimental models of inflammatory bowel diseases. Food Funct. 2015, 6, 1773–1786. [Google Scholar] [CrossRef]
- Hiebert, P.; Werner, S. Targeting NRF2 to promote epithelial repair. Biochem. Soc. Trans. 2023, 51, 101–111. [Google Scholar] [CrossRef]
- Glorieux, C.; Enríquez, C.; González, C.; Aguirre-Martínez, G.; Buc Calderon, P. The Multifaceted Roles of NRF2 in Cancer: Friend or Foe? Antioxidants 2024, 13, 70. [Google Scholar] [CrossRef]
- Hu, M.; Yuan, L.; Zhu, J. The Dual Role of NRF2 in Colorectal Cancer: Targeting NRF2 as a Potential Therapeutic Approach. J. Inflamm. Res. 2024, 17, 5985–6004. [Google Scholar] [CrossRef]
- O’Cathail, S.M.; Wu, C.-H.; Thomas, R.; Hawkins, M.A.; Maughan, T.S.; Lewis, A. NRF2 Mediates Therapeutic Resistance to Chemoradiation in Colorectal Cancer through a Metabolic Switch. Antioxidants 2021, 10, 1380. [Google Scholar] [CrossRef]
- Li, W.; Zhao, T.; Wu, D.; Li, J.; Wang, M.; Sun, Y.; Hou, S. Colorectal Cancer in Ulcerative Colitis: Mechanisms, Surveillance and Chemoprevention. Curr. Oncol. 2022, 29, 6091–6114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.W.; Harpaz, N.; Itzkowitz, S.H.; Parsons, R.E. Molecular mechanisms in colitis-associated colorectal cancer. Oncogenesis 2023, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Bardelčíková, A.; Šoltys, J.; Mojžiš, J. Oxidative Stress, Inflammation and Colorectal Cancer: An Overview. Antioxidants 2023, 12, 901. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, D.; Liu, Y.; Zhou, Y. Role of NRF2 in Colorectal Cancer Prevention and Treatment. Technol. Cancer Res. Treat. 2022, 21, 15330338221105736. [Google Scholar] [CrossRef]
- Pouremamali, F.; Pouremamali, A.; Dadashpour, M.; Soozangar, N.; Jeddi, F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Commun. Signal. 2022, 20, 100. [Google Scholar] [CrossRef]
- Garufi, A.; Pistritto, G.; D’Orazi, V.; Cirone, M.; D’Orazi, G. The Impact of NRF2 Inhibition on Drug-Induced Colon Cancer Cell Death and p53 Activity: A Pilot Study. Biomolecules 2022, 12, 461. [Google Scholar] [CrossRef]
- Song, C.H.; Kim, N.; Nam, R.H.; Choi, S.I.; Kang, C.; Jang, J.Y.; Nho, H.; Shin, E.; Lee, H.N. Nuclear Factor Erythroid 2-related Factor 2 Knockout Suppresses the Development of Aggressive Colorectal Cancer Formation Induced by Azoxymethane/Dextran Sulfate Sodium-Treatment in Female Mice. J. Cancer Prev. 2021, 26, 41–53. [Google Scholar] [CrossRef]
- Yokoo, Y.; Kijima, A.; Ishii, Y.; Takasu, S.; Tsuchiya, T.; Umemura, T. Effects of Nrf2 silencing on oxidative stress-associated intestinal carcinogenesis in mice. Cancer Med. 2016, 5, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, W.I.; Bae, J.H.; Cho, M.K.; Lee, S.H.; Nam, H.S.; Choi, I.H.; Cho, S.W. Overexpression of Nrf2 promotes colon cancer progression via ERK and AKT signaling pathways. Ann. Surg. Treat. Res. 2020, 98, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.M.; Lin, P.L.; Wu, D.W.; Wang, L.; Huang, C.C.; Lee, H. PSMD4 is a novel therapeutic target in chemoresistant colorectal cancer activated by cytoplasmic localization of Nrf2. Oncotarget 2018, 9, 26342–26352. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Kang, H.K.; Chang, W.Y.; Park, I.C.; Keum, Y.S.; Surh, Y.J.; Hyun, J.W. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: Involvement of TET-dependent DNA demethylation. Cell Death Dis. 2014, 5, e1183. [Google Scholar] [CrossRef]
- Taheri, Z.; Asadzadeh Aghdaei, H.; Irani, S.; Modarressi, M.H.; Zahra, N. Evaluation of the Epigenetic Demethylation of NRF2, a Master Transcription Factor for Antioxidant Enzymes, in Colorectal Cancer. Rep. Biochem. Mol. Biol. 2020, 9, 33–39. [Google Scholar] [CrossRef]
- Ji, L.; Wei, Y.; Jiang, T.; Wang, S. Correlation of Nrf2, NQO1, MRP1, cmyc and p53 in colorectal cancer and their relationships to clinicopathologic features and survival. Int. J. Clin. Exp. Pathol. 2014, 7, 1124–1131. [Google Scholar]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef]
- Rieder, F.; Mukherjee, P.K.; Massey, W.J.; Wang, Y.; Fiocchi, C. Fibrosis in IBD: From pathogenesis to therapeutic targets. Gut 2024, 73, 854–866. [Google Scholar] [CrossRef]
- Fousekis, F.S.; Mpakogiannis, K.; Mastorogianni, I.N.; Lianos, G.D.; Christodoulou, D.K.; Katsanos, K.H. Intestinal Fibrosis in Crohn’s Disease: Pathophysiology, Diagnosis, and New Therapeutic Targets. J. Clin. Med. 2025, 14, 4060. [Google Scholar] [CrossRef]
- Sputa-Grzegrzolka, P.; Socha-Banasiak, A.; Dziegiel, P.; Kempisty, B. Molecular Basis of Chronic Intestinal Wall Fibrosis in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2025, 26, 5754. [Google Scholar] [CrossRef]
- Xin, S.; Liu, X.; He, C.; Gao, H.; Wang, B.; Hua, R.; Gao, L.; Shang, H.; Sun, F.; Xu, J. Inflammation accelerating intestinal fibrosis: From mechanism to clinic. Eur. J. Med. Res. 2024, 29, 335. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Huda, M.N.; Shin, Y.; Han, S.; Akter, S.; Kang, I.; Ha, J.; Choe, W.; Choi, T.G.; Kim, S.S. Correlation between Oxidative Stress and Transforming Growth Factor-Beta in Cancers. Int. J. Mol. Sci. 2021, 22, 13181. [Google Scholar] [CrossRef]
- Guan, Y.; Tan, Y.; Liu, W.; Yang, J.; Wang, D.; Pan, D.; Sun, Y.; Zheng, C. NF-E2-Related Factor 2 Suppresses Intestinal Fibrosis by Inhibiting Reactive Oxygen Species-Dependent TGF-β1/SMADs Pathway. Dig. Dis. Sci. 2018, 63, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Pompili, S.; Sferra, R.; Gaudio, E.; Viscido, A.; Frieri, G.; Vetuschi, A.; Latella, G. Can Nrf2 Modulate the Development of Intestinal Fibrosis and Cancer in Inflammatory Bowel Disease? Int. J. Mol. Sci. 2019, 20, 4061. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowski, G.; Pasternak, G.; Aebisher, D.; Dynarowicz, K.; Myśliwiec, A.; Bartusik-Aebisher, D.; Sosna, B.; Cieślar, G.; Kawczyk-Krupka, A.; Filip, R. An Analysis of the Content of Metalloproteinases in the Intestinal Wall of Patients with Crohn’s Disease. Life 2023, 13, 2013. [Google Scholar] [CrossRef]
- Lee, S.H.; Sohn, D.H.; Jin, X.Y.; Kim, S.W.; Choi, S.C.; Seo, G.S. 2′,4′,6′-Tris(methoxymethoxy) chalcone protects against trinitrobenzene sulfonic acid-induced colitis and blocks tumor necrosis factor-α-induced intestinal epithelial inflammation via heme oxygenase 1-dependent and independent pathways. Biochem. Pharmacol. 2007, 74, 870–880. [Google Scholar] [CrossRef]
- Luther, J.; Gala, M.; Patel, S.J.; Dave, M.; Borren, N.; Xavier, R.J.; Ananthakrishnan, A.N. Loss of Response to Anti-Tumor Necrosis Factor Alpha Therapy in Crohn’s Disease Is Not Associated with Emergence of Novel Inflammatory Pathways. Dig. Dis. Sci. 2018, 63, 738–745. [Google Scholar] [CrossRef]
- Barberio, B.; D’Incà, R.; Facchin, S.; Dalla Gasperina, M.; Fohom Tagne, C.A.; Cardin, R.; Ghisa, M.; Lorenzon, G.; Marinelli, C.; Savarino, E.V.; et al. Matrix Metalloproteinase 3 Predicts Therapeutic Response in Inflammatory Bowel Disease Patients Treated with Infliximab. Inflamm. Bowel Dis. 2020, 26, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Muneer, P.M.A. Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death. J. Mol. Med. 2019, 97, 1627–1641. [Google Scholar] [CrossRef] [PubMed]
- Geertsema, S.; Bourgonje, A.R.; Fagundes, R.R.; Gacesa, R.; Weersma, R.K.; van Goor, H.; Mann, G.E.; Dijkstra, G.; Faber, K.N. The NRF2/Keap1 pathway as a therapeutic target in inflammatory bowel disease. Trends Mol. Med. 2023, 29, 830–842. [Google Scholar] [CrossRef]
- Ma, Q.-S.; Chen, Q.-L.; Wu, G.-P.; Yao, Y.-W.; Fan, Y.-X.; Linghu, K.-G.; Chen, J.-M.; Xiong, W.; Yu, H. Sigesbeckia pubescens makino alleviates ulcerative colitis in mice by modulating the Nrf2/Keap1 pathway. Front. Pharmacol. 2025, 16, 1588525. [Google Scholar] [CrossRef] [PubMed]
- Aslam, N.; Lo, S.W.; Sikafi, R.; Barnes, T.; Segal, J.; Smith, P.J.; Limdi, J.K. A review of the therapeutic management of ulcerative colitis. Ther. Adv. Gastroenterol. 2022, 15, 17562848221138160. [Google Scholar] [CrossRef]
- Kang, S.; Kim, W.; Jeong, S.; Lee, Y.; Nam, J.; Lee, S.; Jung, Y. Oxidized 5-aminosalicylic acid activates Nrf2-HO-1 pathway by covalently binding to Keap1: Implication in anti-inflammatory actions of 5-aminosalicylic acid. Free Radic. Biol. Med. 2017, 108, 715–724. [Google Scholar] [CrossRef]
- El-Baz, A.M.; Khodir, A.E.; Adel El-Sokkary, M.M.; Shata, A. The protective effect of Lactobacillus versus 5-aminosalicylic acid in ulcerative colitis model by modulation of gut microbiota and Nrf2/Ho-1 pathway. Life Sci. 2020, 256, 117927. [Google Scholar] [CrossRef]
- Wu, W.; Wang, S.; Liu, Q.; Shan, T.; Wang, Y. Metformin Protects against LPS-Induced Intestinal Barrier Dysfunction by Activating AMPK Pathway. Mol. Pharm. 2018, 15, 3272–3284. [Google Scholar] [CrossRef]
- Lu, M.C.; Ji, J.A.; Jiang, Y.L.; Chen, Z.Y.; Yuan, Z.W.; You, Q.D.; Jiang, Z.Y. An inhibitor of the Keap1-Nrf2 protein-protein interaction protects NCM460 colonic cells and alleviates experimental colitis. Sci. Rep. 2016, 6, 26585. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, F.; Lu, S.; Ren, L.; Bian, S.; Liu, M.; Zhao, D.; Wang, S.; Wang, J. Ginseng root extract attenuates inflammation by inhibiting the MAPK/NF-κB signaling pathway and activating autophagy and p62-Nrf2-Keap1 signaling in vitro and in vivo. J. Ethnopharmacol. 2022, 283, 114739. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Xue, K.; Liu, J.; Habotta, O.A.; Hu, L.; Abdel Moneim, A.E. Anticolitic Effect of Berberine in Rat Experimental Model: Impact of PGE2/p38 MAPK Pathways. Mediat. Inflamm. 2020, 2020, 9419085. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Hu, L.; Chen, Y. Rutaecarpine may improve neuronal injury, inhibits apoptosis, inflammation and oxidative stress by regulating the expression of ERK1/2 and Nrf2/HO-1 pathway in rats with cerebral ischemia-reperfusion injury. Drug Des. Dev. Ther. 2019, 13, 2923–2931. [Google Scholar] [CrossRef]
- Chen, G.; Yang, Y.; Liu, M.; Teng, Z.; Ye, J.; Xu, Y.; Cai, X.; Cheng, X.; Yang, J.; Hu, C.; et al. Banxia xiexin decoction protects against dextran sulfate sodium-induced chronic ulcerative colitis in mice. J. Ethnopharmacol. 2015, 166, 149–156. [Google Scholar] [CrossRef]
- Pan, X.; Luo, L.; Wang, M.; Yu, H.; Zheng, Z. Feasibility of galectin-3 as a diagnostic biomarker and therapeutic target for inflammatory bowel disease. Eur. J. Pharmacol. 2025, 1005, 178041. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).